

Abstract
A critical feature of obesity is enhanced insulin secretion from pancreatic β-cells, enabling the majority of individuals to maintain glycaemic control despite adiposity and insulin resistance. Surprisingly, the factors coordinating this adaptive β-cell response with adiposity have not been delineated. Here we show that fatty acid binding protein 4 (FABP4/aP2) is an adipokine released from adipocytes under obesogenic conditions, such as hypoxia, to augment insulin secretion. The insulinotropic action of FABP4 was identified using an in vitro system that recapitulates adipocyte to β-cell endocrine signalling, with glucose-stimulated insulin secretion (GSIS) as a functional readout, coupled with quantitative proteomics. Exogenous FABP4 potentiated GSIS in vitro and in vivo, and circulating FABP4 levels correlated with GSIS in humans. Insulin inhibited FABP4 release from adipocytes in vitro, in mice and in humans, consistent with feedback regulation. These data suggest that FABP4 and insulin form an endocrine loop coordinating the β-cell response to obesity.
Keywords: Obesity, Adipokine, Adipocyte, Beta-cell, Insulin secretion, FABP4
Abbreviations: T2D, type 2 diabetes; NEFA, non-esterified fatty acid; GSIS, glucose-stimulated insulin secretion; SILAC, stable-isotope labelling by amino acids in cell culture; ELISA, enzyme-linked immunosorbant assay; BMI, body mass index; cAMP, cyclic-AMP; IBMX, 3-Isobutyl-1-methylxanthine
Graphical abstract
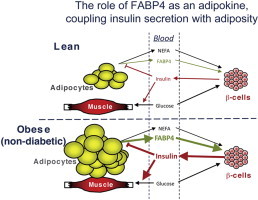
1. Introduction
The current global obesity epidemic is associated with a cluster of diseases including type 2 diabetes (T2D) [1,2], cardiovascular disease [3,4] and cancer [5,6]. Obesity is often accompanied by insulin resistance and enhanced insulin secretion from pancreatic β-cells [7,8]. If GSIS remains tightly coupled to insulin resistance, normal glycaemic control is maintained. In contrast, failure of GSIS to compensate for insulin resistance leads to hyperglycaemia and in some cases T2D [9]. The molecular mechanisms that coordinate enhanced GSIS with increased adiposity to maintain glycaemic control in the pre-diabetic state are not understood.
Blood glucose, although a major determinant of insulin secretion, appears largely uncoupled from the pronounced enhancement of insulin secretion in obese mice [10] and humans [8] that display blood glucose levels within the normal range. Non-esterified fatty acids (NEFAs) potentiate GSIS [11], yet increases in adiposity often occur independently from changes in plasma NEFA [12] suggesting that NEFA are not a common driver of compensatory insulin secretion. Adipokines regulate β-cell function, however the two most well characterised, leptin and adiponectin, do not adequately explain enhanced insulin secretion during obesity. Leptin, which increases with adiposity [13], inhibits insulin release [14] while adiponectin, a potentiator of GSIS [15,16], decreases with obesity [17]. The adipose enriched, secreted enzyme eNampt/visfatin augments GSIS in mice [18], although only subtle alterations in serum concentrations accompany obesity and correlate negatively with insulin secretion in humans [19]. In the current study, we sought to address the hypothesis that another adipokine plays an integral role coupling β-cell function with adiposity, thereby facilitating enhanced insulin secretion during obesity.
2. Materials and methods
2.1. Cell culture and islet isolation
Cell culture reagents were from Life Technologies unless otherwise indicated. 3T3-L1 fibroblasts were differentiated into adipocytes as described [20] and used for experiments 8 days post-differentiation. For signalling experiments, 3T3-L1 adipocytes were stimulated with β-3 adrenergic agonists CL316243 and BRL37344 (Sigma) at 0.1 μg/ml in PBS, or 3-Isobutyl-1-methylxanthine (IBMX; Sigma) at 500 μmol/l in 0.1% DMSO. Human insulin (Actrapid; Novo Nordisk) was diluted in PBS. Islet isolation, culture and insulin secretion experiments were performed as described [21].
2.2. Conditioned media experiments
We conditioned serum-free islet media (RPMI1640 containing penicillin, streptomycin and l-glutamine) by culturing with 3T3-L1 adipocytes for 18 hours under normoxic (21% O2, 5% CO2, 74% N2) or hypoxic (1% O2, 5% CO2, 94% N2) atmospheres, followed by 0.45 μm filtration to remove detached cells. From this conditioned media, a >10 kDa protein fraction was dialysed against fresh islet media using centrifugal filtration (Amicon/Merck Millipore), then reconstituted at a 3× concentration (i.e. 33% of starting volume of conditioned media) in fresh islet media containing 10% foetal calf serum, before being used to culture isolated mouse islets for 24 h. Insulin secretion was subsequently assessed for 1 h. Replicate insulin secretion data were averaged to give a single data point (n) for each batch of conditioned media.
2.3. Proteomics
Stable-isotope labelling by amino acids in cell culture (SILAC) [22] labelling of 3T3-L1 fibroblasts was performed as described [23], using “heavy” amino acid isotopes arginine U-13C6 U-15N4 (CNLM-539) and lysine U-13C6 U-15N2 (CNLM-291) from Cambridge Isotope Laboratories, and non-labelled “light” equivalents from Sigma. To reduce amino acid conversion to proline [24], arginine was used at 0.021 g/L, and lysine at 0.0365 g/L. Differentiated labelled 3T3-L1 adipocytes were used to condition media, with “light” cells cultured under normoxia and “heavy” cells under hypoxia. Conditioned media were collected and mixed in a 1:1 ratio to reduce subsequent inter-sample handling variability. Secreted proteins were enriched, precipitated, resolved by SDS-PAGE and subjected to in-gel trypsin digestion, as described [25]. Resulting peptides were subjected to liquid chromatography and tandem mass spectrometry using a Waters Ultima instrument at the Bioanalytical Mass Spectrometry Facility at the University of New South Wales. Data were analysed using Mascot Distiller v2.3.1.0. The SwissProt reference database was queried using Mascot Server 2.2 to identify peptide spectra. Only proteins represented by >5 unique peptide spectra were retained for further analysis. Potential for secretion was predicted using SecretomeP2.0 [26].
