

Abstract
Reductions in the blood supply produce considerable injury if the duration of ischemia is prolonged. Paradoxically, restoration of perfusion to ischemic organs can exacerbate tissue damage and extend the size of an evolving infarct. Being highly metabolic organs, the heart and brain are particularly vulnerable to the deleterious effects of ischemia/reperfusion (I/R). While the pathogenetic mechanisms contributing to I/R-induced tissue injury and infarction are multifactorial, the relative importance of each contributing factor remains unclear. However, an emerging body of evidence indicates that the generation of reactive oxygen species (ROS) by mitochondria plays a critical role in damaging cellular components and initiating cell death. In this review, we summarize our current understanding of the mechanisms whereby mitochondrial ROS generation occurs in I/R and contributes to myocardial infarction and stroke. In addition, mitochondrial ROS have been shown to participate in preconditioning by several pharmacologic agents that target potassium channels (e.g., ATP-sensitive potassium (mKATP) channels or large conductance, calcium-activated potassium (mBKCa) channels) to activate cell survival programs that render tissues and organs more resistant to the deleterious effects of I/R. Finally, we review novel therapeutic approaches that selectively target mROS production to reduce postischemic tissue injury, which may prove efficacious in limiting myocardial dysfunction and infarction and abrogating neurocognitive deficits and neuronal cell death in stroke.
Introduction
Acute coronary artery disease and stroke are the number one and third leading causes, respectively, of death and disability among Americans and in most westernized cultures. Ischemia caused by vascular obstruction in the cerebral circulation is the most common cause of stroke, although increased microvascular permeability and intracerebral hemorrhage can also result in decreased perfusion. If diagnosed in a timely manner, significant I/R injury can be avoided by prompt treatment with thrombolytic agents or by physical removal of the obstruction using angioplasty approaches. Neurons or myocytes that are supplied by vessels downstream from the occlusion die from prolonged ischemia and comprise the infarcted region of the tissue that is termed the ischemic core. The cells in this region never regain function and are dead prior to therapeutic intervention in the brain or progress irreversibly to death in the case of severe cardiac ischemia. Of greater clinical interest are cells that die in a delayed manner after reperfusion is initiated. This population of neural or myocyte cells surrounds the ischemic core and is referred to as the penumbra in stroke and area-at-risk in myocardial I/R. These jeopardized cells are not fully reliant on blood flow from the occluded artery, with modest perfusion maintained by collateral blood vessels that provides resistance to ischemic damage. Although penumbral neurons or at-risk myocytes do not succumb to the initial ischemia-induced cell death, they progress to death during reperfusion in a delayed manner that resembles apoptosis. The delayed onset of death provides a window of opportunity for therapeutic intervention. Recognition of this initial penumbral resistance to cell death led to the concept that treatments targeting these cells should be initiated prior to or in the first hours after recanalization of the obstructed vessel.
While significant progress has been made with regard to identifying ROS as key mediators of both detrimental and protective responses in I/R, therapeutic antioxidant management of I/R syndromes such as myocardial infarction, stroke, and circulatory arrest has proven disappointingly ineffective [1–4]. This is most likely due to a number of factors including the fact that untargeted application of antioxidants may not differentiate between detrimental vs beneficial ROS generation. However, recent breakthroughs regarding use of targeted antioxidant therapies to enhance therapeutic efficacy of treatments to ameliorate oxidative stress in I/R injury have rekindled interest in use of agents that modify oxidative stress in I/R. Particularly promising developments have arisen with regard to targeted delivery of therapeutic agents to the mitochondria, as a means to reduce ROS-dependent I/R injury. The aim of this review is to summarize evidence supporting a role for mitochondrially-derived ROS in the pathogenesis of I/R injury and for their participation in the beneficial protective actions of preconditioning. We begin with a generalized review of the multifactorial pathogenetic mechanisms of I/R injury, followed by a brief description of mitochondrial sources of ROS, before moving on to a review of the evidence supporting a pivotal role for mitochondrial oxidants in heart and brain injury induced by I/R. We conclude this review with a brief discussion regarding the roles for mitochondrial ROS as redox signaling molecules that underlie metabolic and flow-dependent vasodilation and finish with a summary of work implicating mitochondrial oxidant generation as an essential trigger for activation of cell survival programs that enhance tolerance to I/R, which again emphasizes the Jekyll and Hyde nature of mitochondrial ROS production. From this discussion, it will be apparent that mitochondrial ROS can exert detrimental or beneficial effects, a double-edged sword that is probably explained by the type of oxidant, the amount of ROS produced, and the subcellular site of their production in these two vary different situations (preconditioning vs I/R).
General Concepts of I/R Injury
In tissue subjected to ischemia followed by reperfusion (I/R), pathologic mechanisms are elicited that produce reversible cell injury and dysfunction, which can progress to irreversible damage if the nature and extent of ischemia is prolonged or if the pathologic sequelae to reperfusion are of sufficient magnitude (reviewed in [5]). This damage is referred to as I/R injury and can be divided into three phases (Fig. 1). During ischemia (the first phase of injury), interruption of the blood supply to an organ causes a reduction in oxygen and nutrient delivery to the affected tissues. This disrupts ATP generation via oxidative phosphorylation, causing cells to alter their metabolism and impairs energy-dependent cellular function. Reduced ATP availability limits ion pumps in cell membranes, resulting in calcium overload, structural disorganization, and apoptotic, necroptotic, and necrotic cell death. In addition, ischemia induces conformational changes in enzymes such as xanthine oxidase and elicits the formation of proinflammatory mediators and expression of adhesion molecules that promote leukocyte/endothelial cell adhesive interactions. These latter processes do not directly contribute to injury during the ischemic phase, but rather set the stage for the second stage of I/R injury (ie, that due to reperfusion), wherein tissue injury is exacerbated when the blood supply is re-established (Fig. 1). Paradoxically, the lack of oxygen during ischemia and the replenishment of oxygen during reperfusion both contribute to the total injury sustained by tissues subjected to I/R. The clinical outcome is also determined by a third phase of ROS production that occurs during post-reperfusion repair that is characterized by tissue remodeling and adaptation (Fig. 1).
Fig. 1.
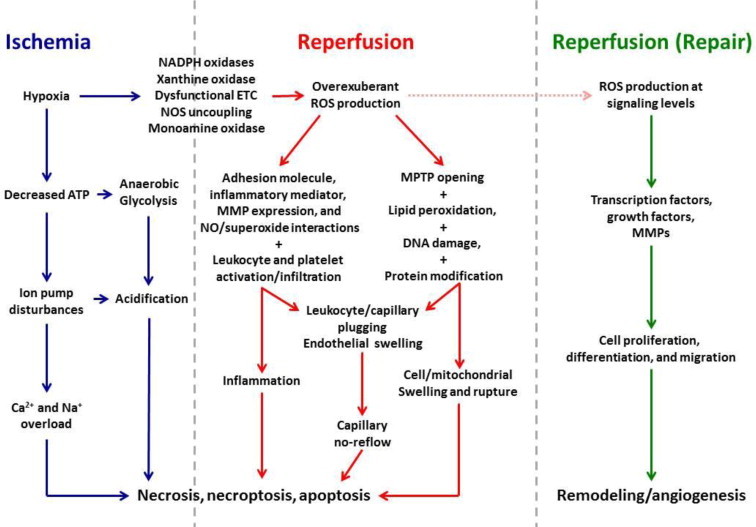
Mechanisms contributing to tissue injury in ischemia/reperfusion (I/R). Cellular hypoxia secondary to ischemia results in decreased ATP production, which in turn, disrupts ion pump function, leading to accumulation of Na+, Ca2+, and H+, with cellular acidification further promoted by a shift to anaerobic glycolysis for energy production. Activation and upregulated expression of enzymes capable of producing reactive oxygen species (ROS) and electron transport chain (ETC) dysfunction are also initiated during ischemia. These events set the stage for a burst of ROS generation when molecular oxygen is reintroduced to ischemic tissues when the blood supply is re-established. ROS-dependent expression of proinflammatory stimuli and expression of adhesion molecules by endothelial cells and leukocytes precipitates the infiltration and activation of neutrophils, T cells and monocytes. Phagocytic Nox2 activation results the respiratory burst of superoxide production that further magnifies the massive oxidative stress that directly damages virtually every biomolecule found in cells and induces the programmed cell death responses, apoptosis and necroptosis. Postischemic ROS generation also activates matrix metalloproteinases (MMPs) and other proteases that act to cleave proteins and receptors, thereby impairing their function. The net impact of these ROS-dependent events is opening of mitochondrial permeability transition pores (MPTPs), which contributes to swelling and lysis of cells. Increases in leukocyte stiffness induced by hypoxia and acidosis during ischemia lead to impaction of these cells in capillaries, an effect that is exacerbated by ROS-dependent endothelial cell swelling which in turn reduces their diameter when the blood supply is re-established. Thus, a nutritive perfusion impairment becomes prominent during reperfusion, despite repair of the precipitating ischemic event. In direct contrast to these catastrophic effects of ROS generation secondary to events occurring in ischemia and early reperfusion, oxidant production also occurs at later stages of reperfusion as tissue repair is initiated. However, ROS production occurs at lower levels that allow oxidant species to serve as signaling molecules that participate in transcriptional activation of growth factors and promote cell proliferation, differentiation and migration. The net effect of these processes is tissue and vascular remodeling, including angiogenesis. While some of these repair processes help restore organ function, others such as tissue fibrosis contribute over time to eventual organ failure. The mechanisms depicted in this figure emphasize the concept that ROS generation play key roles in all three phases of ischemia/reperfusion injury and cell death.
Increased generation of reactive oxygen species (ROS) has been suggested as a major contributor to the pathogenetic mechanisms underlying ischemia, reperfusion, and the later post-reperfusion phase of I/R injury (Fig. 1). As a second paradox, ROS appear to exert both detrimental and beneficial effects in I/R, causing damage that leads to neurocognitive defects in stroke and contribute to the expansion of infarct size as a result of their production during ischemia and reperfusion while subserving a signaling function to promote fibrosis, angiogenesis, and vascular remodeling during the repair phase (Fig. 1). Moreover, ROS signaling promotes the activation of cell survival programs when the heart or brain (or other organs) are exposed to preconditioning stimuli (such as short bouts of ischemia, moderate ethanol ingestion, or a wide variety of pharmacologic agents) prior to the onset of prolonged I/R. The double-edged sword effects of I/R-induced ROS generation may be related to species of ROS produced, the amount of oxidants generated, and the subcellular location and cellular source of their production under a given set of conditions, as well as at what time during the three phases of responses to I/R they are formed.
