

Abstract
Interleukin-33 (IL-33) is a recently described member of the interleukin-1 (IL-1) family. It is produced by diverse cell types in response to a variety of stresses including hemorrhage and increased mechanical load. Though only relatively recently discovered, IL-33 has been shown to participate in several pathological processes including promoting type 2 T helper cell-associated autoimmune diseases. In contrast, IL-33 has been also found to have protective effects in cardiovascular diseases. Recent studies have illustrated that IL-33 attenuates cardiac fibrosis induced by increased cardiovascular load in mice (transaortic constriction). Since cardiac fibrosis is largely dependent on increased production of extracellular matrix by cardiac fibroblasts, we hypothesized that IL-33 directly inhibits pro-fibrotic activities of these cells. Experiments have been carried out with isolated rat cardiac fibroblasts to evaluate the effects of IL-33 on the modulation of cardiac fibroblast gene expression and function to test this hypothesis. The expression of the IL-33 receptor, interleukin-1 receptor-like 1 (ST2), was detected at the mRNA and protein levels in isolated adult rat cardiac fibroblasts. Subsequently, the effects of IL-33 treatment (0–100 ng/ml) on the expression of extracellular matrix proteins and pro-inflammatory cytokines/chemokines were examined as well as the effects on rat cardiac fibroblast activities including proliferation, collagen gel contraction and migration. While IL-33 did not directly inhibit collagen I and collagen III production, it yielded a dose-dependent increase in the expression of interleukin-6 and monocyte chemotactic protein-1. Treatment of rat cardiac fibroblasts with IL-33 also impaired the migratory activity of these cells. Further experiments illustrated that IL-33 rapidly activated multiple signaling pathways including extracellular signal-regulated kinases, p38 mitogen-activated protein kinase, c-Jun N-terminal kinases and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) in a dose-dependent manner. Experiments were carried out with pharmacological inhibitors to determine the role of specific signaling pathways in the response of fibroblasts to IL-33. These experiments illustrated that the activation of p38 mitogen-activated protein kinase and extracellular signal-regulated kinases are critical to the increased production of interleukin-6 and monocyte chemotactic protein-1 in response to IL-33. These studies suggest that IL-33 has an important role in the modulation of fibroblast function and gene expression. Surprisingly, IL-33 had no effect on the expression of genes encoding extracellular matrix components or on proliferation, markers typical of fibrosis. The major effects of IL-33 detected in these studies included inhibition of cell migration and activation of cytokine/chemokine expression. The previously reported inhibition of cardiac fibrosis may include more complicated mechanisms that involve other cardiac cell types. Future studies aimed at determining the effects of IL-33 on other cardiac cell types are warranted.
Keywords: Cardiac fibroblast, Interleukin-33, Collagen, Inflammation, Fibrosis
1. Introduction
Heart disease has long been recognized as the number one health threat for the western world [1]. Despite their various physiological and pathological etiologies, several major categories of heart disease including hypertension, atherosclerosis and valve disease, share cardiac fibrosis as a common pathological feature [2–4]. Cardiac fibrosis is characterized by the interstitial accumulation of fibrous extracellular matrix (ECM) proteins including collagens and the alteration of the mechanical properties of cardiac tissue [5, 6]. Eventually this pathological change will lead to diastolic or even systolic dysfunction and heart failure [7, 8]. During cardiac fibrosis, cardiac fibroblasts are of particular importance because these cells produce the bulk of the ECM components and secrete enzymes responsible for ECM turnover. These cells respond rapidly to alterations in the biochemical and mechanical environment such as seen during disease situations [9]. While a number of studies have addressed the role of cardiac fibroblasts in heart disease, many questions remain regarding the mechanisms through which they are regulated [10, 11].
Inflammation has classically been described from the perspective of a defensive response to invasion by pathogenic organisms or as an initial step in healing following tissue damage [12]. The more recent point of view considers inflammation as an adaptive process to perturbed homeostasis. The inflammatory process can be initially a beneficial adaptation but if the pathological stimulus causing the response persists too long or is too strong, the inflammatory response may also develop into a detrimental disease condition [13]. Inflammation is a highly complicated process that involves numerous cell types as well as extensive groups of cytokines and chemokines as mediators [14, 15]. Numerous cytokines and chemokines have been discovered and in-depth research has been carried out to elucidate their roles in cardiac diseases as well as in other pathologies [16, 17]. Our previous research demonstrated that one of the interleukin-1 (IL-1) family cytokines, interleukin-18 (IL-18), can promote alterations in cultured cardiac fibroblast behavior consistent with a pro-fibrotic response [18]. In recent years, a newly discovered IL-1 family cytokine, IL-33, has been demonstrated to play interesting roles in several cardiovascular disease processes.
IL-33 was first cloned in 1999 from canine vasospastic cerebral arteries after subarachnoid hemorrhage [19]. IL-33 was originally given several names including DVS-27 gene encoding protein and nuclear factor from high endothelial venules (NF-HEV) [19, 20]. In 2005, this protein was determined to be the ligand of the orphan receptor ST2 (protein from the ST2 cDNA clone) or interleukin-1 receptor-like 1 receptor [21–24] of the IL-1 receptor family and thereafter called IL-33 [25]. Upon binding to IL-33, the ST2 receptor together with interleukin-1 receptor accessory protein (RAcP), form a functional signaling receptor complex [25–27]. There are two forms of ST2, the soluble ST2 (sST2) and the membrane bound ST2 (ST2L). Serum sST2 levels correlate with the severity of heart failure in humans and have recently been suggested as a potential predictor of sudden cardiac death [28–30]. IL-33 can trigger pro- type 2 T helper (Th2) responses in several autoimmune diseases such as arthritis and asthma [31, 32]. IL-33 appears to have more functions besides modulating immune system status. The critical role of IL-33 in regulating cardiac myocyte activities and its protective role in cardiac fibrotic diseases have been suggested [33–35]. In addition, the in vivo administration of IL-33 significantly decreased cardiac interstitial fibrosis in wild type mice that had undergone transaortic constriction (TAC) surgery to increase cardiovascular load [33]. The decrease in cardiac fibrosis was not seen in ST2 receptor gene knock-out mice. These studies suggest that IL-33 plays a protective role in response of the heart to increased mechanical load. While IL-33 has been suggested to play a beneficial role in cardiac fibrosis and arthrosclerosis [36], it was also found to be harmful in diseases such as arthritis and asthma by activating Th2 type immune responses [31, 32]. In contradiction to studies in animal models of heart disease, IL-33 also has been reported to worsen skin fibrosis [37].
As discussed above, IL-33 is a relatively newly discovered cytokine whose expression is enhanced in response to increased cardiovascular load (cyclic stretch and TAC surgery). This cytokine appears to play an important protective role in attenuating load-induced cardiac hypertrophy and fibrosis; however, the mechanisms of this protective role are far from understood. To better elucidate the function of IL-33 in cardiac disease, experiments have been carried out to elucidate the effects that exposure to this cytokine has on gene expression and activity of rat cardiac fibroblasts.
2. Material and Methods
2.1. Rat cardiac fibroblast isolation
Adult rat cardiac fibroblasts were isolated from eight- week- old male Sprague Dawley rats (200–250 gram body weight). Animals were housed in the University of South Carolina animal facility and provided food and water ad libitum. Experiments were conducted according to Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), National Institutes of Health (NIH) and University of South Carolina Institutional Animal Use and Care Committee (IACUC) regulations. Hearts were removed from euthanized animals, rinsed and minced under sterile conditions. Liberase 3 digestion of minced cardiac tissue was conducted as described previously [38, 39] and fibroblasts plated in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% neonatal calf serum, 5% fetal bovine serum (FBS) and antibiotics (hereafter referred to as normal fibroblast medium) for purification by selective attachment to tissue culture dishes. Rat cardiac fibroblasts were cultured in normal fibroblast medium and passaged by incubation in a solution containing 0.1% ethylenediaminetetraacetic acid (EDTA) and 0.25% trypsin before confluence. Cells from passage three to passage five were used for all the assays and at least three independent experiments were conducted for each assay. Cells were incubated in low serum medium (DMEM with 1.5% FBS) for 24 hours prior to treatment with IL-33. Subsequently rat cardiac fibroblasts were incubated in varying doses of IL-33 (0–100 ng/ml). The doses of IL-33 used here were determined based on previous published in vitro IL-33 treatment doses [32, 33]. After 24 hours of cytokine treatment, samples of cell mRNAs, proteins, as well as, conditioned medium were collected. Cells were treated for shorter periods (15 and/or 30 minutes) for evaluation of activation of signal transduction pathways.
2.2. Reverse transcriptase-polymerase chain reaction (RT-PCR)
mRNA expression of specific ECM molecules and cytokines/chemokines were evaluated by semi-quantitative RT-PCR. Isolated male adult rat cardiac fibroblasts were kept in normal fibroblast medium until 24 hours before IL-33 treatment. Fibroblast medium was changed to low serum medium, followed by 24 hours of IL-33 treatment (0–100 ng/ml). Rat cardiac fibroblast conditioned medium was collected for western blot or enzyme-linked immunosorbent assay (ELISA) analysis. Cells were rinsed with phosphate-buffered saline (PBS) and total RNA was extracted in TRIzol Reagent (Invitrogen; Carlsbad, CA). RNA was precipitated in isopropanol, resuspended in nuclease-free water and the concentration of each sample was determined spectrophotometrically. Two micrograms of RNA from each sample was used to produce cDNA by using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA). PCR was carried out by using primers pairs (Table 1) specific to IL-33 functional receptors, ECM proteins (collagen type I, collagen type III, periostin) as well as the cytokines/chemokines interleukin-6 (IL-6) and monocyte chemoattractant protein -1 (MCP-1). Preliminary experiments were carried out to determine appropriate amplification cycle numbers (25–35) for each target of interest and expression of each tested target RNA was normalized to the acidic ribosomal phosphoprotein PO (Arbp) RNA expression. The RNA expression of Arbp was comparatively stable under all conditions evaluated. The final quantification of RNA expression was expressed as the ratio of the integrated density value of target mRNAs to Arbp mRNA [40, 41].