2.4. Recombinant FABP4 production
Recombinant FABP4 (rFABP4) used in Figure 2A and B was from Cayman Chemicals. rFABP4 used in all other figures was produced as follows. Mouse Fabp4 (GenBank BC054426.1) was cloned into His-tag expression vector pDEST17, transformed into BL21 Escherichia coli, and 0.5 ocular density cultures induced to produce rFABP4 by 1 mmol/l IPTG treatment. After 6 h, cultures were DNAseI and lysozyme treated, before addition of 1% Triton X-100. Clarified lysates were applied to Co2+ coated agarose bead columns and washed. For in vivo rFABP4–linoleate experiments, 0.4 mmol/l linoleate was passed over the column, followed by washing, to enable coupling with rFABP4. rFABP4 was eluted with 300 mmol/l imidazole and dialysed against PBS. Size, identity and purity of rFABP4 were confirmed by SDS-PAGE with Western blotting and Coomassie blue (Sigma) protein staining (Figure S1A and B). Our rFABP4 preparations contain very low endotoxin levels <0.02 EU/μg protein (Lonza LAL assay).
Figure 2.
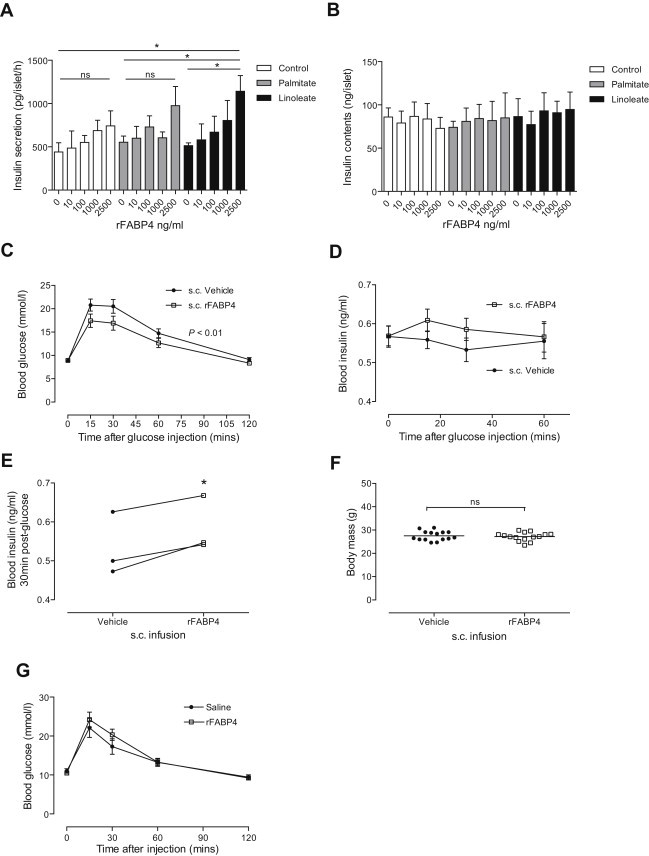
FABP4 potentiates glucose-stimulated insulin secretion in vitro and in vivo. A, B, Isolated mouse islets were exposed to recombinant FABP4 (rFABP4) for 24 h, with or without the fatty acids palmitate or linoleate, before 20 mM GSIS was assessed for 1 h (A), and islet insulin content assayed after lysis (B); n = 3–4. C–F, Osmotic mini-pumps containing rFABP4–linoleate (1:1) or saline were implanted (subcutaneously) in mice for 7 days providing continuous rFABP4 infusion (1.22 μg/h), before glucose tolerance (C), insulin secretion (D) and body mass (F) were assessed; n = 15 mice. To account for day-to-day variation in insulin assay results, mean insulin levels 30 min post-glucose from each experimental day were presented as paired treatment groups (n = 3) (E). G, Intraperitoneal co-injection of an rFABP4 bolus (80 μg), or PBS control, along with glucose (2 g/kg) revealed that acute administration of FABP4 does not alter glucose tolerance; n = 5 mice. Data presented as mean ± SEM. *P < 0.05 by unpaired (A) or paired (E) t-test. P < 0.01 by 2-way ANOVA for treatment effect indicated in C. See also Figure S1.
2.5. Preparation of fatty acids
Linoleate (Nu-Chek prep) was stored at −80 °C under nitrogen. Immediately before use, 0.4 mmol/l linoleate or sodium-palmitate (Sigma) were solubilised by coupling to 0.92% BSA (Sigma).
2.6. Mouse studies
2.6.1. Welfare
12-week old C57Bl/6J mice were purchased from Australian BioResources and housed in groups of 2–5, within a barrier facility with 12 h light/dark cycles. Environmental enrichment was provided and mice had free access to a standard lab diet [27] and water. Approval for mouse studies was issued to J.Cantley by the Garvan Institute/St.Vincent's Hospital Animal Ethics Committee.
2.6.2. Diet-induced obesity
10-week old male mice were fed either a standard lab diet (lean) or high-fat diet (obese) for 40 weeks. Diet composition was described previously [27].
2.6.3. rFABP4 treatment
Mice were treated with rFABP4–linoleate via i.p. injection (80 μg) or by subcutaneous implantation of osmotic mini-pumps (Alzet) providing a 0.5 μl/h flow rate and mean rFABP4 dose of 1.22 μg/h. Mini-pumps were loaded, primed and implanted (mid-scapular) into mice under gaseous general anaesthesia (1–4% isoflurane in oxygen), with ketoprofen (5 mg/kg) and marcaine (8 mg/kg) used as local analgesics.
2.6.4. Metabolic testing
Glucose tolerance testing (GTT) was performed after a 6 h fast (with free access to water), by intraperitoneal (i.p.) injection of sterile 20% glucose (2 g/kg). Insulin action was assessed by i.p. injection of insulin (0.75 U/kg) following a 6 h fast. Blood was obtained from the tail tip for glucose monitoring (Roche Accu-Chek Performa glucometer) and insulin assay (Crystal Chem ELISA). Circulating FABP4 was assayed by ELISA (Circulex/MBL).
2.7. Human studies
2.7.1. Participants
Sedentary, non-diabetic Caucasian participants were recruited (n = 17, 4 males). Mean age and body mass index (BMI) ± standard error (SEM) were 56 ± 2 years and 26 ± 1 kg/m2, respectively. Exclusion criteria were: weight instability (>2 kg change in the preceding 6 months), exercising more than 60 min per week, taking medications known to affect glucose homeostasis, or a personal history of type 2 diabetes or cardiovascular disease. The study protocol was approved by the Human Research and Ethics Committee at St Vincent's Hospital, Sydney. Participants provided informed written consent before commencement.
2.7.2. Metabolic testing
Participants attended the Clinical Research Facility at 8 am after a 12-h fast. Weight, height and blood pressure were measured and fasting blood samples drawn. Glucose tolerance testing (0.3 g/kg intravenous glucose dose; max 25 g) was performed, followed by a 2-h hyperinsulinaemic (60 mU/m2/min)–euglycemic (5.0 mmol/L) clamp. Human serum FABP4 levels were determined by ELISA (Biovendor) and insulin levels determined by RIA (Merck Millipore).