Reperfusion represents the second phase of I/R injury and precipitates the generation of ROS that is fueled by the reintroduction of molecular oxygen to the tissues (Fig. 1). Xanthine oxidase- and phagocyte NADPH oxidase-derived oxidants can damage virtually every biomolecule found in cells and tissues. Although essential for cell survival, re-establishing the blood supply to ischemic tissues also delivers blood-borne formed elements (platelets and leukocytes), which become activated and establish adhesive interactions with the walls of postcapillary venules (Fig. 1). Upon transmigration into the tissues, these activated leukocytes release their cytotoxic arsenal of ROS and hydrolytic enzymes to exacerbate parenchymal cell injury. ROS induce tissue dysfunction by directly damaging cells via a number of mechanisms including peroxidation of cell membrane and organelle lipids, oxidizing DNA, activation of matrix metalloproteinases and calpains, producing osmotic cell lysis, induction of no-reflow, and causing opening of the mitochondrial permeability transition pore (Fig. 1). ROS may also induce cell dysfunction and death by indirect mechanisms by interacting with NO, fatty acids or free iron to form peroxynitrite, peroxyl radicals, and hydroxyl radicals, respectively, each of which are capable of producing even more cellular damage than superoxide or hydrogen peroxide. Oxygen-derived ROS also act to enhance the inflammatory response to reperfusion via formation of oxidant-dependent proinflammatory mediators and upregulation of cytokine/chemokine and adhesion molecule expression (Fig. 1). Thus, while there is cellular demand for replenishment of oxygen which is met by re-establishing the blood supply, the reintroduction of molecular oxygen to the tissues results in ROS formation that is detrimental to the reperfused tissues. The divergent roles of oxygen in the first two phases of I/R injury are referred to as the oxygen paradox.
A second ROS paradox arises in later phases of reperfusion, where ROS generation affects several tightly regulated processes that promote organ repair and survival (Fig. 1). This third phase constitutes the reparative phase of I/R injury and involves ROS-dependent generation of growth factors that promote angiogenesis, induce proliferation and differentiation of vascular smooth muscle cells to effect vascular remodeling, and promote the activation of matrix metalloproteinases and other factors that contribute to fibrosis, tissue remodeling and formation of scar tissue. Interestingly, hypoxia induces transport of mitochondria to perinuclear regions by a microtubule-dependent mechanism, where they increase nuclear ROS levels to enhance the expression of VEGF, a growth factor important in angiogenesis [6].
Cells are equipped with a wide variety of oxidant-producing enzymes including xanthine oxidase, NADPH oxidases (Nox), and cyclooxygenases/lipoxygenases. The mitochondria also produce ROS from respiratory chain components, as well as by activation of mitochondrially localized monoamine oxidase, the growth factor adaptor Shc (p66Shc), cytochrome b5 reductase, dihydroorotate dehydrogenase, mitochondrial ATP-sensitive potassium (mKATP) and large-conductance, calcium activated potassium (BKCa) channels, and the Nox isoform designated Nox4. Superoxide normally produced in mitochondria is scavenged by manganese-superoxide dismutase (Mn-SOD or SOD-2) localized in the matrix. In addition, copper/zinc-SOD (Cu/ZN-SOD or SOD-1), which is typically considered a cytoplasmic isoform, is also located in mitochondria between its inner and outer membranes. These SODs dismutate superoxide to less reactive hydrogen peroxide, which can be further metabolized to water and oxygen by the catalytic activity of catalase and glutathione peroxidase. Mitochondrial uncoupling proteins also serve to reduce the production of ROS by causing mitochondrial depolarization, which reduces the potential driving electron transfer and by allowing protons to reenter the matrix, thereby bypassing ATP synthase.
Mitochondria play a central role in the development of reperfusion injury because the recovery of pH, oxidative stress, and calcium overload induce abrupt opening of mitochondria permeability transition pores (mPTPs), high conductance megachannels that are localized to contact sites between the inner and outer mitochondrial membranes [7]. When opened, mPTPs permit communication between the cytoplasm and the mitochondrial matrix. While low pH during ischemia prevents opening of the megachannel, oxidative opening of the mPTP is critical to reperfusion injury (Fig. 1). Depending on a complex balance among cellular inducers and antagonists, the open probability of the mPTP can be transient or long-lived. Short-term opening is involved in cardioprotection that involves transient ROS formation (see below). In contrast, long-lasting mPTP opening, which is facilitated by restoration of pH, calcium overload and the burst of ROS formation at the onset of reperfusion, is followed by profound and irreversible alterations in cellular bioenergetics. Sustained pore formation results in increased mitochondrial permeability to ions and other solutes up to molecular weights of 1.5 kD and collapse of the mitochondrial membrane potential. This is rapidly followed by ATP and NAD+ depletion, release of accumulated mitochondrial calcium, matrix swelling and outer mitochondrial membrane rupture, which in turn results in loss of pyridine nucleotides, release of pro-apoptotic factors, and further inhibits electron flow through the electron transport chain. The massive release of ROS during reperfusion requires the involvement of the mPTP in a ROS-induced ROS release positive feedback loop [8]. It is widely believed that mPTP is thus a major causative event in reperfusion injury and cell death. This concept is consistent with the observation that cardioprotective interventions all seem to intersect at inhibition of the mPTP as an end-effector of enhanced tolerance to I/R.
Mitochondrial sources of ROS
Mitochondria are the main source of cellular ROS [9] and contain a number of enzymes that convert molecular oxygen to superoxide or its derivative hydrogen peroxide (H2O2) (Fig. 2). However, the mitochondrial electron transport chain in the inner membrane, also known as the respiratory chain, is the most important source of the intracellular ROS production. Indeed, it has been estimated that 95% of ROS generated in normal (i.e., non-ischemic) cells are derived from electron leak from the respiratory chain enzyme complexes [10]. The proton motive force represents the potential energy driving proton movement into the mitochondrial matrix for ATP synthesis. It describes an electrochemical gradient that consists of a mitochondrial membrane potential and a proton gradient, which represent sources for mitochondrial ROS generation via electron transfer to oxygen. For an extensive and excellent discussion of factors involved in superoxide formation by the complexes comprising the electron transport chain, the reader is directed to a recent review of this topic by Chen and Zweier [11]. Importantly, the electron transport chain proteins are rich in iron sulfur clusters and heme groups, providing cofactors necessary to form highly reactive hydroxyl radicals from hydrogen peroxide [12].
Fig. 2.
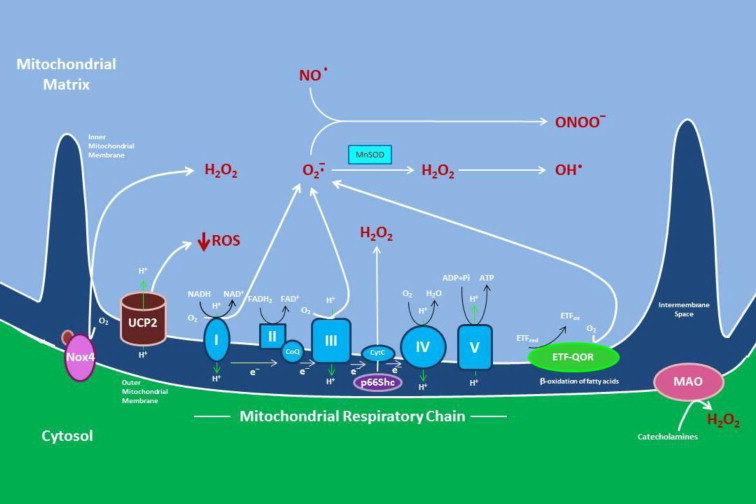
Sources of reactive oxygen species (ROS) in mitochondria. The activity of the electron transport chain generates a relatively small flux of ROS under normal conditions, but its production can be greatly magnified by events occurring during ischemia and reperfusion. Complex I (NADH dehydrogenase) and complex III (coenzyme Q (CoQ) and cytochrome C oxidoreductase) produce superoxide (O2-), which leads to hydrogen peroxide (H2O2) formation by spontaneous dismutation or via the enzymatic action of manganese superoxide dismutase (MnSOD). In the presence of transition metals, H2O2 can form the highly reactive hydroxyl radical (OH•) superoxide can also interact with nitric oxide (NO) to form reactive nitrogen oxide species such as peroxynitrite (ONOO-), which produce cellular dysfunction by S-nitrosylating proteins. ROS generated by complex I are released into the mitochondrial matrix, while superoxide produced by complex III can occur in both the mitochondrial matrix and the intermembrane space between the outer and inner mitochondrial membranes. Other sources of mitochondrial superoxide are enzymes glycerol-3-phosphate dehydrogenase (G3PD), the growth factor adaptor p66Shc, and NADPH oxidase-4 (Nox4). ß-oxidation of fatty acids can also result in mitochondrial superoxide generation secondary to oxidation of electron transferring protein (ETF) by the catalytic activity of the electron transferring flavoprotein ubiquinone oxidoreductase (ETF-QOR), another enzyme expressed on the mitochondrial inner membrane. Monoamine oxidase (MAO), which is localized to the outer mitochondrial membrane, catalyzes the formation of H2O2 secondary to catecholamine metabolism. Not depicted are the mitochondrial enzymes aconitase and dihydroorotate, which can produce superoxide, but their role in ischemia/reperfusion is uncertain.The mitochondrial anion carrier, uncoupling protein-2 (UCP2), functions to separate oxidative phosphorylation from ATP synthesis with energy dissipated as heat, a phenomenon referred to as the mitochondrial proton leak. UCP2 acts to facilitate the transfer of anions from the inner to the outer mitochondrial membrane and the return transfer of protons from the outer to the inner mitochondrial membrane. They also reduce the mitochondrial membrane potential in mammalian cells. Although it was originally thought to play a role in nonshivering thermogenesis, obesity, diabetes and atherosclerosis, it now appears that the main function of UCP2 is the control of mitochondria-derived ROS.
Most oxygen consumed by aerobic eukaryotic cells is reduced to water through 4 steps of single electron reduction by the terminal cytochrome c oxidase of mitochondrial respiratory chain. A small proportion (0.1–2%) of consumed oxygen can also be reduced partially by one or two electrons in the mid-pathway of respiratory chain to generate superoxide or H2O2 as normal metabolic products of oxygen during respiration [13,14]. Superoxide is generated mainly in complexes I and III [15,16] (Fig. 2). Ubisemiquinone at the Qo site of Q cycle in complex III appears to be a major site of superoxide production [17,18]. Although electron leak also occurs at complex I of the respiratory chain, the precise generating site(s) for superoxide formation within this supercomplex has not been established [12,13,19].
In addition to complexes I and III of the electron transport chain, other mitochondrial components are likely to contribute to ROS generation, such as growth factor adaptor Shc (p66Shc), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-4 (Nox4), monoamine oxidase (MAO), mitochondrial BKCa and mKATP channels, cytochrome b5 reductase, and dihydroorotate dehydrogenase (Fig. 2). However, the contribution of these enzymes to total mitochondrial ROS production is significantly lower than that of the respiratory chain in the inner mitochondrial membrane [20].