Table 1.
The primers used for semi-quantitative RT-PCR are listed in table 1.
| Gene Name | Sequence 5′-3′ | Accession Number |
|---|---|---|
|
| ||
| RAcP | (Forward) aaaggtggtaggctcaccaaagga (Reverse) aacagggacgtgatcaggcttctt |
NM_012968 |
| sST2 | (Forward) gcccttcatctgggctacact (Reverse) gcaatggcacaggaaggtaac |
NM_013037 |
| ST2L | (Forward) gggtgaaatcccaagctacact (Reverse) gcaatggcacaggaaggtaac |
NM_001127689 |
| Col I | (Forward) tggttctcctggcaaagatggact (Reverse) atcggtcatgctctctccaaacca |
NM_053304 |
| Col III | (Forward) tcctaaccaaggctgcaagatgga (Reverse) aggccagctgtacatcaaggacat |
NM_032085 |
| Periostin | (Forward) aatgccaacagttactatgac (Reverse) cttgataccagttcttacagg |
NM_001108550 |
| IL-6 | (Forward) caaagccagagtcattcagagc (Reverse) ctgaccacagtgaggaatgtcgc |
NM_012589 |
| MCP-1 | (Forward) tgctgtctcagccagatgcagtta (Reverse) acctgctgctggtgattctcttgt |
NM_031530 |
| Arbp | (Forward) ccacgttcccccggatgtga (Reverse) taaaccccgcgtggcaatc |
NM_022402 |
2.3. Western blots
Western blots were used to detect the expression of functional IL-33 receptors and to evaluate the accumulation of ECM proteins in the conditioned medium of rat cardiac fibroblasts treated with varying doses of IL-33 and for analysis of IL-33 induced activation of signaling pathways. Briefly, isolated male adult rat cardiac fibroblasts were maintained in normal fibroblast medium. As described above, medium was replaced with low serum medium 24 hours prior to IL-33 treatment. For the evaluation of ECM protein secretion, cells were treated with various doses of IL-33 (0–100 ng/ml) for 24 hours, and conditioned medium samples were collected. Total protein concentrations were determined by bicinchoninic acid assay (BCA) (Pierce; Rockford, IL). Medium samples containing equal amount of protein (25–40 μg) were loaded on polyacrylamide gels and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After proteins were separated in the gels by their molecular weight, they were transferred from the gel onto nitrocellulose membranes. Fast green staining was used to demonstrate efficient protein transfer and also used as loading control for fibroblast ECM western blots. Membranes were then incubated with diluted primary antibodies against collagen type I (Santa Cruz Biotechnology, Inc., Santa Cruz, CA catalog number: sc-8784), collagen type III (Santa Cruz Biotechnology, Inc., Santa Cruz, CA catalog number: sc-28888) or periostin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA catalog number: sc-49480). The collagen I primary antibody was raised against a peptide mapping to the C-terminus of collagen I (α1) of human origin and the collagen III primary antibody was raised against amino acids mapping near the C-terminus of collagen III (α1) of human origin. The procollagen form of α1(I) collagen (COL1A1) and α1(III) collagen (COL3A1) were quantified from the western blots [42]. After primary antibody incubation, blots were then incubated with secondary antibodies (GE Healthcare UK Limited, Buckingham shire, UK), developed in SuperSignal reagent (Pierce; Rockford, IL) and exposed to X-ray films. Both images of the X-ray film and images of the fast green stained membrane were subjected to image analysis by using an Alpha Innotech gel analysis system. (Alpha Innotech Corp.; San Leandro, CA).
For the detection of IL-33 receptors, the primary antibodies used for detecting ST2 and RAcP are both from Santa Cruz Biotechnology. (Santa Cruz Biotechnology, Inc., Santa Cruz, CA catalog number: sc-18687, Santa Cruz Biotechnology, Inc., Santa Cruz, CA catalog number: sc-47056). Fibroblasts were growing in normal fibroblast medium until confluence, then cells were put on ice and one milliliter ice-cold non-denaturing lysis buffer (containing 1% Triton X-100, 50 mM Tris-Cl, 300 mM NaCl, 5 mM EDTA, 0.02% sodium azide and protease inhibitors) was added to the plate and plate rotated by hand to cover the plate bottom. Subsequently, cells were scraped off from plates and transferred into 1.5 ml conical microcentrifuge tubes, vortexed gently for three seconds. Following 30 minutes incubation on ice, cell lysates were cleared by spinning at 4°C under maximum speed (16,000g) for 15 minutes. Cell lysates of rat cardiac fibroblasts and RBL 2H3 cells were incubated with anti-ST2 serum and then secondary antiserum as mentioned above. This primary antibody is an affinity purified goat polyclonal antibody. It is commercially available from Santa Cruz Biotechnology and was raised against a peptide mapping within an internal region of ST2 of human origin. Because both mast cells and basophils have been documented to express functional IL-33 receptors, extracts from the rat basophilic leukemia cell line, RBL 2H3 cells, were used as positive controls.
For the evaluation of the activation of signaling pathways by IL-33, cells were pre-treated in low serum medium for 24 hours. Cells were then treated with various doses of IL-33 for 15 minutes. Following treatment, cells were rinsed in PBS and extracted in lysis buffer (composed of 10 mM Tris Base (pH 7.4), 1% sodium dodecyl sulfate, and 1 mM sodium orthovanadate as well as protease inhibitors). Protein lysates (20–25 μg) were separated on polyacrylamide gels and transferred to nitrocellulose as described above and Insulin-like growth factor 1 stimulated cell lysates were used as positive controls (data not shown). Other western blots were performed with the following primary antibodies: anti-phosphorylated extracellular signal-regulated kinases (Erk) (Signaling Technology, Inc.; Danvers, MA catalog number: 9101s), anti-phosphorylated c-Jun N-terminal kinases (JNK) (Signaling Technology, Inc.; Danvers, MA catalog number: 9251s), anti-phosphorylated p38 mitogen-activated protein kinase (p38)(Signaling Technology, Inc.; Danvers, MA catalog number: 92112), anti-total Erk (Signaling Technology, Inc.; Danvers, MA catalog number: 912), anti-total JNK (Signaling Technology, Inc.; Danvers, MA catalog number: 9252) and anti-total p38 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA catalog number: sc535). Western blots were initially performed with antibodies specific for the phosphorylated protein of interest and x-ray films quantified as indicated above. Western blots were subsequently stripped and reprobed with antiserum to the corresponding “total” protein (non-phosphorylated and phosphorylated forms). Signals obtained with the phospho-specific antisera were normalized to those with the corresponding total protein antiserum.
2.4. ELISA
The concentrations of IL-6 and MCP-1 in cell culture supernatants following IL-33 treatment were measured by commercially available ELISA kits according to the manufacturer’s protocols (Pierce; Rockford, IL). Conditioned medium from treated cells was collected and the total protein concentrations were determined by the BCA assay as described earlier in western blots. Samples were then diluted and adjusted to the same total protein amount (MCP-1 125μg; IL-6 190μg) and subjected to ELISA measurements. The concentration of IL-6 and MCP-1 were represented as ng/g and μg/g total protein, respectively and calculated based on the standard curves.
2.5. Fibroblast proliferation assay
5′-bromo-2′-deoxyuridine (BrdU) incorporation assay was used to evaluate the proliferation of cultured rat cardiac fibroblasts [43]. Isolated cells were cultured on coverslips coated with 10 μg/ml collagen I for 24 hours in normal fibroblast medium. Cells then were cultured an additional 24 hours in low serum medium as described above. Following culture in low serum medium, various doses of IL-33 were used to treat cells for 24 hours. BrdU (20 μM) was added to the medium for the final 24 hours of IL-33 treatment. After rinsing the coverslips with PBS, cells were fixed in absolute ethanol containing 10 mM glycine (pH 2.0) at −20°C for 30 minutes. The incorporation of BrdU was detected by immunocytochemical staining (Roche Applied Science; Indianapolis, IN). Following immunocytchemical staining with anti-BrdU, cells were subjected to 4′-6-diamidino-2-phenylindole (DAPI) staining to visualize all cell nuclei. Cells were examined by using a Nikon E600 fluorescent microscope and the ratio of BrdU positive cells to total cell numbers were determined. Three independent experiments were repeated. For each slide ten random fields were examined and stained cells counted.
2.6. Fibroblast collagen gel contraction assay
The three-dimensional collagen gel system is widely used for evaluating fibroblast and collagenous matrix interactions [44–46]. Briefly, cells were kept in normal fibroblast medium followed by culture in low serum medium for 24 hours before treatment. Cells were then detached by trypsin/EDTA digestion, pelleted by centrifugation and resuspended in low serum medium. A cell-collagen mixture was made by mixing an equal volume of resuspended cells with a collagen solution which contained 1.25 mg/ml collagen to reach a final concentration of 100,000 cells per milliliter. Then one milliliter of this mixture was added into each well of 24-well plates that had been pre-coated with bovine serum albumin to prevent the collagen from attaching to the culture plastic. Cell/collagen mixtures were allowed one hour at 37°C to polymerize to form the collagen gels and one milliliter of low serum medium was added to each well. The collagen gels were gently detached from the wells to allow the gels to float in the medium. Various doses of IL-33 (0–100 ng/ml) were administrated for 24 hours. Pictures of collagen gels were taken and the relative contraction of collagen gels was represented by the ratio of the final collagen gel surface area to the original collagen gel surface area. Each condition was measured in triplicate and three independent experiments were conducted.