2.7.3. Body composition
Fat mass and fat-free mass were assessed by dual energy X-ray absorptiometry (DXA; Lunar DPX-Lunar Radiation, GE Healthcare).
2.8. Western blotting
Cell lysates were prepared in RIPA buffer and western blotting performed as described [21] using 12% polyacrylamide gels. Extracellular released proteins were detected by western blotting proteins from equal volumes of filtered conditioned media. Antibodies used were: FABP4 (Cell Signaling Technology), FABP5 (gift from Prof. Charles Mackay), Adiponectin (Merck Millipore or Sigma), complement C3 (MP Biomedicals), 14-3-3β (Santa Cruz Biotechnology).
2.9. Statistical analysis
All data are reported as mean ± SEM. Area under the curves (AUC) and incremental AUC (iAUC) of blood glucose and serum insulin were calculated using the trapezoidal method. Differences between groups were tested using 2-tailed unpaired t-tests or ANOVA with Tukey's multiple comparisons test as indicated. Correlation analyses were carried out using Pearson correlations. Stepwise regression was used to determine whether adjustment for baseline serum FABP4 and body fat mass or BMI could explain GSIS. Two-tailed P <0.05 was taken to be statistically significant. Analyses were performed using SPSS Statistics 21 (SPSS) and Prism 6 (GraphPad Software) software.
3. Results
3.1. Proteins released from hypoxic adipocytes modulate insulin secretion
Recent evidence indicates that nutrient excess in humans and mice is accompanied by a rapid expansion of fat mass that exceeds vascular capacity leading to transient adipose tissue hypoxia [28–30]. We reasoned that this scenario might provide an ideal experimental model system with which to identify insulinotropic adipokines released during obesity. We established an in vitro culture system, using proteins recovered from media conditioned by 3T3-L1 adipocytes under normoxic (21% O2) or hypoxic (1% O2) atmospheres, to model adipocyte to β-cell signalling under lean or obese conditions (Figure 1A). There was a significant potentiation of GSIS from isolated mouse islets cultured with proteins released from hypoxic adipocytes, whereas proteins from normoxic adipocytes did not alter GSIS (Figure 1B). This indicated that an adipocyte-derived protein(s) enriched in hypoxic conditioned media modulates β-cell function.
Figure 1.
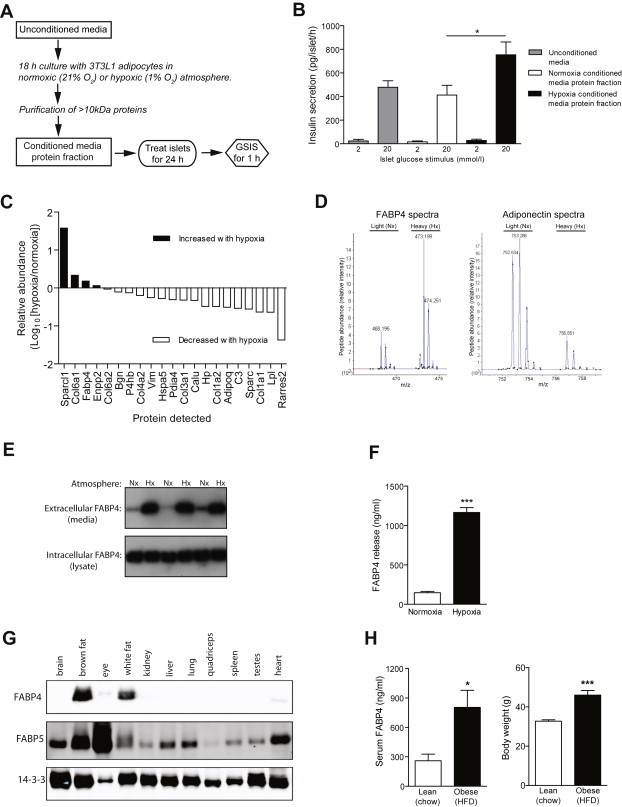
Identification of FABP4 as a candidate insulinotropic protein released from adipocytes. A, B, Media were conditioned by 18 h culture with 3T3-L1 adipocytes under normoxia (21% O2) or hypoxia (1% O2), before a >10 kDa protein fraction was purified. Pancreatic islets isolated from mice were cultured for 24 h in media enriched with this conditioned media protein fraction, or unconditioned media, before insulin secretion were measured for 1 h. n = 9 batches of conditioned media. C, Relative abundance of proteins released from hypoxic vs normoxic 3T3L1 adipocytes, detected using SILAC labelling and mass spectrometry. D, Fatty acid binding protein 4 (FABP4) and adiponectin spectra. E, Western blots showing increased FABP4 release (extracellular), and stable intracellular FABP4 levels, during hypoxia; n = 6. F, Quantification of FABP4 release by ELISA; n = 5. G, FABP4 and FABP5 were detected by western blotting protein lysates from a range of mouse tissues. FABP4 protein was restricted to brown and white adipose tissue. H, Mice were fed a standard lab diet (chow/lean) or high-fat diet (HFD/obese) for 40 weeks. Serum FABP4 concentrations were significantly increased during obesity. Data presented as mean ± SEM. *P < 0.05, ***P < 0.001 by t-test. See also Table S1 and data file S1.
3.2. Adipocyte FABP4 release is enhanced under hypoxic conditions
To identify proteins enriched in hypoxic 3T3-L1 conditioned media we utilised SILAC labelling to perform a mass spectrometry-based quantitative proteomic screen (Supplementary data file 1). From this screen, we identified 21 proteins that had the potential for secretion based on gene sequence analysis (Figure 1C and Table S1). Of these proteins, 4 were enriched in hypoxic 3T3-L1 media thus correlating with enhanced GSIS in our in vitro system (Figure 1A and B). Sparcl1 and Col6a1 were excluded as they are extracellular matrix proteins. Enpp2 is a soluble lysophospholipaseD secreted from adipose tissue [31], however its release was only increased subtly by hypoxia and it is ubiquitously expressed. The candidate protein that we decided to pursue was fatty acid binding protein 4 (FABP4/aP2). This FABP is primarily expressed in adipocytes and macrophages [32] and regulates whole body glucose metabolism [33], in part via its intracellular role in adipocyte lipolysis [34,35]. FABP4 is present in the serum of humans, with levels increasing during obesity [36,37]. Intriguingly, FABP4 has recently been shown to play a role in the metabolic response to fasting possibly via a role in FA uptake into muscle [38] as well as in regulating hepatic glucose production [37]. However, a physiological role for circulating FABP4 in regulating β-cell function has not been described. Importantly, Fabp4/aP2KO mice display an 80% reduction in circulating insulin levels under high-fat feeding [39], consistent with a role for circulating FABP4 in modulating β-cell function. FABP4 is therefore a prime candidate for an insulinotropic adipokine: an adipocyte specific protein, enriched in both hypoxic-adipocyte conditioned media and in the serum of obese subjects.