Recent studies have suggested a novel pathway of mitochondrial ROS production and oxidant-induced apoptosis involving that the 66-kDa isoform of the growth factor adaptor protein, Shc (p66Shc) [21,22] (Fig. 2). Several chronic stimuli have been shown to activate protein kinase C ßII (PKCßII) isoform to induce Ser-36 phosphorylation of p66Shc, allowing transfer of the adaptor protein from the cytosol to the mitochondrial intermembrane space [23,24]. Once translocated, p66Shc uses reducing equivalents from the electron transport chain through the oxidation of cytochrome c to make H2O2 directly without formation of superoxide. Redox-defective mutants of p66Shc are unable to induce mitochondrial ROS generation and swelling in vitro or to mediate mitochondrial apoptosis in vivo [21,25,26].
Nox enzymes generate ROS as the product of their natural catalytic cycle. When fully assembled, they are the membrane-bound proteins catalyzing electron transfer from NAD(P)H to oxygen, thus producing superoxide [27]. Nox4 is the most ubiquitous of these oxidases outside of phagocytic leukocytes (which use the Nox2 isoform to generate ROS in the respiratory burst) and is a major source of ROS in many cell types, but its role in physiologic signaling and pathologic states is perhaps the most controversial (Fig. 2). This Nox isoform is localized to the outer mitochondrial membrane, and like p66Shc, it produces H2O2 [28,29]. As has been recently reviewed, Nox4 may function to control the redox set-point and thus influence cellular metabolism. This Nox isoform has also been shown to play important roles in cell differentiation, migration, growth, apoptosis, and senescence, as well as participating in proinflammatory responses and oxygen sensing [30].
MAO is another prominent source of mitochondrial ROS. This enzyme is localized to the outer mitochondrial membrane, where it is involved in the oxidative breakdown of key neurotransmitters (catecholamines) and generates H2O2 [31] (Fig. 2). MAO plays the major role in the development of oxidative stress of the nervous system and heart. When the heart is subjected to chronic neurohumoral and/or peripheral hemodynamic stress, the attendant abundance of circulating/tissue monoamines fuels MAO-derived H2O2 production and has been shown to play in I/R injury [26,32,33]. Recent results demonstrate that MAO is an important determinant of redox balance in human atrial myocardium as well [34].
Mitochondrial ROS and the pathogenesis of I/R
Overproduction of ROS by mitochondria plays a role in the pathogenesis of myocardial I/R and stroke via impaired endothelium-dependent vasodilator mechanisms, disrupted excitation–contraction coupling, generation of arrhythmias, direct damage to biomolecules that result in necrosis, necroptosis and apoptosis, induction of mitochondrial permeability transition, and contributions to cardiac fibrosis, remodeling and hypertrophy (Figs. 1 and 3). Overexuberant liberation of ROS secondary to enhanced electron leak at complexes I and III are major oxidant sources in I/R [9,35–38]. Both ischemia- and reperfusion-induced defects in the electron transport chain leads to increased ROS production in both subsarcolemmal and interfibrillary mitochondria of the heart [39,40]. This appears to be mediated by oxidant-induced disruption of cardiolipin-respiratory chain superassemblies in complexes I-III, which further increases electron leakage fueling superoxide generation [9,36,41–45] (Figs. 2 and 3). This is exacerbated by peroxynitrite-mediated protein nitration of complexes I and III at specific sites [9,43,44]. ROS leakage from the electron transport chain can also activate a mitochondrial anion channel (IMAC) that releases ROS into the cytoplasm and simultaneously contributes to mitochondrial permeability transition [46,47]. Aquaporin-8 is expressed on the inner mitochondrial membrane in hepatic (and perhaps other) cells, where it also acts to facilitate the diffusional transport of H2O2 [48].
Fig. 3.
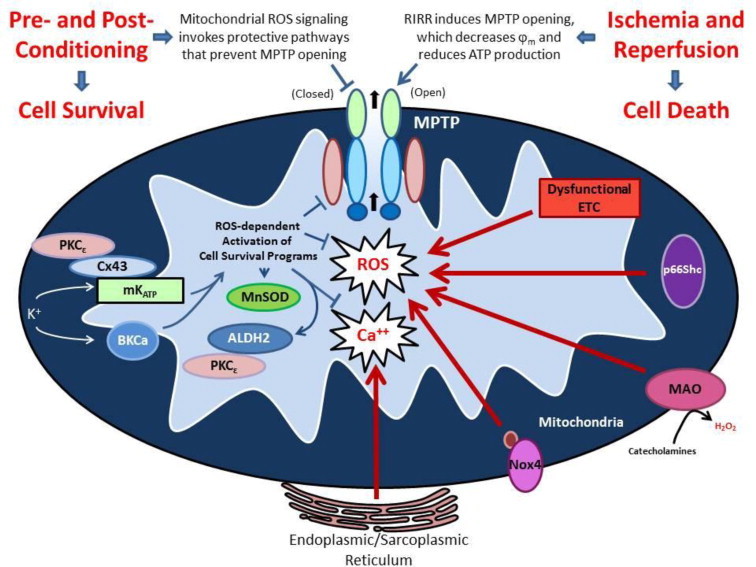
Generation of reactive oxygen species (ROS) by mitochondria (mitoROS) is a nexus for both activation of cell survival programs that mediate the effect of conditioning stimuli to enhance tolerance to ischemia/reperfusion (I/R) and serves as a focal point for overexuberant ROS-induced ROS release that contributes to the pathogenesis of cell injury in I/R. On the one hand, ROS triggers the activation of cell survival programs in responses to a number of mildly noxious stimuli such as short bouts of ischemia or antecedent ethanol exposure or pharmacologic agents (ethanol or activators of mitochondrial ATP-sensitive potassium (mKATP) or large conductance, calcium-activated potassium (BKCa) channels). The enhanced tolerance to ischemia invoked by these mitoROS-dependent conditioning stimuli, which can be delivered before (preconditioning), during (preconditioning) or at the onset of reperfusion (postconditioning), activate protective protein kinases such as PKCe, the expression of prosurvival genes (e.g., heme oxygenase-1) and mitochondrial antioxidant defenses (e.g., MnSOD, aldehyde dehydrogenase-1 or ALDH2), as well as targeting the mitochondrial permeability transition pore (MPTP) to maintain the channel in a closed state. On the other hand, overexuberant ROS generation at the onset of reperfusion, driven by ROS-induced ROS release that is fueled by electron transport chain dysfunction, especially at complexes I and III, and enhanced activities of p66Shc, monoamine oxidase (MAO), and NADPH oxidase-4 (Nox4) in mitochondria, causes the MPTP to open, leading to swelling, cell disruption and death. Not depicted is the effect of oxidants to alter the balance of mitochondrial fission and fusion in conditioning and I/R, which emerging evidence has implicated as contributory to both processes. The dual nature of ROS as protective vs damaging species relates to the type of ROS generated in particular circumstances, their concentration, and/or compartmental localization of their production.
In addition to complexes I and III, insufficient oxygen delivery during ischemia results in increased electron leakage that is mediated by hypoxia- and ß1-adrenergic-mediated, mitochondrial protein kinase A/cAMP-dependent reduction in the activity of complex IV of the electron transport chain, which can interact with residual oxygen to produce superoxide [9,11,49,50]. Owing to the significant depletion of ADP during early reperfusion, the reintroduction of molecular oxygen greatly enhances electron leakage thereby markedly increasing ROS production that overwhelms oxidant scavenging capacity, peroxidizes membrane lipid components, damages mitochondrial DNA, and inactivates electron transport chain proteins, effects that are markedly reduced by treatment with a free radical spin trap prior to ischemia [51]. Overproduction of ROS via the electron transport chain and other sources establishes a vicious positive feedback loop that induces mitochondrial permeability transition and ROS-induced ROS production [8,52,53].
Global and focal cerebral ischemia induce different modes of cell death. Brief periods of global cerebral ischemia cause delayed neuronal cell death by apoptosis. In contrast, most cells in the ischemic core die by necrosis after focal cerebral ischemia, while cell death in the ischemic penumbral region occurs by mechanisms dependent on activation of apoptotic signaling. Like cardiac myocytes, neurons are long-lived cells that are highly dependent upon oxidative metabolism. As a consequence, neural tissue is predisposed to cumulative injury by endogenously produced ROS, even under non-pathological conditions, as evidenced by progressive accumulation of lipofuscin granules, a pigment composed of a mixture of proteins and lipid oxidation products, in neuronal lysosomes as we age [54]. Moreover, long term treatment with dinitrophenol, administered at a dose that produces mild mitochondrial uncoupling, has been shown to reduce brain markers of accumulated oxidative stress and increase survival times [55].
Following induction of focal stroke, cell death in the ischemic penumbral region becomes evident between 24 and 72 h after reperfusion is initiated. During reperfusion, mitochondria (and other oxidant producing enzyme systems) use oxygen as a substrate to generate ROS, resulting in an immediate increase of multiple markers of oxidative damage, which then remain elevated for several days [56]. More definitive support for the importance of ROS generation in penumbral cell death is derived from studies demonstrating that antioxidant treatment ameliorates cell injury and death in animal models of stroke [57–61]. In addition to well-established roles for phagocytic NADPH oxidases, NOS, and cyclooxygenase/lipoxygenase pathways for ROS/RNOS generation in cerebral I/R injury, a growing body of evidence supports formation of mitochondrial oxidants as another contributing factor in neuronal cell death in stroke. For example, ROS generation is enhanced in mitochondria after cerebral I/R and occurs at time points prior to overt neuronal death [56,62–64]. Postischemic impaired function of respiratory chain complexes I-IV and ATP synthase is a likely major cause of enhanced mitochondrial ROS production in stroke [65,66]. Increased calcium concentrations in the mitochondrial matrix induce oxidative stress that contributes to the MPT in brain mitochondria exposed to hypoxia/reoxygenation and high calcium [67]. Moreover, high calcium exposure, as occurs in I/R, reversibly inhibits the elimination of hydrogen peroxide in brain mitochondria [68].
In addition to the respiratory chain complexes, it also appears that NADPH oxidases (Nox) represent important oxidant sources in postischemic tissues (Figs. 2 and 3). Pharmacologic Nox inhibition prevents glial cell activation, neuronal cell degeneration, and behavioral deficits induced by experimentally-induced cerebral I/R and reduces myocardial infarct size, effects most often attributed to phagocytic Nox2 [69]. Reductions in ventricular dysfunction, cardiac remodeling, blood–brain-barrier disruption, lipid peroxidation, protein nitration, and oxidative DNA damage after I/R have been reported in mice deficient in Nox2 or p47phox expression after exposure to cerebral I/R [70–78]. Similar findings have been reported for non-selective Nox inhibitors, apocynin and diphenyliodonium, in adult brain. On the other hand, inhibition of Nox activity fails to prevent perinatal brain injury in newborn animals [79]. Inhibitor studies using apocynin and/or diphenyliodonium should be interpreted with caution because the former exhibits antioxidant properties while the latter is a general flavoprotein inhibitor [80–84]. However, use of a more selective general Nox inhibitor, VAS2870, administered at 2 and 12 hours after restoration of cerebral blood flow, decreased infarct size, edema, ROS levels, tissue nitration, and apoptosis in wild-type mice subjected to middle cerebral artery occlusion and reperfusion [85]. On the other hand, more recent work has shown that this agent produces significant off-target effects as well, such as thiol alkylation, thereby modifying thiol redox status [86].