2.7. Fibroblast migration assay
A scratch assay described in previous papers [47] was used to evaluate the effect of IL-33 on relative rat cardiac fibroblast migration. Briefly, cells were grown in normal fibroblast medium until confluence in 6-well plates followed by 24 hours of culture in low serum medium. A scratch was gently introduced in the center of the cell culture with a pipette tip. After rinsing gently with Moscona’s saline solution, fresh low serum medium was added and various doses of IL-33 were added. Cells were allowed to migrate into the denuded area for 24 hours. The relative migration distance in 24 hours by rat cardiac fibroblasts was measured using ImageJ (National Institutes of Health; Bethesda, MD). The relative migration of rat cardiac fibroblasts was represented as the ratio of the distance migrated by IL-33- treated cells to that of untreated control cells. Each condition was carried out in triplicate and three independent experiments were conducted.
2.8. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) immunocytochemistry and quantification
Experiments were performed to evaluate the effects of IL-33 on the translocation of NF-kB from the cytoplasm to the nucleus of rat cardiac fibroblasts. Isolated rat cardiac fibroblasts were cultured on glass coverslips coated with collagen I (10 μg/ml) and serum starved as described above until cytokine treatment. Various doses of IL-33 were administrated into the low serum medium for 15 and 30 minutes. The cells were fixed with 3% paraformaldehyde at 37°C for 20 minutes, rinsed in PBS, then incubated with 0.1% Triton X-100 for 10 minutes at room temperature. Antibodies used for imnnunocytochemistry staining were primary anti-NF-kB p65 subunit antibody (Rockland Immunochemicals, Inc.; Gilbertsville, PA catalog number: 100-401-264) and fluorescein isothiocyanate conjugated secondary goat anti-rabbit antibody (invitrogen, Carlsbad, CA) [48]. They were both used at 1:50 dilution and DAPI was used to stain all cell nuclei. Before incubation with the primary antibody, samples were blocked with normal donkey serum. Coverslips were mounted onto glass slides using glycerol/PBS (1:1) as mounting medium and visualized using a Nikon E600 fluorescent microscope.
NF-kB activation was represented by the ratio of cytoplasmic to nuclear localization of the NF-kB p65 subunit. Quantification was accomplished by using ImageJ software (National Institutes of Health; Bethesda, MD) [49]. Briefly, photographs were taken of ten random fields from each sample. The cytoplasm and nuclear regions were determined by using ImageJ binary mask and image subtraction calculation. Then histograms of NF-kB p65 subunit staining in the cytoplasm and nuclei of each field were created and data exported into Excel files. All samples in the same experiment were recorded under the same microscopic settings to make pictures comparable to each other. Each sample had duplicate slides and five independent experiments were conducted.
2.9. Statistical analysis
Results were shown as average ± SEM. Statistical significances (P<0.05) were determined by applying one way- ANOVA followed by Dunnett’s Post-hoc test or t test as appropriate. Data were analyzed and graphed by using Prism 3 software. (GraphPad Software, Inc.; San Diego, CA)
3. Results
3.1. Rat cardiac fibroblast IL-33 receptor expression
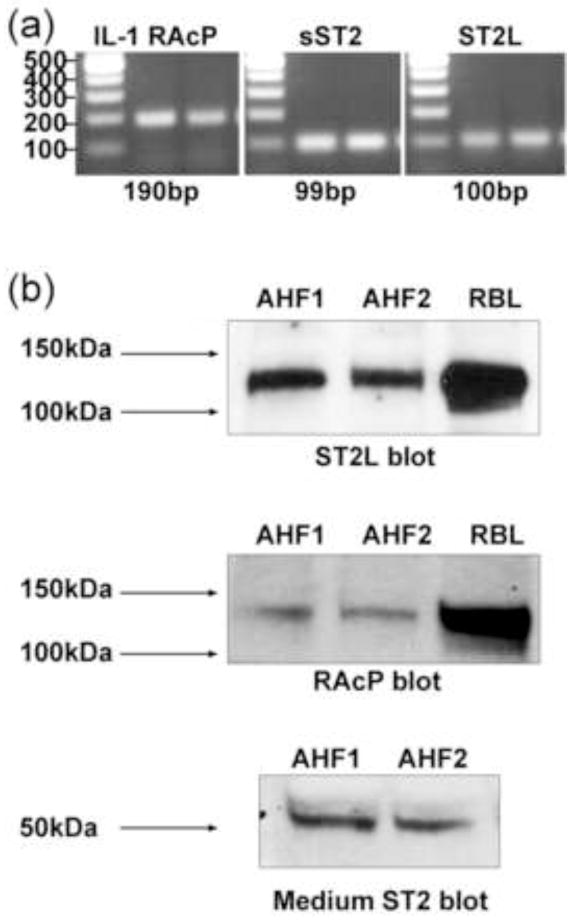
The expression of IL-33 receptors has not been examined in cardiac fibroblasts, thus we first examined the expression of the functional IL-33 receptor, ST2L, the soluble isoform, sST2, and the co-receptor RAcP in isolated adult rat cardiac fibroblasts. For detecting mRNA expression, RT-PCR with receptor- specific primers was conducted. This analysis showed that cultured adult rat cardiac fibroblasts yielded positive products for RAcP, sST2 and ST2L (Fig. 1a). PCR products were not seen in control samples lacking reverse transcriptase in the cDNA synthesis step (not shown). Western blots showed that rat cardiac fibroblasts also express the ST2L protein (Fig. 1b). The size of the predominant protein detected by the anti-ST2L serum (approximately 130–140 kDa) is larger than the previously published size (60–70 kDa) [50, 51]. We speculate that the larger size detected by western blots is due to the interaction of ST2L (60–70 kDa) and RAcP (66 kDa) in rat cardiac fibroblasts [52]. We further confirmed that a RAcP antibody recognized this same protein band by western blot analysis, which further suggests that this is a complex containing ST2L and RAcP. To further tests the ST2 antiserum, conditioned medium from rat cardiac fibroblasts was assayed by western blots. The secreted sST2 was detected in conditioned medium illustrating the specificity of the antiserum for ST2 and indicating that rat cardiac fibroblasts release the sST2 isoform (Fig. 1b). Because both mast cells and basophils have been documented to express functional IL-33 receptors, extracts from the rat basophilic leukemia cell line, RBL 2H3, were used as positive controls [53–55]. We further confirmed the ST2L is on the rat cardiac fibroblast cell surface by biotin-labeling cell surface proteins and immunoprecipitating with anti-ST2L antibodies. The immunoprecipitation data showed that there was cell surface expression of ST2L in both RBL 2H3 cells and rat cardiac fibroblasts (data not shown).
Fig. 1.
3.2. IL-33 effects on rat cardiac fibroblast ECM expression
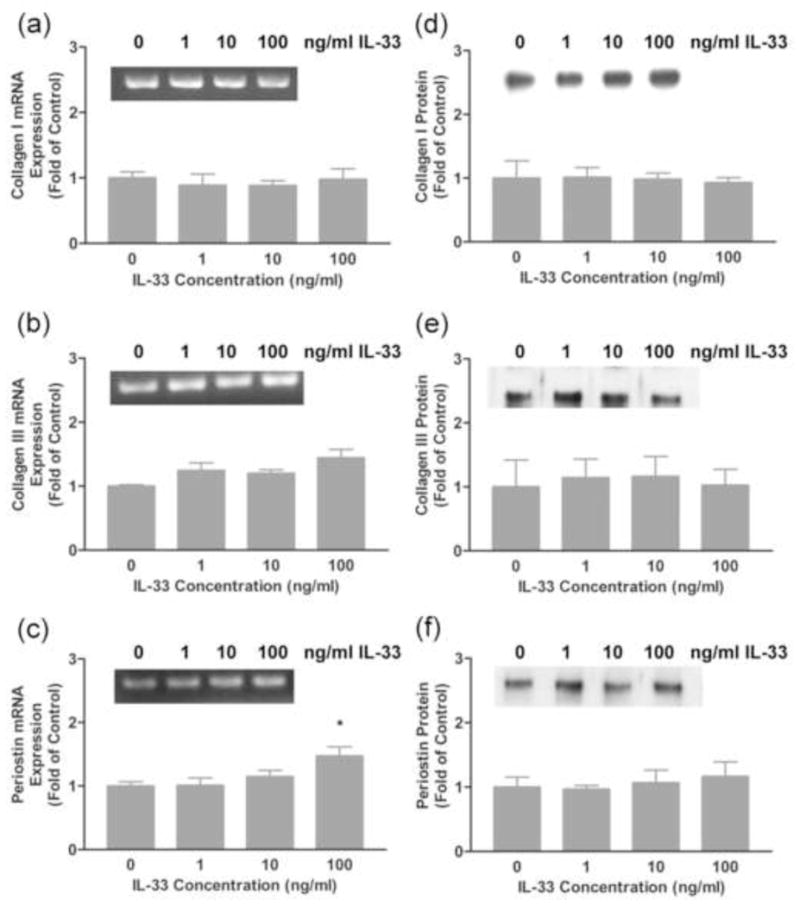
Previous studies showed that administration of IL-33 attenuated cardiac fibrosis in mice following transaortic banding [33]. These results prompted us to evaluate the possible effects of IL-33 on the expression of ECM components by rat cardiac fibroblasts. Cells were treated with various doses of IL-33 for 24 hours and mRNA expression of the major cardiac ECM components was evaluated by semi-quantitative RT-PCR. Expression of the α1(I) collagen or α1(III) collagen mRNA, components of type I and type III collagens, respectively, were not significantly altered following 24 hours of IL-33 treatment (Fig. 2a–b). In contrast, the expression of periostin mRNA, a recently described ECM component that is elevated in fibrosis, was slightly increased upon 24 hours treatment with the highest dose of IL-33 tested (Fig. 2c). The secretion of procollagen forms of collagen I (α1), collagen III (α1) and perisotin proteins by rat cardiac fibroblasts was evaluated in conditioned medium by western blots. Consistent with the mRNA expression data, collagen type I and collagen type III content in the medium were not changed upon IL-33 treatment; however, periostin was slightly increased upon treatment of cells with IL-33 even though this increase did not reach statistical significance (Fig. 2d–f).