FABP4 release from 3T3L1 adipocytes was confirmed by Western blotting (Figure 1E), with an 8-fold increase during hypoxia quantified by ELISA (Figure 1F). In contrast, 3T3L1 adipocyte necrosis increased only 1.6-fold with hypoxia (Roche cell death detection ELISA, data not shown). FABP5 was also enriched in hypoxic conditioned media, but was 5-fold less abundant than FABP4 (Supplementary Data File 1), indicating that FABP4 is the dominant isoform released from adipocytes. Moreover, the FABP4 protein is restricted to brown and white adipose tissue, consistent with a potential role as an adipokine, whereas FABP5 was present in all tissues tested (Figure 1G). We detected a significant increase in circulating FABP4 in the serum of mice fed a high-fat diet for 40 weeks to induce obesity, relative to chow-fed lean control mice (Figure 1H). The physiological range of circulating FABP4 concentrations in mice (Figure 1H) were similar to those detected in our 3T3L1 conditioned media experiments (Figure 1F).
Adiponectin has insulinotropic properties [15,16], however, release of this adipokine decreased with hypoxia (Figure 1C and D and Table S1), consistent with reduced adiponectin secretion in obesity [17] and from hypoxic human adipocytes [40]. Therefore, adiponectin is unlikely to contribute to enhanced GSIS in our hypoxia conditioned media model or during obesity in vivo. Leptin is not expressed in 3T3L1 adipocytes and was not detected in our mass spectrometry experiments.
3.3. FABP4 modulates insulin secretion from β-cells in vitro and in vivo
To determine if FABP4 can directly potentiate insulin secretion, we cultured isolated mouse islets with recombinant FABP4 (rFABP4) in vitro for 24 h, with or without linoleate, a fatty acid that binds FABP4 in a 1:1 ratio and enhances FABP4 activation of nuclear receptors in adipocytes [41,42], or palmitate, prior to measuring GSIS. rFABP4–linoleate potentiated GSIS in a dose-dependent manner, whereas linoleate alone had no effect (Figure 2A). At higher doses, rFABP4 supplied alone potentiated GSIS, with addition of linoleate further enhancing activity (Figure S1C). Islet insulin content was unaltered after 24 h rFABP4 treatment (Figure 2B, Figure S1D). We next sought to test the effects of exogenous FABP4 on GSIS and glucose homeostasis in vivo by implanting subcutaneous osmotic mini-pumps containing either PBS or rFABP4–linoleate into mice. rFABP4–linoleate treatment for 1 week significantly improved glucose tolerance (Figure 2C) with a moderate enhancement of GSIS (Figure 2D and E), without altering body weight (Figure 2F). In contrast, acute co-injection of mice with a supra-physiological bolus of rFABP4–linoleate along with glucose did not alter glucose tolerance (Figure 2G). These results demonstrate that FABP4 acts as an insulinotropic adipokine but, unlike the incretin effect [43], this activity does not occur acutely and manifests over a longer period.
3.4. Serum FABP4 levels predict the GSIS response in humans
To investigate if the insulinotropic effects of FABP4 could also be observed in humans, we tested whether circulating FABP4 levels predict other metabolic parameters in a cohort of non-diabetic humans with a range of adiposity (BMI 19–36 kg/m2). This cohort demonstrated tightly regulated glucose tolerance in response to an i.v. glucose tolerance test but with highly variable GSIS (Figure 3A–C), providing a spectrum of β-cell compensation. Serum FABP4 was significantly increased in overweight individuals (BMI >25; Figure 3D) and correlated positively and significantly with total body fat (Figure 3E; Table S2a), consistent with previous reports [36]. Moreover, circulating FABP4 concentrations correlated significantly with GSIS (Figure 3F) supporting a role for FABP4 in coordinating the β-cell response during obesity in humans. As adiposity may explain increased GSIS per se, we performed linear regression with GSIS (AUC or iAUC) as the dependent variable and serum FABP4 and fat mass (by DXA) as the explanatory variables (Table S2b). Serum FABP4 explained 29.8% (AUC, P = 0.017) and 27.7% (iAUC, P = 0.021) of GSIS and body fat was not retained in the model (P = 0.711 and P = 0.931 for AUC and iAUC, respectively). Similar findings were obtained when BMI replaced fat mass in the regression model (Table S2b). Overall, the regression data suggest that FABP4 plays a major role in determining GSIS in humans.
Figure 3.
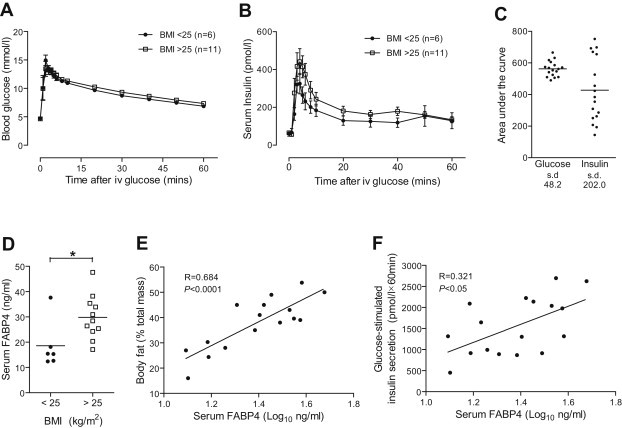
Serum FABP4 levels predict the glucose-stimulated insulin secretion response in humans. Non-diabetic participants were stratified based on body mass index <25 (n = 6) or >25 (n = 11) kg/m2. Intravenous glucose tolerance (0.3 g/kg) was tightly regulated (A) whereas glucose-stimulated insulin secretion was highly variable (B, C). Serum FABP4 levels increased with body mass index (D) and positively correlated with both adiposity (E, P < 0.0001) and glucose-stimulated insulin secretion (F, P < 0.05) using Pearson's correlation coefficients (n = 17). Data presented as mean ± SEM. *P < 0.05 by t-test. See also Table S2.