Mitochondrial Nox4 may also participate in stroke, with some evidence supporting a pre-eminent role for this isoform (Figs. 2 and 3). This is in part related to the fact that Nox4 is the most abundantly expressed isoform in the brain and is found in endothelial cells and vascular smooth muscle cells, with particularly prominent expression and activity exhibited by these cells in the cerebral vasculature that varies by gender [87–90]. This isoform is unique among the Nox family in that it only interacts with p22phox, is constitutively active but can be further activated by angiotensin II, TNF-a, certain growth factors and the binding protein PolDip2, and produces hydrogen peroxide as its major product [91]. Since angiotensin II and TNFa are expressed during I/R, it is likely that these proinflammatory stimuli participate in upregulating Nox4 activity during I/R. In addition, siRNA-mediated downregulation of HIF-1a expression limits the effect of hypoxia to increase Nox4 mRNA while HIF1a overexpression increases Nox4 mRNA and protein levels, observations which suggest that hypoxia should increase Nox4. Indeed, the rapid stabilization and activation of HIFs under conditions of low oxygen may be the primary initiating event for the upregulation on Nox4 and other isoforms in ischemia, which is augmented by proinflammatory signaling molecules described above. Importantly, Nox4 appears to be localized in mitochondria (as well as the plasma membrane, endoplasmic reticulum, and nucleus) [92], and as such could represent an important source of mitochondrial ROS in I/R.
More direct support for a role for the Nox4 isoform in stroke is provided by the observation that Nox4 mRNA is upregulated in stroke models. Importantly, Nox4 knockout mice demonstrated the same reductions in injury as VAS2870-treated wild-type mice, and the degree of protection afforded by Nox4 deficiency was not augmented by VAS2870 treatment. Taken together, these latter observations strongly support the notion that Nox4 is a major contributor to the reperfusion component of cerebral I/R injury. In addition to this evidence, recent work indicates that Nox4 overexpression specifically in endothelium exacerbates stroke-induced infarct size by a mechanism that involves Nox4-dependent suppression of eNOS [93]. On the other hand, infarct size was smaller in endothelial-specific Nox4-deficient mice compared to wild-type animals [76]. As noted above, Nox4 is expressed not only in mitochondria, but also in the plasma membrane, endoplasmic reticulum, and nucleus) [92]. Thus, any or all of these organelles may be a source of Nox4-derived ROS in I/R, not just mitochondria.
There is evidence suggesting that Nox4-derived oxidants may also play a role in the repair processes that arise days after reperfusion is established following cerebral ischemia. This notion is based on the observation that ROS derived from Nox4 are key signaling molecules mediating cell proliferation and differentiation [94–98]. For example, siRNA knockdown of Nox4 inhibits tube formation and wound healing responses in cultured endothelial cells and limits cell migration and proliferation induced by VEGF [99,100]. Thus, activation of Nox4 during latter stages of reperfusion may promote angiogenesis to facilitate perfusion of previously ischemic tissues. Moreover, Nox4 activity may influence vascular smooth muscle differentiation and proliferation in response to hypoxia [101,102]. Because Nox4-dependent ROS production stimulates proliferation and differentiation of cardiac fibroblasts into myofibroblasts, which may lead to fibrosis, cardiac remodeling and heart failure, activation of this Nox isoform may not be entirely beneficial in the setting of recovery in myocardial I/R [97,103,104]. Thus, while Nox4-derived ROS are important for postischemic angiogenesis, these oxidants may also be detrimental owing to effects to promote hypertrophy or dilatation of the ventricles. These divergent effects probably arise as a result of site and/or amount of their production. Since the effects of Nox4 deficiency vs overexpression, induced during the recovery phase of reperfusion, have not been evaluated in postischemic heart and brain, it is unclear whether this mitochondrial oxidant source contributes in beneficial or detrimental ways. Clearly, much additional work will be required to address this important and intriguing question and will likely require site-specific ablation or overexpression studies.
Interplay between different ROS sources also appears to play a role in I/R [95,105,106]. Nox-dependent ROS production can trigger mitochondrial dysfunction which in turn provokes oxidant production. The converse is also true, mitochondrial ROS production can activate NADPH oxidases [8,106,107]. Thus, mitochondrial ROS-induced ROS release synergistically increases postischemic oxidative stress. The generation of ROS by mitochondria also demonstrates spatial heterogeneity within the organelle and among mitochondria in a cell [108]. Following oxidative insult, as occurs in I/R, ROS generation preferentially localizes in mitochondrial regions that progress to swelling and eventually rupture. In addition, fatal oxidative insults result in massive augmentation of ROS formation in nearly all mitochondria in the affected cell. However, mild oxidative stress, as might occur with preconditioning stimuli, induces a more heterogeneous ROS formation that is limited to small numbers of mitochondria [108], consistent with the signaling function of compartmentalized elements of the cell survival program that is activated by these stimuli.
Excessive ROS formation during reperfusion facilitates opening of the mPTP which in turn favors ROS formation by inhibiting the respiratory chain secondary to mPTP-induced loss of cytochrome C and pyridine nucleotides [109]. Mitochondrial DNA is also particularly vulnerable to the deleterious effects of ROS-induced ROS release during reperfusion owing to the lack of protection of mitochondrial DNA by histones, limited capacity of DNA repair mechanisms, and proximity to the production site for ROS-induced ROS. Thus, I/R-induced ROS production induces mitochondrial DNA rearrangement and fragmentation followed by disruption of mitochondrial structure, function, and eventual lysis.
The susceptibility of mitochondrial DNA to oxidative modification in circulating leukocytes is of considerable potential interest as this may provide a reliable marker of mitochondrial dysfunction that could be exploited as a diagnostic indicator for I/R. Oxidative inactivation of mitochondrial aconitase, which predisposes the enzyme to produce hydroxyl radicals and thus contribute to I/R injury, suggests that disrupted activity of this enzyme could also be used as a marker for myocardial infarction. Oxidants generated by mitochondria (and other sources) interact with NO to produce peroxynitrite and other reactive nitrogen species which in turn contribute to the formation of nitrotyrosine residues on proteins, which not only contribute to cell dysfunction by disrupting protein function, but also serve as a surrogate marker for oxidative stress.
Mitochondrially-directed antioxidants reduce I/R injury
Discovery of the important roles for mitochondrial ROS generation in the pathogenesis of I/R injury fueled an intense interest in development of pharmacologic agents and other therapeutic approaches that specifically target the mitochondria and effectively reduce postischemic tissue and organ damage [40,108,110–121]. Treatment with mitoquinone (MitoQ) or mito-phenyl tert-butylnitrone (Mito-PBN), which contain the antioxidants coenzyme Q (quinone) or PBN, respectively, attached to a lipophilic triphenylphosphonium cation that allows these derivatives to accumulate in mitochondria owing to its negative ??m, limits postischemic inflammation and injury. Mimetics of Mn-SOD target mitochondria to inhibit ROS-dependent damage and apoptosis. A series of NO-based and vitamin E molecules, which are linked to the triphenylphosphonium mitochondrial targeting moiety, have been developed to actively sequester these antioxidants in mitochondria and exert cardioprotection in I/R models. A more recent focus for protection is aldehyde dehydrogenase-2, a mitochondrial enzyme that detoxifies aldehydes that have been implicated in myocardial I/R. A small molecule activator of this enzyme, Alda1, has been shown to reduce postischemic injury. Gene therapy approaches that target upregulation of mitochondrial antioxidant enzymes such as Mn-SOD or matrix peroxiredoxins or overexpressing pro-survival molecules such as aldehyde dehydrogenase-2 (in addition to other extramitochondrial targets) hold particular promise as novel approaches to render cells, tissues and organs resistant to the deleterious effects of I/R as does use of microRNAs to modulate mitochondrial redox status in postischemic tissues [122,123].
Mitochondrial ROS as signaling molecules: preconditioning and cell survival in I/R
While excess mitochondrial oxidant generation is well known to exert detrimental effects via mitochondrial dysfunction and energetic decline, a growing body of recent evidence has established that generation of ROS at low levels by this organelle can serve as signals mediating physiologic responses. For example, acetylcholine and flow-induced coronary vasodilation is mediated by activation of BKCa channels that are triggered by H2O2 derived from complexes I and III in the endothelial electron transport chain [124–130]. In addition, graded production of H2O2 by myocardial mitochondria serves as a metabolic vasodilator to couple oxygen consumption to coronary blood flow by activation of redox- and 4-aminopyridine-sensitive voltage-dependent potassium (Kv) channels and has also been implicated in the regulation of coronary collateral flow [131–133].
Recent work has established that mitochondrially centered mechanisms play an important role in promoting the activation of cell survival programs mediating preconditioning via mechanisms that involve ROS signaling (Fig. 3). Preconditioning refers to a phenomenon where transient exposure of cells, tissues, or organs to a sublethal stress (such as short bouts of ischemia prior to or at the onset of reperfusion, or ischemic pre- or postconditioning, respectively) or a variety of pharmacologic agents (e.g., ethanol, KATP and BKCa channel activators) enhances tolerance to potentially lethal I/R. Mitochondrial specific targets include respiratory chain enzyme complexes, oxidative phosphorylation, and KATP and BKCa channels localized on the inner mitochondrial membrane. For example, NOX-derived oxidants play a role in stroke, which can be prevented by antecedent exposure to ethanol or grape polyphenol extracts [134–136]. Moreover, ROS production by the respiratory chain has been shown to activate mitochondrial ATP-sensitive potassium channels (mKATP) [137], which play a role in protection against stroke-induced damage in the brain [137–143]. When activated by preconditioning stimuli, these mKATP channels allow potassium to flow into mitochondria to produce depolarization and alkalinization of the mitochondrial matrix, which in turn appears to activate cell survival signaling in both acute (early) and late preconditioning in MI and stroke by inducing downstream ROS production [138,144,145]. While ROS scavenging and mKATP channel inhibitors reduce the cardioprotection afforded by ischemic pre- and postconditioning or that induced by the mitoKATP opener diazoxide, exogenous generation of ROS at the onset of reperfusion fails to confer protection [146]. These latter results suggest that the type of ROS, their concentration, and/or compartmental localization of their production is critical to triggering tolerance to I/R, consistent with a mitochondrial site as a source for ROS generation that precipitates preconditioning.