Fig. 2.
3.3. Effects of IL-33 on fibroblast activity
Cardiac fibrosis is frequently associated with an enhanced proliferative activity and increased density of interstitial fibroblasts [44, 45]. BrdU incorporation assays were used to evaluate the effects of IL-33 on cultured rat cardiac fibroblast proliferation. The results showed that administration of various doses of IL-33 for 24 hours had no effects on the proliferation of fibroblasts isolated from adult rat hearts (Fig. 3a). The averaged percentage of cells incorporating BrdU was approximately 15% under all of the treatment conditions compared in this study.
Fig. 3.
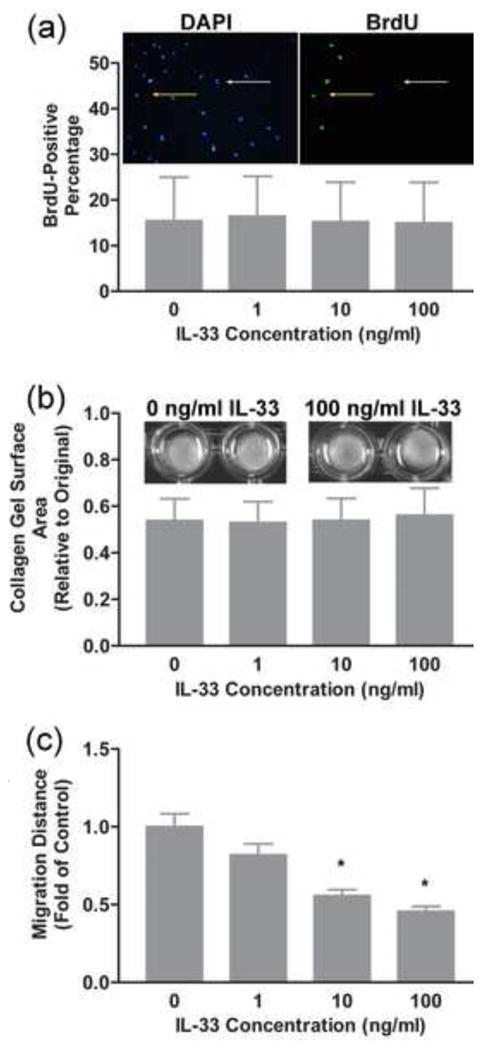
Collagen gel contraction assays have been widely used to evaluate the interaction of fibroblasts with collagenous matrix and their ability to remodel this matrix [46, 56, 57]. Over time, fibroblasts interact with the collagenous fibers and contract the gel, thus reducing the gel surface from its original size. A number of factors including transforming growth factor-beta (TGF-β), platelet-derived growth factor (PDGF) and IL-18 [18, 57, 58] have been shown to enhance contraction of three-dimensional collagen gels by fibroblasts. Treatment of rat cardiac fibroblasts with IL-33 had no effect on the contraction of collagen gels (Fig. 3b). Under the conditions used in the present study, all of the fibroblasts contracted collagen gels to approximately 50% of their original size over the 24 hour period examined.
In some conditions, cardiac fibrosis is associated with increased migration of cardiac fibroblasts. A scratch assay was used to evaluate the potential modulation of the migratory ability of cardiac fibroblasts by IL-33 [47, 59]. A denuded area was introduced in the center of confluent cultured fibroblasts and the ability of fibroblasts to migrate into the denuded area measured following treatment of the cells with different doses of IL-33 (Fig. 3c). Over a 24 hour period, rat cardiac fibroblasts demonstrated a significant decrease in migratory ability following treatment with the higher doses of IL-33 used in the present study (10 ng/ml and 100 ng/ml).
3.4. Effects of IL-33 on rat cardiac fibroblast cytokine expression
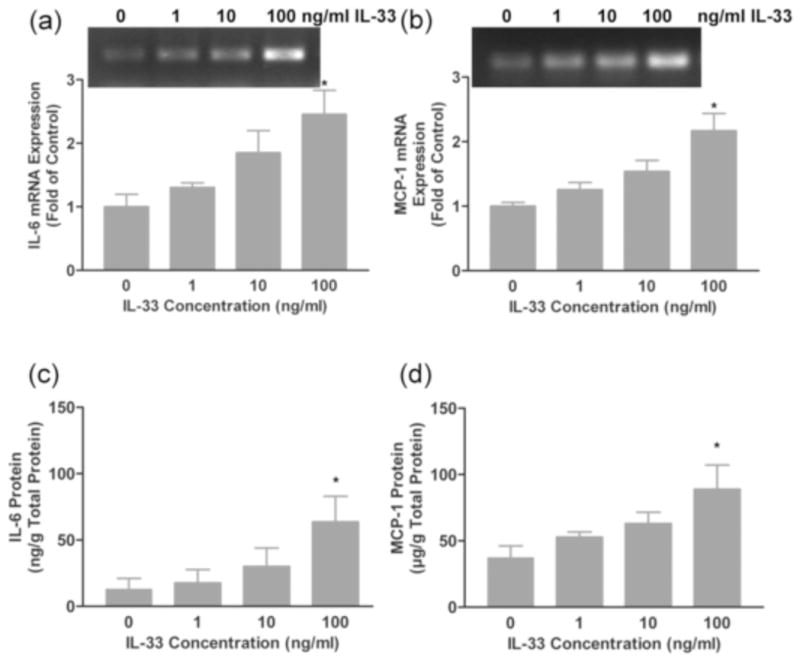
A number of recent studies have illustrated that cardiac fibroblasts play an important role in the heart as sentinel cells [9, 60]. In response to various perturbations, these cells are able to secrete cytokines and other bioactive molecules that modulate functions of other cells in the myocardium [10]. Experiments were performed to elucidate the effects of IL-33 on the expression of other cytokines by fibroblasts. Rat cardiac fibroblasts were treated for 24 hours with varying doses of IL-33 and the expression of TGF-β, IL-6, interleukin-13 (IL-13) and MCP-1 were assayed by semi-quantitative RT-PCR. There were no detectable changes in TGF-β, nor the Th2- associated cytokine IL-13 following treatment of fibroblasts with IL-33 (data not shown). In contrast, the expression of IL-6 and MCP-1 mRNAs was elevated in a dose-dependent manner by IL-33 treatment (Fig. 4 a–b). Analysis of IL-6 and MCP-1 protein accumulation in conditioned medium was evaluated by ELISA. These analyses illustrated increases in IL-6 and MCP-1 protein accumulation in response to IL-33 treatment of rat cardiac fibroblasts similar to the increases seen in mRNA levels (Fig. 4 c–d).
Fig. 4.
3.5. IL-33-induced signaling pathways
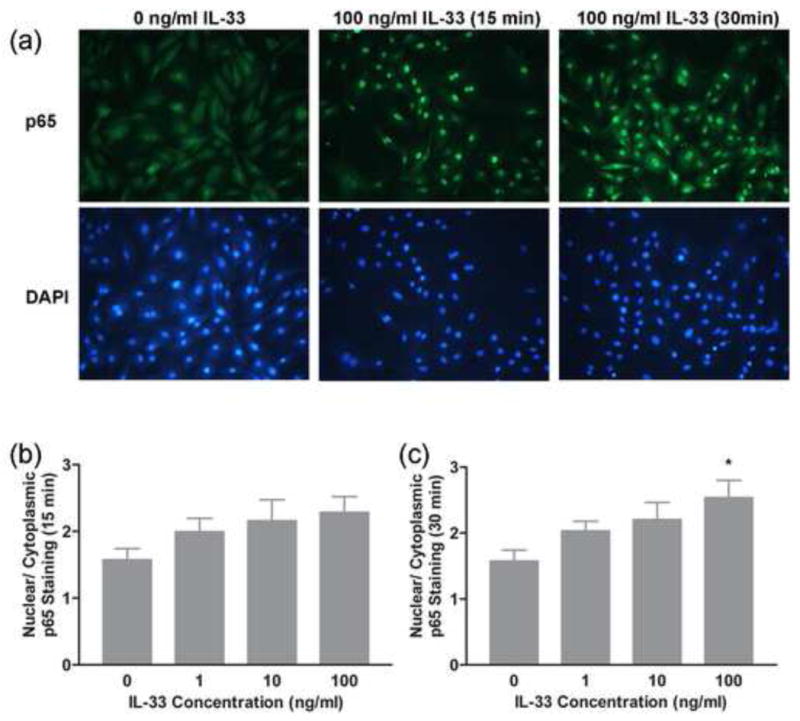
The activation of NF-kB and mitogen activated protein kinase (MAPK) pathways has been reported in multiple cell types following IL-33 treatment but this has not been examined in cardiac fibroblasts [25, 33, 61]. Western blots and immunocytochemistry were utilized to evaluate the activation of NF-kB and MAPK pathways, respectively following IL-33 treatment. Western blots were performed with antisera specific to the phosphorylated forms of Erk, JNK and p38 MAPKs to evaluate their activation following IL-33 treatment. The western blot data showed that all three MAPKs examined here were activated by IL-33 significantly and in a dose-dependent manner (Fig. 5a–c). For examination of NF-kB activation, following 15 to 30 minutes of IL-33 treatment, rat cardiac fibroblasts were fixed and stained immunocytochemically with an NF-kB antibody to evaluate the translocation of NF-kB. Cells demonstrated significant NF-kB activation indicated by enhanced translocation of NF-kB from the cytoplasm to the nucleus of the cells after 30 minutes of IL-33 treatment. This was a dose-dependent response (Fig. 6a–c). Thus, we demonstrated that IL-33 treatment can rapidly activate multiple MAPKs as well as the NF-kB pathway in adult rat cardiac fibroblasts.
Fig. 5.