3.5. Insulin inhibits FABP4 secretion in vitro, in mice and in humans
Most physiological endocrine systems exhibit feedback regulation. To determine if the FABP4-insulin system also possesses feedback regulation we assessed the effect of insulin on FABP4 release from adipocytes. Circulating FABP4 levels were rapidly suppressed by exogenous insulin treatment, within 30 min in mice (Figure 4A) and 2 h in humans (Figure 4B), and treatment of 3T3-L1 adipocytes with insulin potently inhibited FABP4 release in vitro (Figure 4C). Conversely, insulin potentiated adiponectin release under hypoxic conditions (Figure 4C), indicating that the regulation of the release of these two hormones occurs via distinct pathways. These data demonstrate that insulin provides rapid feedback to regulate FABP4 release from adipocytes.
Figure 4.
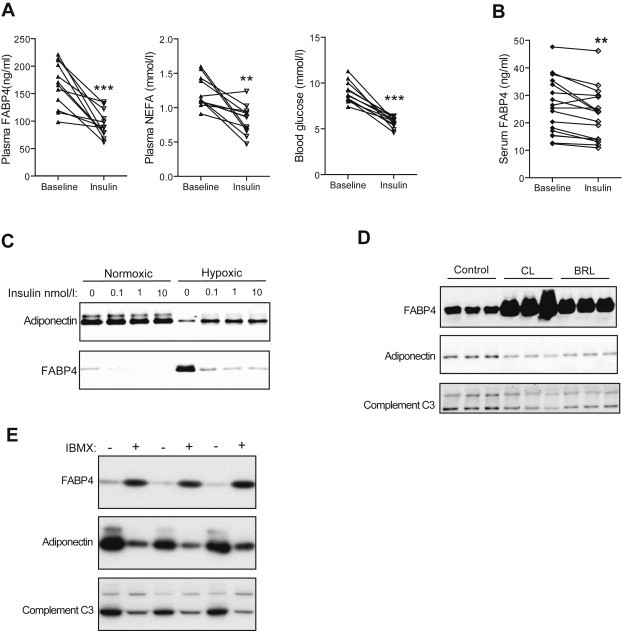
Reciprocal regulation of FABP4 release by insulin. A, In mice, plasma FABP4, NEFA and blood glucose were suppressed 30 min after i.p. insulin injection (0.75 U/kg); n = 12. B, In humans, serum FABP4 was suppressed following 2 h insulin infusion (60 mU/body surface area/min), during a hyperinsulinaemic–euglycaemic clamp; n = 17. C, Western blots showing insulin suppression of FABP4 release from normoxic (21% O2) and hypoxic (1% O2) 3T3-L1 adipocytes during 18 h cultures, with adiponectin secretion enhanced by insulin. D, Western blots of 3T3-L1 adipocytes treated with beta-3 adrenoreceptor agonists (CL316243 or BRL37344; 0.1 μg/ml). E, Western blots of 3T3-L1 adipocytes treated with 3-Isobutyl-1-methylxanthine (IBMX; 500 μmol/l) to enhance cellular cyclic-AMP levels. Data presented as mean ± SEM. **P < 0.01, ***P < 0.001 by paired t-test.
3.6. FABP4 secretion is regulated by β-3-adrenoreceptor signalling
As insulin signalling antagonises β-3 adrenergic signalling [44], we sought to test the involvement of this pathway in FABP4 release. Stimulation of adipocytes with β-3-adrenergic agonists induced a robust increase in FABP4 secretion (Figure 4D) consistent with the potent effect these compounds have on insulin secretion in vivo [45], which is blunted by Fabp4 deletion [34]. To confirm this mode of regulation, we next examined the role of cAMP since this is a major conduit of β-3 adrenergic signalling. The cyclic-AMP elevating drug IBMX markedly induced FABP4 release from adipocytes (Figure 4E). In contrast, both β-3 adrenergic agonists and IBMX suppressed adiponectin and complement C3 release (Figure 4D and E). These data reveal that a cyclic-AMP activated pathway drives FABP4 release from adipocytes independently of other adipokines.
4. Discussion
Our study suggests that FABP4 may play a role in coordinating GSIS with adiposity to maintain glucose homeostasis, thereby providing a potentially parsimonious explanation for the development of enhanced insulin secretion during obesity. In some respects this insulinotropic activity resembles the actions of Glp1 [46], which is released from the gut with feeding to acutely enhance insulin secretion. Hence, this reinforces the notion that multiple organs, including the gut and adipose tissue, play important roles in relaying peripheral metabolic status to the β-cell, thereby enabling adjustments in secretory tone to fulfil the appropriate output.
FABP4 has a plethora of roles in metabolic regulation. As one of the most abundant proteins in the adipocyte it plays a crucial role in fatty acid uptake, as well as promoting transcriptional activity in response to specific ligands. Recently it has been shown to play a role in FA uptake into muscle [38] and ovarian cancer cells [47], although the mechanistic details of this are not clear. The important role of FABP4 and its close homolog FABP5 in metabolism is underscored by the marked alteration in tissue lipid profiles in Fabp4/Fabp5KO mice, which are protected against diet-induced obesity and hyperglycaemia, and show reduced insulin levels [39]. Moreover, the recent description of specific roles for secreted FABP4 in potentiating hepatic glucose production [37] and now insulin secretion, reinforces the role of this protein in metabolic homeostasis. Despite the array of metabolic effects attributed to FABP4, its mode of action remains to be determined. One feature that may consolidate many of these observations is the lipid binding property of FABP4: nuclear localisation and transcriptional activity in adipocytes are regulated by lipid binding [41,42]; liver steatosis in Fabp4/Fabp5KO mice is attributed to a role for dysregulated NEFA uptake into heart and skeletal muscle [38]; and we show here in the case of the β-cell, that the most prominent effects of rFABP4 on potentiating GSIS occur in the presence of linoleate. Moreover, neither rFABP4 nor linoleate supplied alone at physiological concentrations significantly altered GSIS. Therefore, the effect of FABP4 and linoleate on β-cell function is likely due to synergistic actions, such as the transport of linoleate (an essential fatty acid) to a specific cellular location or pathway, or activation of a receptor by the FABP4/linoleate complex, akin to the role of intracellular FABP4 in activating adipocyte nuclear receptors [41].
Hypoxia modulates leptin expression [28,48] and secretion [48,49] although, as 3T3L1 adipocytes do not express leptin, this adipokine was absent from our conditioned media experiments and proteomic screen. As leptin inhibits insulin secretion [14], this raises the question as to whether leptin antagonises the effects of FABP4. However, as 1 week rFABP4 infusion enhanced glucose tolerance and GSIS in mice, this suggests that FABP4 remains active in the presence of endogenous adipokines (including leptin) in vivo. This is consistent with the hyper-secretion of insulin in the presence of elevated leptin during obesity.