Abundant evidence supports a role for PKG- and Akt-dependent phosphorylation in the opening of mitoKATP and subsequent ROS formation secondary to matrix alkalinization in pharmacologic preconditioning. According to this scenario, acetylcholine- or bradykinin-induced PKG activation results in phosphorylation of a protein on the outer mitochondrial membrane which is coupled to mKATP activation on the inner mitochondrial membrane by a step that involves PKC-e activation. ROS generation secondary to mitoKATP channel activation then activates a second pool of PKC which inhibits opening of the mitochondrial permeability transition pore (MPTP) to prevent cell death [145,147–149].
The demonstration that preconditioning with diazoxide, a mitoKATP activator, invoked protection against subsequent I/R-induced neuronal injury and death provided some of the first evidence that mitoKATP channels participate in activation of cell survival programs in stroke [150]. This agent causes generation of ROS at low levels by mechanism which may be related to its ability to inhibit complexes I and III of the electron transport chain to provoke preconditioning [151–157]. Others have presented evidence that mitochondrial connexin-43 (Cx43) plays an important role in mKATP-dependent oxidant signaling invoked by pharmacologic preconditioning with diazoxide, bradykinin or acetylcholine in the heart [147,155,158–160]. Mitochondrial oxidant production is also induced by preconditioning with a d-opioid agonist or endothelin-1. However, Cx43 knockdown did not affect ROS production by mitochondria induced by these preconditioning stimuli, but did abolish downstream cardioprotective signaling events [161].
ROS production has also been shown to enhance mitoKATP channel activation and thus enhance mitochondrial membrane depolarization and confer preconditioning [156,157]. While these observations clearly indicate that ROS signaling can activate mitoKATP to enhance preconditioning, treatment with BMS-191095, which activates mitoKATP but does not increase mitochondrial ROS production, also effectively reduces cerebral I/R injury [138]. The latter results indicate that mitoKATP channel activation, independent of ROS production, can invoke tolerance to lethal I/R. However, it is important to note that when the electron transport chain is inhibited, as occurs in I/R, superoxide can interact with NO to produce peroxynitrite. The reactive nitrogen species potently activates mKATP and may trigger preconditioning induced by short bouts of ischemia [138]. Moreover, respiratory chain inhibition results in decreased ATP production, thereby increasing the ADP/ATP ratio, which favors mKATP channel opening and activation of cell survival mechanisms.
The observation that drugs acting on mitoKATP channels can trigger oxidant-dependent cell survival signaling that limits I/R injury suggests the possibility that pharmacologic agents or interventions targeting other mitochondrial potassium channels could also elicit preconditioning. Indeed, it is well-established that activation of large conductance, calcium-activated potassium channels (BKCa), either by pharmacologic agents or secondary to ischemic preconditioning, elicit tolerance to prolonged I/R [162–169]. Recent work indicates that treatment with the BKCa channel activator NS-1619 protected mitochondrial function in cardiac I/R by a mechanism mediated by superoxide generation [167] but is independent of NO production [165]. In one study [167], it was assumed that BKCa channels localized on the inner mitochondrial membrane were responsible for superoxide generation because NS-1619 treatment was dependent on superoxide production and was associated with reduced mitochondrial calcium accumulation and improved redox state (i.e., normalized NADH) in I/R. In a subsequent study, the same group directly demonstrated that NS-1619 increases mitochondrial oxidant production that was driven by increased mitochondrial matrix potassium secondary to channel opening [170]. This in turn elevated matrix H+ by enhanced but submaximal K+/H+ exchange, thereby stabilizing mitochondrial membrane potential despite increased respiration. These conditions allowed increased direct electron transfer to oxygen thereby generating superoxide [170].
Activation of uncoupling proteins, which reduces oxidative stress, has been reported to elicit cardioprotection in I/R [171–175]. It is of interest to note that transient inhibition of complex I at reperfusion also exerts cardioprotective effects via reduced ROS production [176,177]. These observations suggest that postconditioning-induced tolerance to prolonged I/R would not be triggered by mild ROS production during staccato reperfusion. However, ROS have been shown to be important initiators of this form of conditioning [178]. Resolution of these discrepant observations may simply relate to the concentration and/or site of ROS generation.
A key event underlying preconditioning or postconditioning may be prolongation of cellular acidosis during early reperfusion after index ischemia. It is thought that delayed normalization of pH acts to inhibit mPTP opening in the initial minutes of reperfusion, thereby allowing time for ROS-induced activation of cell survival programs. The ROS formed in the early minutes of acidic reperfusion or during the short bouts of intermittent reperfusion during postconditioning activates PKC-e isoforms, critical kinases in the signaling cascade that act to reduce the probability of mPTP opening after pH normalizes as reperfusion progresses [179,180]. In contrast, transient opening of the mPTP may also induce modest and short-lived ROS production that precipitates preconditioning, a concept supported by the observations that antioxidant treatment during bouts of preconditioning I/R and pharmacologic inhibition or genetic ablation of cyclophilin D attenuates both preconditioning induced oxidant production and protection [181–187].
Summary and perspectives
From the aforementioned discussion, it appears the mitochondrial ROS generation plays a critical role in the pathogenesis of myocardial infarction and stroke. Although complexes I and III of the electron transport chain are the best studied sources of mitochondrial ROS generation in I/R and may be responsible for much of the oxidative stress exhibited by postischemic cells, recent evidence indicates that deglutathionylated complex II and phosphorylated complex IV are also important sources for ROS formation in I/R. As discussed by Chen and Zweier [11], identification of specific redox domains in the respiratory chain supercomplexes that are involved in overexuberant ROS generation in mitochondria during ischemia and reperfusion, how oxidant- and nitrosative-dependent post-translational modifications at specific sites may exacerbate ROS production (ROS-induced ROS release) in animal models of I/R, and how electron transport chain driven superoxide production provokes aconitase to produce hydroxyl radicals are important areas for future investigation. Postischemic ROS generation via the electron transport chain may also enhance oxidative stress via mitochondrial ROS-induced ROS release that may also involve induction of hydroxyl radical formation by the Krebs cycle enzyme aconitase and activation of Nox4. I/R can also be associated with an overabundance of circulating/tissue monoamines, which in turn fuels H2O2 production by monoamine oxidase. The growth factor adaptor Shc (p66Shc) is a novel pathway of ROS production in I/R but evaluation of its contribution to postischemic tissue injury has only just begun.
Despite the preponderance of evidence supporting a role for mitochondria as an intracellular source of ROS in the pathogenesis of I/R injury, recent work demonstrating that directed sarcolemmal stabilization uncouples increased oxidative stress from cellular damage and stress challenges this paradigm and provides an attractive explanation for the failure of anti-oxidant therapies to limit postischemic tissue injury. In an elegant series of experiments Martindale and Metzer [188] demonstrated that reperfusion produced intracellular oxidative stress and lipid peroxidation, membrane instability and swelling, which in turn induced Ca2+ influx and mitochondrial membrane depolarization. Directed stabilization of the outer membrane with synthetic copolymers consisting of tandem linear arrays of polyethylene oxide/polypropylene oxide moieties reduced postischemic necrosis, apoptosis, hypercontracture and mitochondrial dysfunction without disrupting intracellular oxidative stress or lipid peroxidation. Moreover, these investigators further demonstrated that dystrophin deficiency (which increases fragility of cardiac and other cell membranes and is associated with increased oxidative stress) [189–193], increased susceptibility to myocardial I/R, an effect that was mitigated by copolymer treatment, while restoration of membrane dystrophin contributeds to the cardioprotective effects of ischemic preconditioning [190,194]. Taken together, these observations suggest that I/R-induced, oxidant-mediated lipid peroxidation was insufficient by itself to induce cardiac injury and cell death when sarcolemmal membrane integrity was maintained, suggesting that ROS production is not the major factor responsible for the Ca2+ dysregulation that ultimately results in cell death in postischemic myocardium. Rather, it appears that sarcolemmal compromise, which limits its ability to maintain a barrier between the intra- and extracellular compartments, may be the decisive irreversible event that leads to cell death in I/R.
Since the copolymers do not exhibit free radical scavenging capacity and are restricted to engaging the sarcolemmal surface by their amphiphilic structure [69], these studies challenge the paradigm that intracellular oxidant generation from sources such as mitochondria play the decisive role in the pathogenesis of I/R injury. However, it is possible that oxidants generated from intracellular sources such as mitochondria may target the extracellular face of sarcolemmal membrane after transmembrane egress via anion channels. Membrane packing or altered mobility induced by copolymer binding could limit diffusion of oxidant species into the bilayer, providing an alternate mechanistic rationale to reconcile a role for intracellular oxidant generation, membrane fragility, and cell death. Alternatively, stabilization of the outer cell membrane may alter cytoskeletal interactions which in turn may modify or disrupt mitochondrial trafficking such that these organelles are directed away from intracellular sites such as the nucleus that are particularly vulnerable to ROS attack, thereby allowing elevated intracellular ROS production to persist but limiting its intracellular consequences. The effectiveness of mitochondrially-targeted antioxidant treatments, which presumably do not act via membrane stabilization, in reducing I/R injury in preclinical I/R models appear to support important roles for mitochondrial ROS as a decisive contributor to postischemic injury, suggesting that alternate explanations for the protective effects afforded by copolymer treatment should be explored.
While it seems clear that overexuberant ROS production by mitochondria may contribute to I/R, it is also appears that production of hydrogen peroxide by this organelle serves as an important signaling element that couples cellular metabolism to blood flow and as a mediator of flow-dependent vasodilation in the coronary circulation. Mitochondrial ROS generation also appears to play a critical role in evoking the expression of cell survival programs that mitigate postischemic tissue injury and cell death by a variety of pre- and postconditioning stimuli. The cardioprotective mechanisms induced by preconditioning clearly involve activation of mitochondrial KATP and BKCa channels, which produce membrane alkalinization and ROS generation by respiratory chain supercomplexes, which in turn activate downstream signaling elements to induce expression of cardioprotective proteins that limit I/R injury and cell death. Thus, production of ROS by mitochondria can exert counterbalancing effects on tissue cells, with a low flux of mitochondrial oxidants serving signaling functions to regulate vascular caliber to meet metabolic demands. In addition, mitochondrial ROS production at signaling levels and perhaps localized at specific subcellular sites within mitochondria or other organelles activate cell survival programs to enhance tolerance to ischemia. On the other hand, overproduction of mitochondrial oxidants leads to ROS-induced ROS release to exacerbate oxidative stress that promotes mitochondrial permeability transition, which in turn precipitates swelling that can progress to cell lysis and death during I/R.
Acknowledgments
Supported by a grant from the National Institutes of Health (PO1 HL095486).