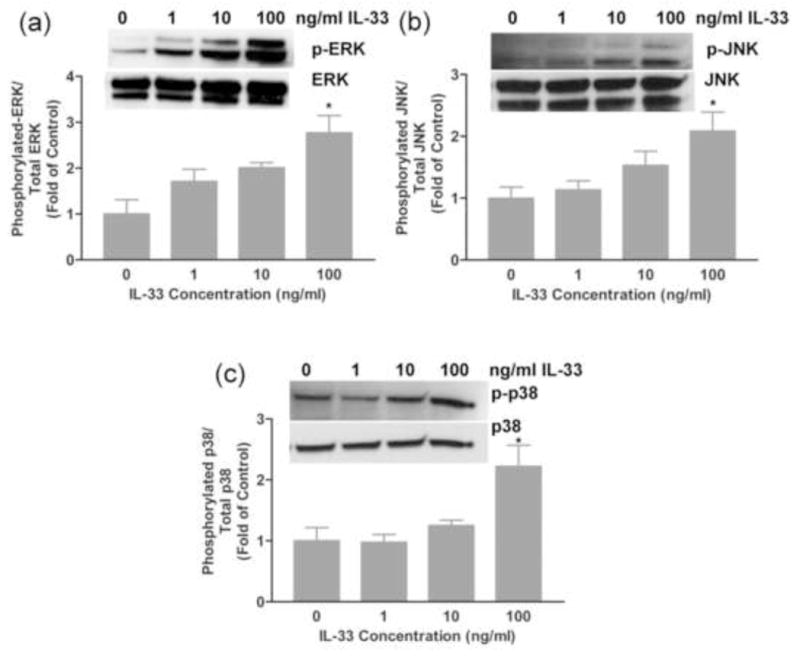
Fig. 6.
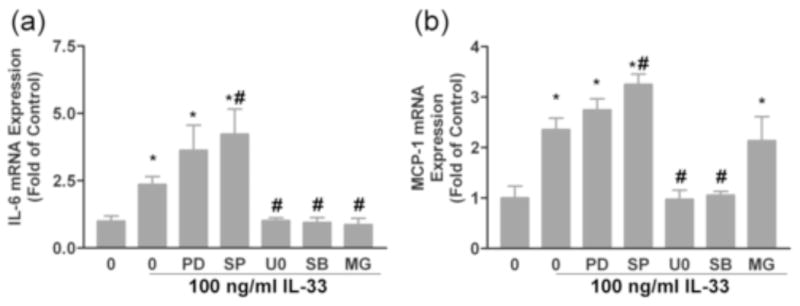
Experiments were performed with pharmacological inhibitors to determine whether specific signaling pathways are involved in the increased IL-6 and MCP-1 expression in response to IL-33 treatment. Cells were treated with inhibitors for 60 minutes prior to treatment with100 ng/ml IL-33. The expression of IL-6 and MCP-1 mRNAs was subsequently evaluated by RT-PCR as performed in Figure 4. As shown by the RT-PCR data, pre-treatment with the p38 inhibitor SB203580 and the mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitor U0126 prevented the stimulation of IL-6 and MCP-1 by IL-33. On the other hand, the mitogen-activated protein kinase kinase 1 (MEK1) and JNK inhibitors, PD98059 and SP600125, did not inhibit the IL-33-induced IL-6 and MCP-1 gene expression and SP600125 even enhanced the expression of these cytokines/chemokines (Fig. 7 a–b). The NF-kB inhibitor, MG132 also inhibited the production of IL-6 following IL-33 treatment (Fig. 7 a). Interestingly, the expression of MCP-1 by cardiac fibroblasts was not significantly attenuated by the NF-kB pathway inhibitor MG132 (Fig. 7 b).
Fig. 7.
4. Discussion
Cardiac fibrosis is a common pathological process shared by multiple major heart diseases. Though the process of cardiac fibrosis has been extensively investigated, many questions remain regarding the mechanisms of this process and there are currently few therapeutic approaches directed at preventing its progression. Because cardiac fibroblasts are the primary cells responsible for ECM deposition and turnover, it is important to understand the regulation of the function and gene expression of these cells. Various biologically- active factors including growth factors, neurohormonal mediators, cytokines and chemokines have been demonstrated to stimulate cardiac fibroblast function and gene expression. However, fewer biochemical factors have been identified that inhibit the pro-fibrotic response. Based upon recent studies using a mouse pressure overload model, IL-33 has emerged as a promising cytokine to inhibit cardiac fibrosis.
IL-33 was first discovered in vascular endothelial cells; however, its functional receptor has been found to be expressed by a number of immune cell types including Th2 lymphocytes, mast cells and basophils. It has been reported that IL-33 is tightly related to Th2 autoimmune diseases such as arthritis, asthma and systemic sclerosis [31, 32, 62]. Further investigations revealed that IL-33 also has significant roles in regulating cardiac function [34–36]. In 2007, IL-33 was reported to inhibit cardiac hypertrophy and fibrosis under pathological pressure overload conditions [33]. It was also reported to inhibit artherosclerotic plaque formation, and the expression of its receptor, ST2, was found to correlate with heart failure severity. Recently, IL-33 levels were found to be significantly elevated upon cyclic stretch of cardiac fibroblasts in vitro, and the administration of IL-33 was shown to inhibit myocyte amino acid incorporation and growth when exposed to hypertrophic stimuli. The direct effect of IL-33 on cardiac fibroblasts has not been addressed and was the basis of the present study.
The functional receptor of IL-33, ST2L, was originally found on Th2 lymphocytes and mast cells, and it is critical to the induction of Th2-associated cytokines from mouse Th2 lymphocytes [54, 63–65]. Besides immune cells, ST2 expression has also been detected in myocytes and endothelial cells [33, 66]. Recent studies also illustrated ST2 expression in activated skin myofibroblasts [67]. We demonstrate here for the first time that fibroblasts isolated from normal adult rat hearts express ST2L at both mRNA and protein levels. Rat cardiac fibroblasts also express the accessory protein RAcP thus suggesting that these cells can form the functional IL-33 receptor complex with RAcP and transduce downstream signaling events upon IL-33 stimulation.
Based on the previous report that IL-33 inhibits pressure overload- induced cardiac hypertrophy and fibrosis we predicted that IL-33 would directly affect ECM production by rat cardiac fibroblasts. Previous research from the mouse TAC model revealed that in vivo administration of IL-33 attenuated myocyte hypertrophy, reduced cardiac interstitial fibrosis and also maintained cardiac wall compliance [33]. They showed a decrease in macrophage infiltration, NF-kB activation and also less atrial natriuretic petide and brain natriuretic peptide production in the wildtype mice heart compared to the ST2 knock out TAC samples. Additional research from the same group demonstrated that IL-33 can inhibit myocyte hypertrophy and apoptosis upon increased mechanical load and myocardial infarction [34]. But there is no evidence yet to demonstrate a direct relationship between IL-33 administration and cardiac fibroblast function [68]. Outside the cardiac system, IL-33 expression has been correlated with liver, lung and skin fibrosis [37, 62, 69]. Interestingly, from our data, IL-33 had no affect on the expression of the major interstitial collagens by isolated rat cardiac fibroblasts (collagen type I and type III). The mRNA and protein expression of collagen I and collagen III were not changed upon 24 hours of IL-33 treatment, and the RNA expression of periostin was even slightly elevated by IL-33 treatment. The discrepancy between the in vitro data presented here and the previous reports illustrating that IL-33 prevents fibrosis in vivo may be due to the differences in the in vivo and in vitro environments. Numerous biochemical and mechanical forces are active in the pressure overloaded heart, many of which are acting to stimulate fibroblast gene expression. In the controlled in vitro environment, these stimulatory factors have been removed and IL-33 did not affect basal ECM expression by the fibroblasts. And IL-33 was found to have limited effects on bone remolding recently [70]. Further experiments are necessary to determine whether IL-33 can counter the actions of pro-fibrotic stimuli like TGF-β or mechanical stretch. An additional explanation for the discrepancy between the in vivo and in vitro results is that IL-33 may affect fibrosis through other cell types. For instance, IL-33 has been shown to inhibit hypoxia-induced myocyte apoptosis following ischemia/reperfusion injury [34]. Preservation of myocyte survival could in turn dampen the fibrotic response in response to ischemia/reperfusion injury. IL-33 has also been shown to affect inflammatory cells such as mast cells, which could in turn impact fibrosis. Further experiments with co-culture models or cell-specific knockouts will be required to address these possibilities.
Besides directly modulating ECM equilibrium, cardiac fibroblasts also sense and integrate various environmental stimuli and response by secreting bioactive molecules such as cytokines and vasoactive peptides and growth factors [10]. In addition to their role in production of the ECM network and secretion of bioactive regulatory molecules, cardiac fibroblasts are tightly associated with other cardiac cells via cell-cell contacts. Thus, cardiac fibroblasts are in a key position to serve as sentinel cells in the myocardium [9]. Here we demonstrated for the first time that IL-33 stimulates IL-6 and MCP-1 production by adult rat cardiac fibroblasts. This is consistent with previous reports that IL-33 promotes IL-6 and/or MCP-1 expression in mast cells, basophils and endothelial cells [54, 66, 71, 72]. The physiological or pathological effects of this cardiac fibroblast response are not yet clear. The role of IL-6 in the heart in general and in cardiac fibrosis in particular is still under considerable debate. IL-6 is a pleiotropic cytokine that plays important roles in cell development, survival, and apoptosis in a cell and organ-specific manner [73]. As to its roles in cardiac physiology and pathophysiology, some research groups have shown that consistent activation of IL-6 signaling causes cardiac fibroblast proliferation, cardiac hypertrophy and induces a pro-fibrotic effect [74, 75]. In contrast, other researchers have argued that IL-6 reduces myocardial damage in mice with viral myocarditis and thus may inhibit cardiac fibrosis indirectly. IL-6 has also been reported to induce ECM degradation by decreasing collagen production and increasing matrix metalloprotease expression in the heart [10, 76]. Recently, the deleterious effect of loss of IL-6 signaling in the heart was demonstrated [77]. The IL-6 knockout mice demonstrated a dilated, functionally impaired fibrotic heart. Thus, IL-6 was proven to be necessary for maintaining basic functions of the cardiac tissue and also has an indispensable protective role under stressed conditions such as infarction and viral myocarditis. MCP-1 on the other hand, appears to have an overall stimulatory role on fibrosis. Under many disease conditions, MCP-1 promotes the attraction of monocytes and lymphocytes, mediates inflammation in cardiac tissue and promotes arthrosclerotic plaque formation [78]. In contrast, MCP-1 has also been demonstrated to have a protective role for cardiac tissue in disease conditions such as myocardial ischemia, ischemic preconditioning and myocardial infarct healing [79–82]. Further in vitro and in vivo experiments are warranted to evaluate the consequences of IL-33-induced IL-6 and MCP-1 expression by heart fibroblasts.