We, and others [36,37], have observed increased circulating FABP4 levels in obese humans and mice. However, Fabp4 does not contain a signal peptide, which would normally be required for targeting to the secretory pathway, consistent with its presence in the cytosol of adipocytes. Despite this, we provide evidence that Fabp4 secretion from the adipocyte is stimulated by hypoxia, β-adrenergic agonists or cAMP, and regulated by insulin. Intriguingly, secretion of adiponectin (which possesses a classical secretory signal peptide) was inversely proportional to FABP4 release with these stimuli. In further support of FABP4 secretion via a non-classical pathway, its regulated release occurs via a golgi-independent (brefeldin A-insensitive) pathway in adipocytes [37]. Taken together, these data suggest that FABP4 is released from the adipocyte by a non-classical secretory pathway as described for a range of growth factors and cytokines including FGF1, FGF2, IL1α, IL1β and Y-box protein-1 [50,51]. An alternative possibility is that FABP4 may be released as a function of adipocyte cell death, an event that may be increased in frequency both with obesity and/or hypoxia. We have found FABP4 to be highly expressed in the adipocyte [23] and the demise of a relatively minor number of cells may generate physiological levels of this protein in the systemic circulation. However, our observations of rapid suppression of systemic FABP4 levels by insulin, along with the relatively subtle increases in necrosis in our conditioned media experiments, favours regulated secretion over cell death as the mechanism of release. In addition, our results suggest that adipocyte insulin resistance may impair insulin suppression of FABP4 release, resulting in enhanced FABP4 secretion and increased reciprocal insulin release, thereby compensating for insulin resistance to maintain glucose and lipid homeostasis.
Our results reveal the therapeutic potential of targeting the FABP4-insulin axis as a treatment for metabolic disease. Reinforcing or enhancing FABP4 action on the β-cell may provide a novel incretin-like therapy to enhance GSIS and improve glucose tolerance. Furthermore, our observation that linoleate enhances the insulinotropic activity of FABP4 provides a potential molecular mechanism that could be targeted to modulate the action of circulating FABP4 on the β-cell. Although our data support the notion that FABP4 is beneficial and protects glucose homeostasis, it remains to be determined what the long-term implications of up-regulating this mechanism may be. Obesity predisposes individuals to diabetes, cardiovascular disease and cancer, and chronic hyperinsulinaemia has been implicated in exacerbating disease progression [52–54], suggesting that up-regulation of circulating FABP4 levels may have both positive and negative consequences. In conclusion, our study places circulating FABP4 in a central role during obesity and related metabolic diseases.
Acknowledgements
We thank Aimee Davenport, Shixiong Tan and Kristen Thomas for technical assistance, Charles Mackay for providing Fabp5 antisera, and Rodrigo Vazquez-Lombardi and Daniela Zinkl for performing endotoxin assays. We also thank Don Chisholm and Jenny Gunton for critical appraisal of our manuscript. This work was supported by the National Health and Medical Research Council of Australia (NHMRC project grant to JC [APP1052782]; program grant to DEJ) and the Diabetes Australia Research Trust and Viertel Charitable Foundation (fellowship support to JC). DEJ is a senior principal research fellow and TJB a senior research fellow of the NHMRC. LEW is supported by an Early Career Fellowship from Cancer Institute NSW, Australia.
Contributor Information
David E. James, Email: d.james@garvan.org.au.
James Cantley, Email: james.cantley@dpag.ox.ac.uk.
Conflict of interest
The authors have no conflicts of interest with the publication of this article.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
All proteins identified during mass spectrometry studies. Complete list of proteins identified as released from 3T3-L1 adipocytes by LC–MS/MS.
Summary of proteins released from 3T3-L1 adipocytes that were identified by mass spectrometry. a, List of the 21 proteins identified as released from 3T3-L1 adipocytes with >5 unique peptides by LC-MS/MS and either presence of a classical secretory signal peptide or potential for non-classical secretion, predicted using SecretomeP. b, Published function and tissue distribution of the 4 proteins showing increased release with hypoxia [1–4].
Table S2: (a) Serum FABP4 levels are associated with adiposity and glucose-stimulated insulin secretion. Pearson correlations of circulating FABP4 concentrations with various metabolic parameters, in a cohort of non-diabetic humans with a range of adiposity (BMI 19–36 kg/m2); n = 17. Serum FABP4 is positively associated with BMI, body fat, abdominal fat and glucose-stimulated insulin secretion, and negatively associated with fat-free mass. Area under the curve (AUC) and incremental AUC (iAUC) was calculated from glucose tolerance test data (intravenous glucose 0.3 g/kg). (b) Linear regression to explain the variability in glucose-stimulated insulin secretion in humans. *Variables were calculated by a linear regression model with glucose-stimulated insulin secretion (insulin AUC or iAUC during 0.3 g/kg i.v.GTT) as outcome and explanatory variables: fasting serum FABP4 and body fat mass. **Variables were calculated by a linear regression model with glucose-stimulated insulin secretion (insulin AUC or iAUC during 0.3 g/kg i.v.GTT) as outcome and explanatory variables: fasting serum FABP4 and BMI. β = beta-estimate of linear regression model; R2 = explained variance.
Fabp4 potentiates glucose-stimulated insulin secretion in vitro. A, Recombinant Fabp4 (rFabp4) produced in E. coli is readily detected by western blot at a comparable size to endogenous 3T3L1 adipocyte Fabp4. B, rFabp4 purity was assessed by SDS-PAGE with Coomassie blue protein stain. C, D, Supra-physiological doses of rFabp4 potentiates glucose-stimulated insulin secretion in vitro without altering insulin content. Isolated mouse islets were treated with 5 μg/ml rFabp4 for 24 h, with or without the fatty acid linoleate, before 20 mM glucose-stimulated insulin secretion was assessed for 1 h (A) and islet insulin content was assayed (B); n = 3. Data presented as mean ± SEM. *P < 0.05, **P = 0.01 by t-test.