References
- 1.Andreadou I., Iliodromitis E.K., Farmakis D., Kremastinos D.T. To prevent, protect and save the ischemic heart: antioxidants revisited. Expert Opinion on Therapeutic Targets. 2009;13:945–956. doi: 10.1517/14728220903039698. 19534573 [DOI] [PubMed] [Google Scholar]
- 2.Bath P.M., Gray L.J., Bath A.J., Buchan A., Miyata T., Green A.R. Effects of NXY-059 in experimental stroke: an individual animal meta-analysis. British Journal of Pharmacology. 2009;157:1157–1171. doi: 10.1111/j.1476-5381.2009.00196.x. 19422398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaxa-Chamiec T., Bednarz B., Herbaczynska-Cedro K., Maciejewski P., Ceremuzynski L. Effects of vitamins C and E on the outcome after acute myocardial infarction in diabetics: aretrospective, hypothesis-generating analysis from the MIVIT study. Cardiology. 2009;112:219–223. doi: 10.1159/000151239. 18698138 [DOI] [PubMed] [Google Scholar]
- 4.Vivekananthan D.P., Penn M.S., Sapp S.K., Hsu A., Topol E.J. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003;361:2017–2023. doi: 10.1016/S0140-6736(03)13637-9. 12814711 [DOI] [PubMed] [Google Scholar]
- 5.Kalogeris T., Baines C.P., Krenz M., Korthuis R.J. Cell biology of ischemia/reperfusion injury. International Review of Cell and Molecular Biology. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. 22878108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Mehdi A.B., Pastukh V.M., Swiger B.M., Reed D.J., Patel M.R., Bardwell G.C., Pastukh V.V., Alexeyev M.F., Gillespie M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Science Signaling. 2012;5:ra47. doi: 10.1126/scisignal.2002712. 22763339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baines C.P. The cardiac mitochondrion: nexus of stress. Annual Review of Physiology. 2010;72:61–80. doi: 10.1146/annurev-physiol-021909-135929. 20148667 [DOI] [PubMed] [Google Scholar]
- 8.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial ROS-induced ROS release: an update and review. Biochimica et Biophysica Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. 16829228 [DOI] [PubMed] [Google Scholar]
- 9.Lee H.L., Chen C.L., Yeh S.T., Zweier J.L., Chen Y.R. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. American Journal of Physiology: Heart and Circulatory Physiology. 2012;302:H1410–H1422. doi: 10.1152/ajpheart.00731.2011. 22268109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turrens J.F. Superoxide production by the mitochondrial respiratory chain. Bioscience Reports. 1997;17:3–8. doi: 10.1023/a:1027374931887. 9171915 [DOI] [PubMed] [Google Scholar]
- 11.Chen Y.R., Zweier J.L. Cardiac mitochondria and reactive oxygen species generation. Circulation Research. 2014;114:524–537. doi: 10.1161/CIRCRESAHA.114.300559. 24481843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carroll J., Fearnley I.M., Skehel J.M., Shannon R.J., Hirst J., Walker J.E. Bovine complex I is a complex of 45 different subunits. Journal of Biological Chemistry. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. 16950771 [DOI] [PubMed] [Google Scholar]
- 13.Nohl H., Gille L., Staniek K. The mystery of reactive oxygen species derived from cell respiration. Acta Biochimica Polonica. 2004;51:223–229. 15094844 [PubMed] [Google Scholar]
- 14.Murphy M.P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417:1–13. doi: 10.1042/BJ20081386. 19061483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navarro A., Boveris A. The mitochondrial energy transduction system and the aging process. American Journal of Physiology: Cell Physiology. 2007;292:C670–C686. doi: 10.1152/ajpcell.00213.2006. 17020935 [DOI] [PubMed] [Google Scholar]
- 16.Chen Q., Moghaddas S., Hoppel C.L., Lesnefsky E.J. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. American Journal of Physiology: Cell Physiology. 2008;294:C460–C466. doi: 10.1152/ajpcell.00211.2007. 18077608 [DOI] [PubMed] [Google Scholar]
- 17.Liu S.S. Cooperation of a “reactive oxygen cycle” with the Q cycle and the proton cycle in the respiratory chain—superoxide generating and cycling mechanisms in mitochondria. Journal of Bioenergetics and Biomembranes. 1999;31:367–376. doi: 10.1023/a:1018650103259. 10665526 [DOI] [PubMed] [Google Scholar]
- 18.Forquer I., Covian R., Bowman M.K., Trumpower B.L., Kramer D.M. Similar transition states mediate the Q-cycle and superoxide production by the cytochrome bc1 complex. Journal of Biological Chemistry. 2006;281:38459–38465. doi: 10.1074/jbc.M605119200. 17008316 [DOI] [PubMed] [Google Scholar]
- 19.Andreyev A.Y., Kushnareva Y.E., Starkov A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry. Biokhimii?a. 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. 15807660 [DOI] [PubMed] [Google Scholar]
- 20.Skulachev V.P. A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry. Biokhimii?a. 2007;72:1385–1396. doi: 10.1134/s0006297907120139. 18205623 [DOI] [PubMed] [Google Scholar]
- 21.Giorgio M., Migliaccio E., Orsini F., Paolucci D., Moroni M., Contursi C., Pelliccia G., Luzi L., Minucci S., Marcaccio M., Pinton P., Rizzuto R., Bernardi P., Paolucci F., Pelicci P.G. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. 16051147 [DOI] [PubMed] [Google Scholar]
- 22.Cosentino F., Francia P., Camici G.G., Pelicci P.G., Luscher T.F., Volpe M. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:622–628. doi: 10.1161/ATVBAHA.107.156059. 18162611 [DOI] [PubMed] [Google Scholar]
- 23.Paneni F., Mocharla P., Akhmedov A., Costantino S., Osto E., Volpe M., Luscher T.F., Cosentino F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circulation Research. 2012;111:278–289. doi: 10.1161/CIRCRESAHA.112.266593. 22693349 [DOI] [PubMed] [Google Scholar]
- 24.Shi Y., Cosentino F., Camici G.G., Akhmedov A., Vanhoutte P.M., Tanner F.C., Luscher T.F. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:2090–2097. doi: 10.1161/ATVBAHA.111.229260. 21817106 [DOI] [PubMed] [Google Scholar]
- 25.Camici G.G., Schiavoni M., Francia P., Bachschmid M., Martin-Padura I., Hersberger M., Tanner F.C., Pelicci P., Volpe M., Anversa P., Luscher T.F., Cosentino F. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5217–5222. doi: 10.1073/pnas.0609656104. 17360381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Lisa F., Kaludercic N., Carpi A., Menabo R., Giorgio M. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Research in Cardiology. 2009;104:131–139. doi: 10.1007/s00395-009-0008-4. 19242637 [DOI] [PubMed] [Google Scholar]
- 27.Grivennikova V.G., Vinogradov A.D. Mitochondrial production of reactive oxygen species. Biochemistry. Biokhimii?a. 2013;78:1490–1511. doi: 10.1134/S0006297913130087. 24490736 [DOI] [PubMed] [Google Scholar]
- 28.Frazziano G., Al Ghouleh I., Baust J., Shiva S., Champion H.C., Pagano P.J. Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension: indications for a seminal role for mitochondrial Nox4. American Journal of Physiology: Heart and Circulatory Physiology. 2014;306:H197–H205. doi: 10.1152/ajpheart.00977.2012. 24213612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Buul J.D., Fernandez-Borja M., Anthony E.C., Hordijk P.L. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxidants and Redox Signaling. 2005;7:308–317. doi: 10.1089/ars.2005.7.308. 15706079 [DOI] [PubMed] [Google Scholar]
- 30.Lassegue B., San Martin A., Griendling K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circulation Research. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. 22581922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaludercic N., Mialet-Perez J., Paolocci N., Parini A., Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. Journal of Molecular and Cellular Cardiology. 2014 doi: 10.1016/j.yjmcc.2013.12.032. 24412580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaludercic N., Carpi A., Menabo R., Di Lisa F., Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochimica et Biophysica Acta. 2011;1813:1323–1332. doi: 10.1016/j.bbamcr.2010.09.010. 20869994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzgerald J.C., Ugun-Klusek A., Allen G., De Girolamo L.A., Hargreaves I., Ufer C., Abramov A.Y., Billett E.E. Monoamine oxidase-A knockdown in human neuroblastoma cells reveals protection against mitochondrial toxins. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2014;28:218–229. doi: 10.1096/fj.13-235481. 24051032 [DOI] [PubMed] [Google Scholar]
- 34.Anderson E.J., Efird J.T., Davies S.W., O’Neal W.T., Darden T.M., Thayne K.A., Katunga L.A., Kindell L.C., Ferguson T.B., Anderson C.A., Chitwood W.R., Koutlas T.C., Williams J.M., Rodriguez E., Kypson A.P. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. Journal of the American Heart Association. 2014;3:e000713. doi: 10.1161/JAHA.113.000713. 24572256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paradies G., Petrosillo G., Pistolese M., Di Venosa N., Federici A., Ruggiero F.M. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: Involvement of reactive oxygen species and cardiolipin. Circulation Research. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. 14656928 [DOI] [PubMed] [Google Scholar]
- 36.Chen J., Chen C.L., Rawale S., Chen C.A., Zweier J.L., Kaumaya P.T., Chen Y.R. Peptide-based antibodies against glutathione-binding domains suppress superoxide production mediated by mitochondrial complex I. Journal of Biological Chemistry. 2010;285:3168–3180. doi: 10.1074/jbc.M109.056846. 19940158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bleier L., Drose S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochimica et Biophysica Acta. 2013;1827:1320–1331. doi: 10.1016/j.bbabio.2012.12.002. 23269318 [DOI] [PubMed] [Google Scholar]
- 38.Pelletier M., Lepow T.S., Billingham L.K., Murphy M.P., Siegel R.M. New tricks from an old dog: mitochondrial redox signaling in cellular inflammation. Seminars in Immunology. 2012;24:384–392. doi: 10.1016/j.smim.2013.01.002. 23391428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Q., Moghaddas S., Hoppel C.L., Lesnefsky E.J. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. Journal of Pharmacology and Experimental Therapeutics. 2006;319:1405–1412. doi: 10.1124/jpet.106.110262. 16990510 [DOI] [PubMed] [Google Scholar]
- 40.Chen C.H., Budas G.R., Churchill E.N., Disatnik M.H., Hurley T.D., Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science (New York, N.Y.) 2008;321:1493–1495. doi: 10.1126/science.1158554. 18787169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gadicherla A.K., Stowe D.F., Antholine W.E., Yang M., Camara A.K. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochimica et Biophysica Acta. 2012;1817:419–429. doi: 10.1016/j.bbabio.2011.11.021. 22178605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zweier J.L., Flaherty J.T., Weisfeldt M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. 3029779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrosillo G., Ruggiero F.M., Di Venosa N., Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2003;17:714–716. doi: 10.1096/fj.02-0729fje. 12586737 [DOI] [PubMed] [Google Scholar]