As discussed earlier, IL-33 has been shown to activate MAPKs and/or NF-kB signaling pathways in mast cells, basophils, T cells, endothelial cells and cardiac myocytes [33, 54, 61, 66, 83, 84]. Exposure of human basophils to IL-33 resulted in the activation of p38, JNK and Erk members of the MAPK family as well as NF-kB [83]. This was followed by enhanced expression and secretion of interleukin-4 (IL-4) and IL-13; however, the dependence of IL-33-induced cytokine expression on specific signaling pathways was not determined. Inhibition studies have been carried out to illustrate that the p38 MAPK pathway was important for IL-33-induced cytokine and chemokine production in several cell types including T cells and endothelial cells [61, 84]. The Erk pathway was shown to be important for IL-33-induced production of interleukin-8 from epithelial cells and also for interleukin-5 (IL-5) and IL-13 production in T cells [61, 84]. JNK signaling was also demonstrated to be indispensable for IL-33- induced IL-5 and IL-13 production in T cells [83]. Our data showed that within 15 minutes, IL-33 treatment activated all three MAPK pathways (Erk, JNK and p38) in cardiac fibroblasts. Interestingly, activation of these signaling pathways did not appear to correlate to alterations in cardiac fibroblast behavior. A similar phenomenon was recently reported following treatment of human cardiac fibroblasts with the High Mobility Box 1 Protein [85]. Administration of this cytokine resulted in activation of multiple signaling pathways and induction of cytokine/growth factor production; however, there was no direct effect on cell behavior (proliferation). Because IL-33 treatment resulted in increased IL-6 and MCP-1 production, we utilized specific MAPK pathway inhibitors to determine whether activation of these pathways is important for IL-33-induced cytokine/chemokine production. Our data showed that inhibition of p38 MAPK totally eliminated the increased production of IL-6 and MCP-1 following IL-33 treatment of adult rat cardiac fibroblasts. This is consistent with the previous report that p38 is specifically important in controlling cell inflammatory responses and survival [86–88]. At the same time, our data showed that Erk pathway inhibition also prevented IL-33-induced IL-6 and MCP-1 production. The Erk pathway was reported previously to be important in the production of IL-6 in cardiac fibroblasts following treatment with angiotensin II and in human colonic myofibroblasts stimulated by interleukin-17 [89, 90]. Collectively, our data and that with other cells types, illustrates that activation of MAPK pathways is essential for enhanced cytokine production following exposure to IL-33. The relative contributions of these pathways are likely to be dependent upon the specific cell type and microenvironment.
The activation of NF-kB by IL-33 stimulation has previously been reported in myocytes, mast cells and basophils [33, 54, 91]. IL-33 binding to ST2L triggers the accumulation of adaptor proteins such as myeloid differentiation primary response gene 88 (MyD88) and TNF receptor associated factor 6 (TRAF6). This is followed by NF-kB translocation to the nucleus and regulation of downstream target gene expression. In the present studies, immunocytochemical staining illustrated rapid translocation (within 15 minutes) of NF-kB to the nucleus of rat cardiac fibroblasts. We used the NF-kB inhibitor MG132 to evaluate the role of NF-kB signaling in the stimulation of IL-6 and MCP-1 expression by IL-33. Our data showed that administration of MG132 attenuated IL-6 production but MG132 did not have effects on IL-33- induced MCP-1 production. Published research had demonstrated that NF-kB activation was upstream of IL-33 induced IL-6 and MCP-1 production in mouse embryonic fibroblasts expressing ST2L [92]. In addition, NF-kB activation was also demonstrated to be necessary for IL-33 induced IL-13 production in a human chronic myelogenous leukemia cell line (KU812) [91]. Interestingly our data illustrating that the activation of NF-kB pathway is not necessary for IL-33 induced MCP-1 expression suggests that different pathways are required for specific downstream effects of IL-33.
Our data showed that administration of IL-33 directly inhibited the ability of rat cardiac fibroblasts to migrate in a wound healing assay. During myocardial fibrosis, cardiac fibroblast number typically increases. The increase in fibroblast number is likely the result of proliferation of resident fibroblasts and the recruitment/differentiation of fibroblast precursors such as fibrocytes [93–95]. The data presented herein suggests that IL-33 may inhibit migration of fibroblasts or their precursors into the stressed myocardium. To our knowledge, this potential role of IL-33 has not been examined in vivo. Studies with IL-33 transgenic mice would be necessary for examining this possibility. Interestingly, IL-33 has been previously shown to be a chemoattractant for Th2 cells and neutrophils [96, 97]. The decreased migration observed in our experiments could illustrate that the effects of IL-33 on migration are cell type-specific. That is, IL-33 may enhance migratory activity of inflammatory cells, but reduce migration of other cell types.
Overall, our data demonstrates for the first time that cardiac fibroblasts express the IL-33 receptors and respond directly to this relatively novel cytokine. Our research begins to shed light on the effects of IL-33 on cardiac fibroblast function and gene expression. Contrary to our original hypothesis, it does not appear that IL-33 affects normal expression of ECM components by rat cardiac fibroblasts. Further experiments will be required to determine whether IL-33 attenuates ECM expression that is stimulated by pro-fibrotic factors. Experiments are also needed to elucidate whether the anti-fibrotic effects of IL-33 are through other cell types. Alternatively, treatment of cardiac fibroblasts with IL-33 resulted in increased expression of IL-6 and MCP-1, two biochemical factors that have been associated with diverse aspects of cardiovascular disease. Future experiments will be aimed at determining the downstream effects of fibroblast-generated cytokines in myocardial function.
IL-33/ST2 signaling was recently reported to have cardiac protective roles.
We detected ST2 expression on rat cardiac fibroblasts.
We investigated the regulatory roles of IL-33 on rat cardiac fibroblasts.
IL-33 induced cell inflammatory cytokine but not the collagen production.
MAPK and NF-kB pathways are important for this induced cytokine production.
Acknowledgments
The study was supported by the NIH grant to W. Carver (HL0803441). We thank Charity F. Dunn for excellent technical assistance. We also would like to thank Cheryl Cook for her great help in cell culture experiments and Dr. John Fuseler for his generous suggestions for the NF-kB immunocytochemistry experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 01.Barringer TA. Mediterranean diets and cardiovascular disease. Curr Atheroscler Rep. 2001;3:437–445. doi: 10.1007/s11883-001-0033-8. [DOI] [PubMed] [Google Scholar]
- 02.Gradman AH, Wilson JT. Hypertension and diastolic heart failure. Curr Cardiol Rep. 2009;11:422–429. doi: 10.1007/s11886-009-0061-5. [DOI] [PubMed] [Google Scholar]
- 03.Heeneman S, Cleutjens JP, Faber BC, Creemers EE, van Suylen RJ, Lutgens E, Cleutjens KB, Daemen MJ. The dynamic extracellular matrix: intervention strategies during heart failure and atherosclerosis. J Pathol. 2003;200:516–525. doi: 10.1002/path.1395. [DOI] [PubMed] [Google Scholar]
- 04.Hess OM, Felder L, Krayenbuehl HP. Diastolic function in valvular heart disease. Herz. 1991;16:124–129. [PubMed] [Google Scholar]
- 05.Khan R, Sheppard R. Fibrosis in heart disease: understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology. 2006;118:10–24. doi: 10.1111/j.1365-2567.2006.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 06.Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265–272. doi: 10.1093/cvr/cvq308. [DOI] [PubMed] [Google Scholar]
- 07.Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. 2002;27:92–98. doi: 10.1007/s00059-002-2354-y. [DOI] [PubMed] [Google Scholar]
- 08.Brower GL, Gardner JD, Forman MF, Murray DB, Voloshenyuk T, Levick SP, Janicki JS. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg. 2006;30:604–610. doi: 10.1016/j.ejcts.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 09.Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 10.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin E, Calvano SE, Lowry SF. Inflammatory cytokines and cell response in surgery. Surgery. 2000;127:117–126. doi: 10.1067/msy.2000.101584. [DOI] [PubMed] [Google Scholar]
- 13.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 14.Curfs JH, Meis JF, Hoogkamp-Korstanje JA. A primer on cytokines: sources, receptors, effects, and inducers. Clin Microbiol Rev. 1997;10:742–780. doi: 10.1128/cmr.10.4.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelso A. Cytokines: principles and prospects. Immunol Cell Biol. 1998;76:300–317. doi: 10.1046/j.1440-1711.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 16.Yndestad A, Damås JK, Øie E, Ueland T, Gullestad L, Aukrust P. Role of inflammation in the progression of heart failure. Curr Cardiol Rep. 2007;9:236–241. doi: 10.1007/BF02938356. [DOI] [PubMed] [Google Scholar]