References
- 1.Yach D., Stuckler D., Brownell K.D. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nature Medicine. 2006;12:62–66. doi: 10.1038/nm0106-62. [DOI] [PubMed] [Google Scholar]
- 2.Zimmet P., Alberti K.G., Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 3.Eckel R.H., Grundy S.M., Zimmet P.Z. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 4.Alberti K.G.M.M., Eckel R.H., Grundy S.M., Zimmet P.Z., Cleeman J.I., Donato K.A. Harmonizing the metabolic syndrome. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 5.Calle E.E., Thun M.J. Obesity and cancer. Oncogene. 2004;23:6365–6378. doi: 10.1038/sj.onc.1207751. [DOI] [PubMed] [Google Scholar]
- 6.Renehan A.G., Tyson M., Egger M., Heller R.F., Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 7.Perley M., Kipnis D.M. Plasma insulin responses to glucose and tolbutamide of normal weight and obese diabetic and nondiabetic subjects. Diabetes. 1966;15:867–874. doi: 10.2337/diab.15.12.867. [DOI] [PubMed] [Google Scholar]
- 8.Polonsky K.S., Given B.D., Van Cauter E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. Journal of Clinical Investigation. 1988;81:442–448. doi: 10.1172/JCI113339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahn S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 10.Winzell M.S., Ahren B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004;53(Suppl. 3):S215–219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- 11.Latour M., Alquier T., Oseid E., Tremblay C., Jetton T., Luo J. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56:1087–1094. doi: 10.2337/db06-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karpe F., Dickmann J.R., Frayn K.N. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60:2441–2449. doi: 10.2337/db11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Considine R.V., Sinha M.K., Heiman M.L., Kriauciunas A., Stephens T.W., Nyce M.R. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. New England Journal of Medicine. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 14.Kulkarni R.N., Wang Z.L., Wang R.M., Hurley J.D., Smith D.M., Ghatei M.A. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. Journal of Clinical Investigation. 1997;100:2729–2736. doi: 10.1172/JCI119818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winzell M.S., Nogueiras R., Dieguez C., Ahrén B. Dual action of adiponectin on insulin secretion in insulin-resistant mice. Biochemical and Biophysical Research Communications. 2004;321:154–160. doi: 10.1016/j.bbrc.2004.06.130. [DOI] [PubMed] [Google Scholar]
- 16.Okamoto M., Ohara-Imaizumi M., Kubota N., Hashimoto S., Eto K., Kanno T. Adiponectin induces insulin secretion in vitro and in vivo at a low glucose concentration. Diabetologia. 2008;51:827–835. doi: 10.1007/s00125-008-0944-9. [DOI] [PubMed] [Google Scholar]
- 17.Arita Y., Kihara S., Ouchi N., Takahashi M., Maeda K., Miyagawa J.-I. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochemical and Biophysical Research Communications. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 18.Revollo J.R., Korner A., Mills K.F., Satoh A., Wang T., Garten A. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metabolism. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin H., Jiang B., Tang J., Lu W., Wang W., Zhou L. Serum visfatin concentrations in obese adolescents and its correlation with age and high-density lipoprotein cholesterol. Diabetes Research and Clinical Practice. 2008;79:412–418. doi: 10.1016/j.diabres.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 20.Shewan A.M., Marsh B.J., Melvin D.R., Martin S., Gould G.W., James D.E. The cytosolic C-terminus of the glucose transporter GLUT4 contains an acidic cluster endosomal targeting motif distal to the dileucine signal. Biochemical Journal. 2000;350(Pt 1):99–107. [PMC free article] [PubMed] [Google Scholar]
- 21.Cantley J., Boslem E., Laybutt D.R., Cordery D.V., Pearson G., Carpenter L. Deletion of protein kinase Cdelta in mice modulates stability of inflammatory genes and protects against cytokine-stimulated beta cell death in vitro and in vivo. Diabetologia. 2011;54:380–389. doi: 10.1007/s00125-010-1962-y. [DOI] [PubMed] [Google Scholar]
- 22.Ong S.E., Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nature Protocols. 2006;1:2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 23.Humphrey S.J., Yang G., Yang P., Fazakerley D.J., Stockli J., Yang J.Y. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metabolism. 2013;17:1009–1020. doi: 10.1016/j.cmet.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong S.E., Kratchmarova I., Mann M. Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC) Journal of Proteome Research. 2003;2:173–181. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 25.Crowe S., Wu L.E., Economou C., Turpin S.M., Matzaris M., Hoehn K.L. Pigment epithelium-derived factor contributes to insulin resistance in obesity. Cell Metabolism. 2009;10:40–47. doi: 10.1016/j.cmet.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Bendtsen J.D., Jensen L.J., Blom N., Von Heijne G., Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Engineering, Design & Selection: PEDS. 2004;17:349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 27.Turner N., Bruce C.R., Beale S.M., Hoehn K.L., So T., Rolph M.S. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes. 2007;56:2085–2092. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- 28.Hosogai N., Fukuhara A., Oshima K., Miyata Y., Tanaka S., Segawa K. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 29.Pasarica M., Sereda O.R., Redman L.M., Albarado D.C., Hymel D.T., Roan L.E. Reduced adipose tissue oxygenation in human obesity. Diabetes. 2009;58:718–725. doi: 10.2337/db08-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun K., Wernstedt Asterholm I., Kusminski C.M., Bueno A.C., Wang Z.V., Pollard J.W. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5874–5879. doi: 10.1073/pnas.1200447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dusaulcy R., Rancoule C., Grès S., Wanecq E., Colom A., Guigné C. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. Journal of Lipid Research. 2011;52:1247–1255. doi: 10.1194/jlr.M014985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Storch J., Thumser A.E. Tissue-specific functions in the fatty acid-binding protein family. Journal of Biological Chemistry. 2010;285:32679–32683. doi: 10.1074/jbc.R110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hotamisligil G.S., Johnson R.S., Distel R.J., Ellis R., Papaioannou V.E., Spiegelman B.M. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 34.Scheja L., Makowski L., Uysal K.T., Wiesbrock S.M., Shimshek D.R., Meyers D.S. Altered insulin secretion associated with reduced lipolytic efficiency in aP2-/- mice. Diabetes. 1999;48:1987–1994. doi: 10.2337/diabetes.48.10.1987. [DOI] [PubMed] [Google Scholar]
- 35.Coe N.R., Simpson M.A., Bernlohr D.A. Targeted disruption of the adipocyte lipid-binding protein (aP2 protein) gene impairs fat cell lipolysis and increases cellular fatty acid levels. Journal of Lipid Research. 1999;40:967–972. [PubMed] [Google Scholar]