- 44.Liu B., Tewari A.K., Zhang L., Green-Church K.B., Zweier J.L., Chen Y.R., He G. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: Mitochondria as the major target. Biochimica et Biophysica Acta. 2009;1794:476–485. doi: 10.1016/j.bbapap.2008.12.008. 19150419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paradies G., Paradies V., Ruggiero F.M., Petrosillo G. Cardiolipin and mitochondrial function in health and disease. Antioxidants and Redox Signaling. 2014;20:1925–1953. doi: 10.1089/ars.2013.5280. 24094094 [DOI] [PubMed] [Google Scholar]
- 46.Aon M.A., Cortassa S., Marban E., O’Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. Journal of Biological Chemistry. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. 12930841 [DOI] [PubMed] [Google Scholar]
- 47.Akar F.G., Aon M.A., Tomaselli G.F., O’Rourke B. The mitochondrial origin of postischemic arrhythmias. Journal of Clinical Investigation. 2005;115:3527–3535. doi: 10.1172/JCI25371. 16284648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marchissio J.M., Frances D.E., Carnovale C.E., Marinelli R.A. Mitochondrial aquaporin-8 knockdown in human hepatoma HepG2 cells causes ROS-induced mitochondrial depolarization and loss of viability. Toxicology and Applied Pharmacology. 2012;264:246–254. doi: 10.1016/j.taap.2012.08.005. 22910329 [DOI] [PubMed] [Google Scholar]
- 49.Spear J.F., Prabu S.K., Galati D., Raza H., Anandatheerthavarada H.K., Avadhani N.G. beta1-Adrenoreceptor activation contributes to ischemia-reperfusion damage as well as playing a role in ischemic preconditioning. American Journal of Physiology: Heart and Circulatory Physiology. 2007;292:H2459–H2466. doi: 10.1152/ajpheart.00459.2006. 17237252 [DOI] [PubMed] [Google Scholar]
- 50.Chandel N.S., Budinger G.R., Choe S.H., Schumacker P.T. Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. Journal of Biological Chemistry. 1997;272:18808–18816. doi: 10.1074/jbc.272.30.18808. 9228055 [DOI] [PubMed] [Google Scholar]
- 51.Zuo L., Chen Y.R., Reyes L.A., Lee H.L., Chen C.L., Villamena F.A., Zweier J.L. The radical trap 5,5-dimethyl-1-pyrroline N-oxide exerts dose-dependent protection against myocardial ischemia–reperfusion injury through preservation of mitochondrial electron transport. Journal of Pharmacology and Experimental Therapeutics. 2009;329:515–523. doi: 10.1124/jpet.108.143479. 19201989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zorov D.B., Filburn C.R., Klotz L.O., Zweier J.L., Sollott S.J. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. Journal of Experimental Medicine. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. 11015441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davidson S.M., Yellon D.M., Murphy M.P., Duchen M.R. Slow calcium waves and redox changes precede mitochondrial permeability transition pore opening in the intact heart during hypoxia and reoxygenation. Cardiovascular Research. 2012;93:445–453. doi: 10.1093/cvr/cvr349. 22198507 [DOI] [PubMed] [Google Scholar]
- 54.Brunk U.T., Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radical Biology and Medicine. 2002;33:611–619. doi: 10.1016/s0891-5849(02)00959-0. 12208347 [DOI] [PubMed] [Google Scholar]
- 55.Caldeira da Silva C.C., Cerqueira F.M., Barbosa L.F., Medeiros M.H., Kowaltowski A.J. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell. 2008;7:552–560. doi: 10.1111/j.1474-9726.2008.00407.x. 18505478 [DOI] [PubMed] [Google Scholar]
- 56.Seet R.C., Lee C.Y., Chan B.P., Sharma V.K., Teoh H.L., Venketasubramanian N., Lim E.C., Chong W.L., Looi W.F., Huang S.H., Ong B.K., Halliwell B. Oxidative damage in ischemic stroke revealed using multiple biomarkers. Stroke: A Journal of Cerebral Circulation. 2011;42:2326–2329. doi: 10.1161/STROKEAHA.111.618835. 21700941 [DOI] [PubMed] [Google Scholar]
- 57.Dirnagl U., Iadecola C., Moskowitz M.A. Pathobiology of ischaemic stroke: an integrated view. Trends in Neurosciences. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. 10441299 [DOI] [PubMed] [Google Scholar]
- 58.Endres M., Namura S., Shimizu-Sasamata M., Waeber C., Zhang L., Gomez-Isla T., Hyman B.T., Moskowitz M.A. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. 9498840 [DOI] [PubMed] [Google Scholar]
- 59.Hall E.D., Pazara K.E., Braughler J.M., Linseman K.L., Jacobsen E.J. Nonsteroidal lazaroid U78517F in models of focal and global ischemia. Stroke: A Journal of Cerebral Circulation. 1990;21:III83–III87. 2237990 [PubMed] [Google Scholar]
- 60.Phillis J.W., Clough-Helfman C. Protection from cerebral ischemic injury in gerbils with the spin trap agent N-tert-butyl-alpha-phenylnitrone (PBN) Neuroscience Letters. 1990;116:315–319. doi: 10.1016/0304-3940(90)90093-o. 2243611 [DOI] [PubMed] [Google Scholar]
- 61.Figueira T.R., Barros M.H., Camargo A.A., Castilho R.F., Ferreira J.C., Kowaltowski A.J., Sluse F.E., Souza-Pinto N.C., Vercesi A.E. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antiox Redox Signal 2013;18: 2029-2074.23244576DOI:10.1089/ars.2012.4729. [DOI] [PubMed]
- 62.Friberg H., Wieloch T., Castilho R.F. Mitochondrial oxidative stress after global brain ischemia in rats. Neuroscience Letters. 2002;334:111–114. doi: 10.1016/s0304-3940(02)01116-3. 12435484 [DOI] [PubMed] [Google Scholar]
- 63.Kim G.W., Kondo T., Noshita N., Chan P.H. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: implications for the production and role of superoxide radicals. Stroke: A Journal of Cerebral Circulation. 2002;33:809–815. doi: 10.1161/hs0302.103745. 11872908 [DOI] [PubMed] [Google Scholar]
- 64.Piantadosi C.A., Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke: A Journal of Cerebral Circulation. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. 8571432 [DOI] [PubMed] [Google Scholar]
- 65.Moro M.A., Almeida A., Bolanos J.P., Lizasoain I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radical Biology and Medicine. 2005;39:1291–1304. doi: 10.1016/j.freeradbiomed.2005.07.010. 16257638 [DOI] [PubMed] [Google Scholar]
- 66.Niatsetskaya Z.V., Sosunov S.A., Matsiukevich D., Utkina-Sosunova I.V., Ratner V.I., Starkov A.A., Ten V.S. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2012;32:3235–3244. doi: 10.1523/JNEUROSCI.6303-11.2012. 22378894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schild L., Reiser G. Oxidative stress is involved in the permeabilization of the inner membrane of brain mitochondria exposed to hypoxia/reoxygenation and low micromolar Ca2+ FEBS Journal. 2005;272:3593–3601. doi: 10.1111/j.1742-4658.2005.04781.x. 16008559 [DOI] [PubMed] [Google Scholar]
- 68.Tretter L., Biagioni Angeli E., Ardestani M.R., Goracci G., Adam-Vizi V. Reversible inhibition of hydrogen peroxide elimination by calcium in brain mitochondria. Journal of Neuroscience Research. 2011;89:1965–1972. doi: 10.1002/jnr.22658. 21541982 [DOI] [PubMed] [Google Scholar]
- 69.Wang J.Y., Marks J., Lee K.Y. Nature of interactions between PEO-PPO-PEO triblock copolymers and lipid membranes: (I) effect of polymer hydrophobicity on its ability to protect liposomes from peroxidation. Biomacromolecules. 2012;13:2616–2623. doi: 10.1021/bm300847x. 22808900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao W., Zhao D., Yan R., Sun Y. Cardiac oxidative stress and remodeling following infarction: role of NADPH oxidase. Cardiovascular Pathology: the Official Journal of the Society for Cardiovascular Pathology. 2009;18:156–166. doi: 10.1016/j.carpath.2007.12.013. 18402834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen H., Song Y.S., Chan P.H. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. 19417757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen H., Kim G.S., Okami N., Narasimhan P., Chan P.H. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiology of Disease. 2011;42:341–348. doi: 10.1016/j.nbd.2011.01.027. 21303700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kahles T., Luedike P., Endres M., Galla H.J., Steinmetz H., Busse R., Neumann-Haefelin T., Brandes R.P. NADPH oxidase plays a central role in blood–brain barrier damage in experimental stroke. Stroke: A Journal of Cerebral Circulation. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. 17916764 [DOI] [PubMed] [Google Scholar]
- 74.KahlesT. and Brandes R.P. Which NADPH oxidase isoform is relevant for ischemic stroke. Antioxid Redox Signal 2013;18: 1400-1417.227462276DOI: 10.1089/ars.2012.4721. [DOI] [PMC free article] [PubMed]
- 75.Walder C.E., Green S.P., Darbonne W.C., Mathias J., Rae J., Dinauer M.C., Curnutte J.T., Thomas G.R. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke: A Journal of Cerebral Circulation. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. 9368573 [DOI] [PubMed] [Google Scholar]
- 76.Radermacher K.A., Wingler K., Kleikers P., Altenhofer S., Jr., Hermans J., Kleinschnitz C., Schmidt H. The 1027th target candidate in stroke: will NADPH oxidase hold up? Experimental and Translational Stroke Medicine. 2012;4:11. doi: 10.1186/2040-7378-4-11. 22625431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Doerries C., Grote K., Hilfiker-Kleiner D., Luchtefeld M., Schaefer A., Holland S.M., Sorrentino S., Manes C., Schieffer B., Drexler H., Landmesser U. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circulation Research. 2007;100:894–903. doi: 10.1161/01.RES.0000261657.76299.ff. 17332431 [DOI] [PubMed] [Google Scholar]
- 78.Looi Y.H., Grieve D.J., Siva A., Walker S.J., Anilkumar N., Cave A.C., Marber M., Monaghan M.J., Shah A.M. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension. 2008;51:319–325. doi: 10.1161/HYPERTENSIONAHA.107.101980. 18180403 [DOI] [PubMed] [Google Scholar]
- 79.Doverhag C., Keller M., Karlsson A., Hedtjarn M., Nilsson U., Kapeller E., Sarkozy G., Klimaschewski L., Humpel C., Hagberg H., Simbruner G., Gressens P., Savman K. Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiology of Disease. 2008;31:133–144. doi: 10.1016/j.nbd.2008.04.003. 18571099 [DOI] [PubMed] [Google Scholar]