- 17.Nakajima H, Takatsu K. Role of cytokines in allergic airway inflammation. Int Arch Allergy Immunol. 2007;142:265–273. doi: 10.1159/000097357. [DOI] [PubMed] [Google Scholar]
- 18.Fix C, Bingham K, Carver W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine. 2011;53:19–28. doi: 10.1016/j.cyto.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Onda H, Kasuya H, Takakura K, Hori T, Imaizumi T, Takeuchi T, Inoue I, Takeda J. Identification of genes differentially expressed in canine vasospastic cerebral arteries after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 1999;19:1279–1288. doi: 10.1097/00004647-199911000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Baekkevold ES, Roussigné M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, Brandtzaeg P, Erard M, Haraldsen G, Girard JP. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. 2003;163:69–79. doi: 10.1016/S0002-9440(10)63631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tominaga S. A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett. 1989;258:301–304. doi: 10.1016/0014-5793(89)81679-5. [DOI] [PubMed] [Google Scholar]
- 22.Klemenz R, Hoffmann S, Werenskiold AK. Serum- and oncoprotein-mediated induction of a gene with sequence similarity to the gene encoding carcinoembryonic antigen. Biochemistry. 1989;86:5708–5712. doi: 10.1073/pnas.86.15.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Tzimas MN, Griswold DE, Young PR. Expression of ST2, an interleukin-1 receptor homologue, is induced by proinflammatory stimuli. Biochem Biophys Res Commun. 1997;235:474–478. doi: 10.1006/bbrc.1997.6810. [DOI] [PubMed] [Google Scholar]
- 24.Bergers G, Reikerstorfer A, Braselmann S, Graninger P, Busslinger M. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. EMBO J. 1994;13:1176–1188. doi: 10.1002/j.1460-2075.1994.tb06367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 26.Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol. 2007;179:2551–2555. doi: 10.4049/jimmunol.179.4.2551. [DOI] [PubMed] [Google Scholar]
- 27.Lingel A, Weiss TM, Niebuhr M, Pan B, Appleton BA, Wiesmann C, Bazan JF, Fairbrother WJ. Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors--insight into heterotrimeric IL-1 signaling complexes. Structure. 2009;17:1398–1410. doi: 10.1016/j.str.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah RV, Januzzi JL., Jr ST2: a novel remodeling biomarker in acute and chronic heart failure. Curr Heart Fail Rep. 2010;7:9–14. doi: 10.1007/s11897-010-0005-9. [DOI] [PubMed] [Google Scholar]
- 29.Pascual-Figal DA, Ordoñez-Llanos J, Tornel PL, Vázquez R, Puig T, Valdés M, Cinca J, de Luna AB, Bayes-Genis A MUSIC Investigators. Soluble ST2 for predicting sudden cardiac death in patients with chronic heart failure and left ventricular systolic dysfunction. J Am Coll Cardiol. 2009;54:2174–2179. doi: 10.1016/j.jacc.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 30.Bhardwaj A, Januzzi JL., Jr ST2: a novel biomarker for heart failure. Expert Rev Mol Diagn. 2010;10:459–464. doi: 10.1586/erm.10.25. [DOI] [PubMed] [Google Scholar]
- 31.Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. 2007;282:26369–26380. doi: 10.1074/jbc.M704916200. [DOI] [PubMed] [Google Scholar]
- 32.Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, Pitman N, Kurowska-Stolarska M, McKenzie AN, McInnes IB, Liew FY. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci U S A. 2008;105:10913–10918. doi: 10.1073/pnas.0801898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest. 2007;117:1538–1549. doi: 10.1172/JCI30634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seki K, Sanada S, Kudinova AY, Steinhauser ML, Handa V, Gannon J, Lee RT. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ Heart Fail. 2009;2:684–691. doi: 10.1161/CIRCHEARTFAILURE.109.873240. [DOI] [PubMed] [Google Scholar]
- 35.Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008;7:827–840. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, Baker AH, McInnes IB, Liew FY. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–346. doi: 10.1084/jem.20071868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rankin AL, Mumm JB, Murphy E, Turner S, Yu N, McClanahan TK, Bourne PA, Pierce RH, Kastelein R, Pflanz S. IL-33 Induces IL-13-Dependent Cutaneous Fibrosis. J Immunol. 2010;184:1526–1535. doi: 10.4049/jimmunol.0903306. [DOI] [PubMed] [Google Scholar]
- 38.Burgess ML, Terracio L, Hirozane T, Borg TK. Differential integrin expression by cardiac fibroblasts from hypertensive and exercise-trained rat hearts. Cardiovasc Pathol. 2002;11:78–87. doi: 10.1016/s1054-8807(01)00104-1. [DOI] [PubMed] [Google Scholar]
- 39.Diaz-Araya G, Borg TK, Lavandero S, Loftis MJ, Carver W. IGF-1 modulation of rat cardiac fibroblast behavior and gene expression is age-dependent. Cell Commun Adhes. 2003;10:155–165. [PubMed] [Google Scholar]
- 40.Zhao C, Zha Y, Wu X, Chen L, Shi J, Cui L. The quantification of ADAMTS4 and 8 expression and selection of reference genes for quantitative real-time PCR analysis in myocardial infarction. Biomed Pharmacother. doi: 10.1016/j.biopha.2010.12.002. In Press. [DOI] [PubMed] [Google Scholar]
- 41.Yndestad A, Marshall AK, Hodgkinson JD, Tham el L, Sugden PH, Clerk A. Modulation of interleukin signalling and gene expression in cardiac myocytes by endothelin-1. Int J Biochem Cell Biol. 2010;42:263–272. doi: 10.1016/j.biocel.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 42.Poobalarahi F, Baicu CF, Bradshaw AD. Cardiac myofibroblasts differentiated in 3D culture exhibit distinct changes in collagen I production, processing, and matrix deposition. Am J Physiol Heart Circ Physiol. 2006;291:H2924–2932. doi: 10.1152/ajpheart.00153.2006. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Stewart JA, Jr, Kane ID, Massey EP, Cashatt DO, Carver WE. Effects of elevated glucose levels on interactions of cardiac fibroblasts with the extracellular matrix. In Vitro Cell Dev Biol Anim. 2007;43:297–305. doi: 10.1007/s11626-007-9052-2. [DOI] [PubMed] [Google Scholar]
- 44.Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 45.Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, Imaizumi T. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004;43:739–745. doi: 10.1161/01.HYP.0000118584.33350.7d. [DOI] [PubMed] [Google Scholar]
- 46.Bell E, Ivarsson B, Merrill C. Production of a tissue-like structure by contraction of collagen lattices by human fibroblasts of different proliferative potential in vitro. Proc Natl Acad Sci U S A. 1979;76:1274–1278. doi: 10.1073/pnas.76.3.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 48.Rogers JA, Fuseler JW. Regulation of NF-kappaB activation and nuclear translocation by exogenous nitric oxide (NO) donors in TNF-alpha activated vascular endothelial cells. Nitric Oxide. 2007;16:379–391. doi: 10.1016/j.niox.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Noursadeghi M, Tsang J, Haustein T, Miller RF, Chain BM, Katz DR. Quantitative imaging assay for NF-kappaB nuclear translocation in primary human macrophages. J Immunol Methods. 2008;329:194–200. doi: 10.1016/j.jim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Espinassous Q, Garcia-de-Paco E, Garcia-Verdugo I, Synguelakis M, von Aulock S, Sallenave JM, McKenzie AN, Kanellopoulos J. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J Immunol. 2009;183:1446–1455. doi: 10.4049/jimmunol.0803067. [DOI] [PubMed] [Google Scholar]
- 51.Beltrán CJ, Núñez LE, Díaz-Jiménez D, Farfan N, Candia E, Heine C, López F, González MJ, Quera R, Hermoso MA. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1097–1107. doi: 10.1002/ibd.21175. [DOI] [PubMed] [Google Scholar]
- 52.Whelan R, Kim C, Chen M, Leiter J, Grunstein MM, Hakonarson H. Role and regulation of interleukin-1 molecules in pro-asthmatic sensitised airway smooth muscle. Eur Respir J. 2004;24:559–567. doi: 10.1183/09031936.04.00133803. [DOI] [PubMed] [Google Scholar]
- 53.Schneider E, Petit-Bertron AF, Bricard R, Levasseur M, Ramadan A, Girard JP, Herbelin A, Dy M. IL-33 activates unprimed murine basophils directly in vitro and induces their in vivo expansion indirectly by promoting hematopoietic growth factor production. J Immunol. 2009;183:3591–3597. doi: 10.4049/jimmunol.0900328. [DOI] [PubMed] [Google Scholar]
- 54.Moulin D, Donzé O, Talabot-Ayer D, Mézin F, Palmer G, Gabay C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine. 2007;40:216–225. doi: 10.1016/j.cyto.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 55.Andrade MV, Iwaki S, Ropert C, Gazzinelli RT, Cunha-Melo JR, Beaven MA. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur J Immunol. 2011;41:760–772. doi: 10.1002/eji.201040718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guidry C, Grinnell F. Contraction of hydrated collagen gels by fibroblasts: evidence for two mechanisms by which collagen fibrils are stabilized. Coll Relat Res. 1987;6:515–529. doi: 10.1016/s0174-173x(87)80050-x. [DOI] [PubMed] [Google Scholar]
- 57.Carver W, Molano I, Reaves TA, Borg TK, Terracio L. Role of the alpha 1 beta 1 integrin complex in collagen gel contraction in vitro by fibroblasts. J Cell Physiol. 1995;165:425–437. doi: 10.1002/jcp.1041650224. [DOI] [PubMed] [Google Scholar]