- 36.Xu A., Wang Y., Xu J.Y., Stejskal D., Tam S., Zhang J. Adipocyte fatty acid-binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clinical Chemistry. 2006;52:405–413. doi: 10.1373/clinchem.2005.062463. [DOI] [PubMed] [Google Scholar]
- 37.Cao H., Sekiya M., Ertunc M.E., Burak M.F., Mayers J.R., White A. Adipocyte lipid chaperone aP2 is a secreted adipokine regulating hepatic glucose production. Cell Metabolism. 2013;17:768–778. doi: 10.1016/j.cmet.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Syamsunarno M.R., Iso T., Hanaoka H., Yamaguchi A., Obokata M., Koitabashi N. A critical role of fatty acid binding protein 4 and 5 (FABP4/5) in the systemic response to fasting. PLoS One. 2013;8:e79386. doi: 10.1371/journal.pone.0079386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda K., Cao H., Kono K., Gorgun C.Z., Furuhashi M., Uysal K.T. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metabolism. 2005;1:107–119. doi: 10.1016/j.cmet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Wang B., Wood I., Trayhurn P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Archiv European Journal of Physiology. 2007;455:479–492. doi: 10.1007/s00424-007-0301-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan N.-S., Shaw N.S., Vinckenbosch N., Liu P., Yasmin R., Desvergne B. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Molecular and Cellular Biology. 2002;22:5114–5127. doi: 10.1128/MCB.22.14.5114-5127.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillilan R.E., Ayers S.D., Noy N. Structural basis for activation of fatty acid-binding protein 4. Journal of Molecular Biology. 2007;372:1246–1260. doi: 10.1016/j.jmb.2007.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nauck M.A., Homberger E., Siegel E.G., Alllen R.C., Eatpm R.P., Ebert R. Incretin Effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. Journal of Clinical Endocrinology & Metabolism. 1986;63:492–498. doi: 10.1210/jcem-63-2-492. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J., Hupfeld C.J., Taylor S.S., Olefsky J.M., Tsien R.Y. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature. 2005;437:569–573. doi: 10.1038/nature04140. [DOI] [PubMed] [Google Scholar]
- 45.Susulic V.S., Frederich R.C., Lawitts J., Tozzo E., Kahn B.B., Harper M.-E. Targeted disruption of the adrenergic receptor gene. Journal of Biological Chemistry. 1995;270:29483–29492. doi: 10.1074/jbc.270.49.29483. [DOI] [PubMed] [Google Scholar]
- 46.Parkes D.G., Pittner R., Jodka C., Smith P., Young A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism. 2001;50:583–589. doi: 10.1053/meta.2001.22519. [DOI] [PubMed] [Google Scholar]
- 47.Nieman K.M., Kenny H.A., Penicka C.V., Ladanyi A., Buell-Gutbrod R., Zillhardt M.R. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature Medicine. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang B., Wood I.S., Trayhurn P. Hypoxia induces leptin gene expression and secretion in human preadipocytes: differential effects of hypoxia on adipokine expression by preadipocytes. Journal of Endocrinology. 2008;198:127–134. doi: 10.1677/JOE-08-0156. [DOI] [PubMed] [Google Scholar]
- 49.Tschop M., Strasburger C.J., Topfer M., Hautmann H., Riepl R., Fischer R. Influence of hypobaric hypoxia on leptin levels in men. International Journal of Obesity and Related Metabolic Disorders: Journal of the International Association for the Study of Obesity. 2000;24(Suppl. 2):S151. doi: 10.1038/sj.ijo.0801309. [DOI] [PubMed] [Google Scholar]
- 50.Nickel W., Seedorf M. Unconventional mechanisms of protein transport to the cell surface of eukaryotic cells. Annual Review of Cell and Developmental Biology. 2008;24:287–308. doi: 10.1146/annurev.cellbio.24.110707.175320. [DOI] [PubMed] [Google Scholar]
- 51.Frye B.C., Halfter S., Djudjaj S., Muehlenberg P., Weber S., Raffetseder U. Y-box protein-1 is actively secreted through a non-classical pathway and acts as an extracellular mitogen. EMBO Reports. 2009;10:783–789. doi: 10.1038/embor.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gallagher E.J., LeRoith D. Minireview: IGF, insulin, and cancer. Endocrinology. 2011;152:2546–2551. doi: 10.1210/en.2011-0231. [DOI] [PubMed] [Google Scholar]
- 53.Shanik M.H., Xu Y., Škrha J., Dankner R., Zick Y., Roth J. Insulin resistance and hyperinsulinemia. Diabetes Care. 2008;31:S262–S268. doi: 10.2337/dc08-s264. [DOI] [PubMed] [Google Scholar]
- 54.Zimmet P.Z. Hyperinsulinemia – how innocent a bystander? Diabetes Care. 1993;16(Suppl. 3):56–70. doi: 10.2337/diacare.16.3.56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All proteins identified during mass spectrometry studies. Complete list of proteins identified as released from 3T3-L1 adipocytes by LC–MS/MS.
Summary of proteins released from 3T3-L1 adipocytes that were identified by mass spectrometry. a, List of the 21 proteins identified as released from 3T3-L1 adipocytes with >5 unique peptides by LC-MS/MS and either presence of a classical secretory signal peptide or potential for non-classical secretion, predicted using SecretomeP. b, Published function and tissue distribution of the 4 proteins showing increased release with hypoxia [1–4].
Table S2: (a) Serum FABP4 levels are associated with adiposity and glucose-stimulated insulin secretion. Pearson correlations of circulating FABP4 concentrations with various metabolic parameters, in a cohort of non-diabetic humans with a range of adiposity (BMI 19–36 kg/m2); n = 17. Serum FABP4 is positively associated with BMI, body fat, abdominal fat and glucose-stimulated insulin secretion, and negatively associated with fat-free mass. Area under the curve (AUC) and incremental AUC (iAUC) was calculated from glucose tolerance test data (intravenous glucose 0.3 g/kg). (b) Linear regression to explain the variability in glucose-stimulated insulin secretion in humans. *Variables were calculated by a linear regression model with glucose-stimulated insulin secretion (insulin AUC or iAUC during 0.3 g/kg i.v.GTT) as outcome and explanatory variables: fasting serum FABP4 and body fat mass. **Variables were calculated by a linear regression model with glucose-stimulated insulin secretion (insulin AUC or iAUC during 0.3 g/kg i.v.GTT) as outcome and explanatory variables: fasting serum FABP4 and BMI. β = beta-estimate of linear regression model; R2 = explained variance.
Fabp4 potentiates glucose-stimulated insulin secretion in vitro. A, Recombinant Fabp4 (rFabp4) produced in E. coli is readily detected by western blot at a comparable size to endogenous 3T3L1 adipocyte Fabp4. B, rFabp4 purity was assessed by SDS-PAGE with Coomassie blue protein stain. C, D, Supra-physiological doses of rFabp4 potentiates glucose-stimulated insulin secretion in vitro without altering insulin content. Isolated mouse islets were treated with 5 μg/ml rFabp4 for 24 h, with or without the fatty acid linoleate, before 20 mM glucose-stimulated insulin secretion was assessed for 1 h (A) and islet insulin content was assayed (B); n = 3. Data presented as mean ± SEM. *P < 0.05, **P = 0.01 by t-test.