- 58.Tingström A, Heldin CH, Rubin K. Regulation of fibroblast-mediated collagen gel contraction by platelet-derived growth factor, interleukin-1 alpha and transforming growth factor-beta 1. J Cell Sci. 1992;102:315–322. doi: 10.1242/jcs.102.2.315. [DOI] [PubMed] [Google Scholar]
- 59.Stewart JA, Jr, Massey EP, Fix C, Zhu J, Goldsmith EC, Carver W. Temporal alterations in cardiac fibroblast function following induction of pressure overload. Cell Tissue Res. 2010;340:117–126. doi: 10.1007/s00441-010-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsueh WA, Law RE, Do YS. Integrins, adhesion, and cardiac remodeling. Hypertension. 1998;31:176–180. doi: 10.1161/01.hyp.31.1.176. [DOI] [PubMed] [Google Scholar]
- 61.Yagami A, Orihara K, Morita H, Futamura K, Hashimoto N, Matsumoto K, Saito H, Matsuda A. IL-33 mediates inflammatory responses in human lung tissue cells. J Immunol. 2010;185:5743–5750. doi: 10.4049/jimmunol.0903818. [DOI] [PubMed] [Google Scholar]
- 62.Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol. 2011;30:825–830. doi: 10.1007/s10067-011-1686-5. [DOI] [PubMed] [Google Scholar]
- 63.Lécart S, Lecointe N, Subramaniam A, Alkan S, Ni D, Chen R, Boulay V, Pène J, Kuroiwa K, Tominaga S, Yssel H. Activated, but not resting human Th2 cells, in contrast to Th1 and T regulatory cells, produce soluble ST2 and express low levels of ST2L at the cell surface. Eur J Immunol. 2002;32:2979–2987. doi: 10.1002/1521-4141(2002010)32:10<2979::AID-IMMU2979>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 64.Meisel C, Bonhagen K, Löhning M, Coyle AJ, Gutierrez-Ramos JC, Radbruch A, Kamradt T. Regulation and function of T1/ST2 expression on CD4+ T cells: induction of type 2 cytokine production by T1/ST2 cross-linking. J Immunol. 2001;166:3143–3150. doi: 10.4049/jimmunol.166.5.3143. [DOI] [PubMed] [Google Scholar]
- 65.Coyle AJ, Lloyd C, Tian J, Nguyen T, Erikkson C, Wang L, Ottoson P, Persson P, Delaney T, Lehar S, Lin S, Poisson L, Meisel C, Kamradt T, Bjerke T, Levinson D, Gutierrez-Ramos JC. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med. 1999;190:895–902. doi: 10.1084/jem.190.7.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aoki S, Hayakawa M, Ozaki H, Takezako N, Obata H, Ibaraki N, Tsuru T, Tominaga S, Yanagisawa K. ST2 gene expression is proliferation-dependent and its ligand, IL-33, induces inflammatory reaction in endothelial cells. Mol Cell Biochem. 2010;335:75–81. doi: 10.1007/s11010-009-0244-9. [DOI] [PubMed] [Google Scholar]
- 67.Manetti M, Ibba-Manneschi L, Liakouli V, Guiducci S, Milia AF, Benelli G, Marrelli A, Conforti ML, Romano E, Giacomelli R, Matucci-Cerinic M, Cipriani P. The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann Rheum Dis. 2010;69:598–605. doi: 10.1136/ard.2009.119321. [DOI] [PubMed] [Google Scholar]
- 68.Miller AM, Liew FY. The IL-33/ST2 pathway - A new therapeutic target in cardiovascular disease. Pharmacol Ther. 2011;131:179–186. doi: 10.1016/j.pharmthera.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 69.Marvie P, Lisbonne M, L’helgoualc’h A, Rauch M, Turlin B, Preisser L, Bourd-Boittin K, Théret N, Gascan H, Piquet-Pellorce C, Samson M. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. 2010;14:1726–1739. doi: 10.1111/j.1582-4934.2009.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saidi S, Bouri F, Lencel P, Duplomb L, Baud’huin M, Delplace S, Leterme D, Miellot F, Heymann D, Hardouin P, Palmer G, Magne D. IL-33 is expressed in human osteoblasts, but has no direct effect on bone remodeling. Cytokine. 2011;53:347–354. doi: 10.1016/j.cyto.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 71.Chow JY, Wong CK, Cheung PF, Lam CW. Intracellular signaling mechanisms regulating the activation of human eosinophils by the novel Th2 cytokine IL-33: implications for allergic inflammation. Cell Mol Immunol. 2010;7:26–34. doi: 10.1038/cmi.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–1030. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 73.Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- 74.Kanda T, Takahashi T. Interleukin-6 and cardiovascular diseases. Jpn Heart J. 2004;45:183–193. doi: 10.1536/jhj.45.183. [DOI] [PubMed] [Google Scholar]
- 75.Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT3 axis. Basic Res Cardiol. 2007;102:279–297. doi: 10.1007/s00395-007-0658-z. [DOI] [PubMed] [Google Scholar]
- 76.Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 77.Banerjee I, Fuseler JW, Intwala AR, Baudino TA. IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am J Physiol Heart Circ Physiol. 2009;296:H1694–1704. doi: 10.1152/ajpheart.00908.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Niu J, Kolattukudy PE. Role of MCP-1 in cardiovascular disease: molecular mechanisms and clinical implications. Clin Sci (Lond) 2009;117:95–109. doi: 10.1042/CS20080581. [DOI] [PubMed] [Google Scholar]
- 79.Tarzami ST, Cheng R, Miao W, Kitsis RN, Berman JW. Chemokine expression in myocardial ischemia: MIP-2 dependent MCP-1 expression protects cardiomyocytes from cell death. J Mol Cell Cardiol. 2002;34:209–221. doi: 10.1006/jmcc.2001.1503. [DOI] [PubMed] [Google Scholar]
- 80.Tarzami ST, Calderon TM, Deguzman A, Lopez L, Kitsis RN, Berman JW. MCP-1/CCL2 protects cardiac myocytes from hypoxia-induced apoptosis by a G(alphai)-independent pathway. Biochem Biophys Res Commun. 2005;335:1008–1016. doi: 10.1016/j.bbrc.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 81.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 82.Martire A, Fernandez B, Buehler A, Strohm C, Schaper J, Zimmermann R, Kolattukudy PE, Schaper W. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice mimics ischemic preconditioning through SAPK/JNK1/2 activation. Cardiovasc Res. 2003;57:523–534. doi: 10.1016/s0008-6363(02)00697-1. [DOI] [PubMed] [Google Scholar]
- 83.Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood. 2009;113:1526–1534. doi: 10.1182/blood-2008-05-157818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, Komai-Koma M, Pitman N, Li Y, Niedbala W, McKenzie AN, Teixeira MM, Liew FY, Xu D. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–4790. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 85.Rossini A, Zacheo A, Mocini D, Totta P, Facchiano A, Castoldi R, Sordini P, Pompilio G, Abeni D, Capogrossi MC, Germani A. HMGB1-stimulated human primary cardiac fibroblasts exert a paracrine action on human and murine cardiac stem cells. J Mol Cell Cardiol. 2008;44:683–693. doi: 10.1016/j.yjmcc.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 86.Li M, Georgakopoulos D, Lu G, Hester L, Kass DA, Hasday J, Wang Y. p38 MAP kinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation. 2005;111:2494–2502. doi: 10.1161/01.CIR.0000165117.71483.0C. [DOI] [PubMed] [Google Scholar]
- 87.Frantz S, Behr T, Hu K, Fraccarollo D, Strotmann J, Goldberg E, Ertl G, Angermann CE, Bauersachs J. Role of p38 mitogen-activated protein kinase in cardiac remodelling. Br J Pharmacol. 2007;150:130–135. doi: 10.1038/sj.bjp.0706963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang W, Elimban V, Nijjar MS, Gupta SK, Dhalla NS. Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp Clin Cardiol. 2003;8:173–183. [PMC free article] [PubMed] [Google Scholar]
- 89.Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, Koyasu S, Matsui H, Yamauchi-Takihara K, Harada M, Saito Y, Ogawa S. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res. 2001;89:661–669. doi: 10.1161/hh2001.098873. [DOI] [PubMed] [Google Scholar]
- 90.Hata K, Andoh A, Shimada M, Fujino S, Bamba S, Araki Y, Okuno T, Fujiyama Y, Bamba T. IL-17 stimulates inflammatory responses via NF-kappaB and MAP kinase pathways in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1035–1044. doi: 10.1152/ajpgi.00494.2001. [DOI] [PubMed] [Google Scholar]
- 91.Tare N, Li H, Morschauser A, Cote-Sierra J, Ju G, Renzetti L, Lin TA. KU812 cells provide a novel in vitro model of the human IL-33/ST2L axis: functional responses and identification of signaling pathways. Exp Cell Res. 2010;316:2527–2537. doi: 10.1016/j.yexcr.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 92.Funakoshi-Tago M, Tago K, Sato Y, Tominaga S, Kasahara T. JAK2 is an important signal transducer in IL-33-induced NF-κB activation. Cell Signal. 2011;23:363–370. doi: 10.1016/j.cellsig.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 93.Keeley EC, Mehrad B, Strieter RM. The role of fibrocytes in fibrotic diseases of the lungs and heart. Fibrogenesis Tissue Repair. 2011;4:2. doi: 10.1186/1755-1536-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Keeley EC, Mehrad B, Strieter RM. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of fibrotic disorders. Thromb Haemost. 2009;101:613–618. [PMC free article] [PubMed] [Google Scholar]
- 96.Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. 2007;37:2779–2786. doi: 10.1002/eji.200737547. [DOI] [PubMed] [Google Scholar]
- 97.Verri WA, Jr, Souto FO, Vieira SM, Almeida SC, Fukada SY, Xu D, Alves-Filho JC, Cunha TM, Guerrero AT, Mattos-Guimaraes RB, Oliveira FR, Teixeira MM, Silva JS, McInnes IB, Ferreira SH, Louzada-Junior P, Liew FY, Cunha FQ. IL-33 induces neutrophil migration in rheumatoid arthritis and is a target of anti-TNF therapy. Ann Rheum Dis. 2010;69:1697–1703. doi: 10.1136/ard.2009.122655. [DOI] [PubMed] [Google Scholar]