- 80.Wind S., Beuerlein K., Eucker T., Muller H., Scheurer P., Armitage M.E., Ho H., Schmidt H.H., Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. British Journal of Pharmacology. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. 20860666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Altenhofer S., Radermacher K.A., Kleikers P.W., Wingler K., Schmidt H.H. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxidants and Redox Signaling. 2014 doi: 10.1089/ars.2013.5814. 24383718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hong H., Zeng J.S., Kreulen D.L., Kaufman D.I., Chen A.F. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. American Journal of Physiology: Heart and Circulatory Physiology. 2006;291:H2210–H2215. doi: 10.1152/ajpheart.01270.2005. 16766636 [DOI] [PubMed] [Google Scholar]
- 83.Kleinschnitz C., Grund H., Wingler K., Armitage M.E., Jones E., Mittal M., Barit D., Schwarz T., Geis C., Kraft P., Barthel K., Schuhmann M.K., Herrmann A.M., Meuth S.G., Stoll G., Meurer S., Schrewe A., Becker L., Gailus-Durner V., Fuchs H., Klopstock T., de Angelis M.H., Jandeleit-Dahm K., Shah A.M., Weissmann N., Schmidt H.H. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biology. 2010;8 doi: 10.1371/journal.pbio.1000479. 20877715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vallet P., Charnay Y., Steger K., Ogier-Denis E., Kovari E., Herrmann F., Michel J.P., Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132:233–238. doi: 10.1016/j.neuroscience.2004.12.038. 15802177 [DOI] [PubMed] [Google Scholar]
- 85.Kleinschnitz C1, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HH. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010 Sep 21;8(9). pii: e1000479. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed]
- 86.Sun Q.A., Hess D.T., Wang B., Miyagi M., Stamler J.S. Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870) Free Radical Biology and Medicine. 2012;52:1897–1902. doi: 10.1016/j.freeradbiomed.2012.02.046. 22406319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lassegue B., Griendling K.K. NADPH oxidases: functions and pathologies in the vasculature. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. 19910640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wingler K., Wunsch S., Kreutz R., Rothermund L., Paul M., Schmidt H.H. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radical Biology and Medicine. 2001;31:1456–1464. doi: 10.1016/s0891-5849(01)00727-4. 11728818 [DOI] [PubMed] [Google Scholar]
- 89.Miller A.A., Drummond G.R., Schmidt H.H., Sobey C.G. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circulation Research. 2005;97:1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. 16210546 [DOI] [PubMed] [Google Scholar]
- 90.Miller A.A., Drummond G.R., Mast A.E., Schmidt H.H., Sobey C.G. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: role of estrogen. Stroke: A Journal of Cerebral Circulation. 2007;38:2142–2149. doi: 10.1161/STROKEAHA.106.477406. 17525399 [DOI] [PubMed] [Google Scholar]
- 91.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. 17237347 [DOI] [PubMed] [Google Scholar]
- 92.Block K., Gorin Y., Abboud H.E. Subcellular localization of Nox4 and regulation in diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. 19706525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arimura K., Ago T., Kuroda J., Ishitsuka K., Nishimura A., Sugimori H., Kamouchi M., Sasaki T., Kitazono T. Role of NADPH oxidase 4 in brain endothelial cells after ischemic stroke. Stroke. 2012;43:A2514. [Google Scholar]
- 94.Griendling K.K., Sorescu D., Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circulation Research. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. 10720409 [DOI] [PubMed] [Google Scholar]
- 95.Cave A.C., Brewer A.C., Narayanapanicker A., Ray R., Grieve D.J., Walker S., Shah A.M. NADPH oxidases in cardiovascular health and disease. Antioxidants and Redox Signaling. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. 16771662 [DOI] [PubMed] [Google Scholar]
- 96.Clempus R.E., Sorescu D., Dikalova A.E., Pounkova L., Jo P., Sorescu G.P., Schmidt H.H., Lassegue B., Griendling K.K. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:42–48. doi: 10.1161/01.ATV.0000251500.94478.18. 17082491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cucoranu I., Clempus R., Dikalova A., Phelan P.J., Ariyan S., Dikalov S., Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circulation Research. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. 16179589 [DOI] [PubMed] [Google Scholar]
- 98.Deliri H., McNamara C.A. Nox 4 regulation of vascular smooth muscle cell differentiation marker gene expression. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:12–14. doi: 10.1161/01.ATV.0000254154.43871.50. 17185622 [DOI] [PubMed] [Google Scholar]
- 99.Craige S.M., Chen K., Pei Y., Li C., Huang X., Chen C., Shibata R., Sato K., Walsh K., Keaney J.F., Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011;124:731–740. doi: 10.1161/CIRCULATIONAHA.111.030775. 21788590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Datla S.R., Peshavariya H., Dusting G.J., Mahadev K., Goldstein B.J., Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2319–2324. doi: 10.1161/ATVBAHA.107.149450. 17717289 [DOI] [PubMed] [Google Scholar]
- 101.Diebold I., Petry A., Hess J., Gorlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Molecular Biology of the Cell. 2010;21:2087–2096. doi: 10.1091/mbc.E09-12-1003. 20427574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sturrock A., Cahill B., Norman K., Huecksteadt T.P., Hill K., Sanders K., Karwande S.V., Stringham J.C., Bull D.A., Gleich M., Kennedy T.P., Hoidal J.R. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. 16227320 [DOI] [PubMed] [Google Scholar]
- 103.Maejima Y., Kuroda J., Matsushima S., Ago T., Sadoshima J. Regulation of myocardial growth and death by NADPH oxidase. Journal of Molecular and Cellular Cardiology. 2011;50:408–416. doi: 10.1016/j.yjmcc.2010.12.018. 21215757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ago T., Kuroda J., Pain J., Fu C., Li H., Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circulation Research. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. 20185797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Elahi M.M., Kong Y.X., Matata B.M. Oxidative stress as a mediator of cardiovascular disease. Oxidative Medicine and Cellular Longevity. 2009;2:259–269. doi: 10.4161/oxim.2.5.9441. 20716913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radical Biology and Medicine. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. 21777669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gustafsson A.B., Gottlieb R.A. Heart mitochondria: gates of life and death. Cardiovascular Research. 2008;77:334–343. doi: 10.1093/cvr/cvm005. 18006487 [DOI] [PubMed] [Google Scholar]
- 108.Jou M.J. Pathophysiological and pharmacological implications of mitochondria-targeted reactive oxygen species generation in astrocytes. Advanced Drug Delivery Reviews. 2008;60:1512–1526. doi: 10.1016/j.addr.2008.06.004. 18692534 [DOI] [PubMed] [Google Scholar]
- 109.Zorov D.B., Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006 May-Jun;1757(5-6):509-17. 16829228. [DOI] [PubMed]
- 110.Mukhopadhyay P., Horvath B., Zsengeller Z., Batkai S., Cao Z., Kechrid M., Holovac E., Erdelyi K., Tanchian G., Liaudet L., Stillman I.E., Joseph J., Kalyanaraman B., Pacher P. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants. Free Radical Biology and Medicine. 2012;53:1123–1138. doi: 10.1016/j.freeradbiomed.2012.05.036. 22683818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dhanasekaran A., Kotamraju S., Karunakaran C., Kalivendi S.V., Thomas S., Joseph J., Kalyanaraman B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radical Biology and Medicine. 2005;39:567–583. doi: 10.1016/j.freeradbiomed.2005.04.016. 16085176 [DOI] [PubMed] [Google Scholar]
- 112.Anders MW1, Robotham JL, Sheu SS. Mitochondria: new drug targets for oxidative stress-induced diseases. Expert Opin Drug Metab Toxicol. 2006 Feb;2(1):71-9.2006. [DOI] [PubMed]
- 113.Adlam V.J., Harrison J.C., Porteous C.M., James A.M., Smith R.A., Murphy M.P., Sammut I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. 15985532 [DOI] [PubMed] [Google Scholar]
- 114.Hobbs C.E., Murphy M.P., Smith R.A., Oorschot D.E. Neonatal rat hypoxia-ischemia: effect of the anti-oxidant mitoquinol, and S-PBN. Pediatrics International: Official Journal of the Japan Pediatric Society. 2008;50:481–488. doi: 10.1111/j.1442-200X.2008.02705.x. 18937752 [DOI] [PubMed] [Google Scholar]
- 115.Walters AM, Porter GA Jr, Brookes P.S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy.Circ Res. 2012 Oct 12;111(9):1222-36. 23065345doi: 10.1161/CIRCRESAHA.112.265660 2012. [DOI] [PMC free article] [PubMed]
- 116.Smith R.A., Hartley R.C., Murphy M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxidants and Redox Signaling. 2011;15:3021–3038. doi: 10.1089/ars.2011.3969. 21395490 [DOI] [PubMed] [Google Scholar]
- 117.Smith R.A., Murphy M.P. Mitochondria-targeted antioxidants as therapies. Discovery Medicine. 2011;11:106–114. 21356165 [PubMed] [Google Scholar]
- 118.Brown D.A., Hale S.L., Baines C.P., del Rio C.L., Hamlin R.L., Yueyama Y., Kijtawornrat A., Yeh S.T., Frasier C.R., Stewart L.M., Moukdar F., Shaikh S.R., Fisher-Wellman K.H., Neufer P.D., Kloner R.A. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. Journal of Cardiovascular Pharmacology and Therapeutics. 2014;19:121–132. doi: 10.1177/1074248413508003. 24288396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kotake Y. Pharmacologic properties of phenyl N-tert-butylnitrone. Antioxidants and Redox Signaling. 1999;1:481–499. doi: 10.1089/ars.1999.1.4-481. 11233146 [DOI] [PubMed] [Google Scholar]
- 120.Nadtochiy S.M., Burwell L.S., Ingraham C.A., Spencer C.M., Friedman A.E., Pinkert C.A., Brookes P.S. In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. Journal of Molecular and Cellular Cardiology. 2009;46:960–968. doi: 10.1016/j.yjmcc.2009.01.012. 19339206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Smith R.A., Hartley R.C., Cocheme H.M., Murphy M.P. Mitochondrial pharmacology. Trends in Pharmacological Sciences. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. 22521106 [DOI] [PubMed] [Google Scholar]
- 122.Navarro-Yepes J., Zavala-Flores L., Anandhan A., Wang F., Skotak M., Chandra N., Li M., Pappa A., Martinez-Fong D., Del Razo L.M., Quintanilla-Vega B., Franco R. Antioxidant gene therapy against neuronal cell death. Pharmacology and Therapeutics. 2014;142:206–230. doi: 10.1016/j.pharmthera.2013.12.007. 24333264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ouyang Y.B., Stary C.M., White R.E., Giffard R.G. The use of microRNAs to modulate redox and immune response to stroke. Antioxidants and Redox Signaling. 2014 doi: 10.1089/ars.2013.5757. 24359188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang D.X., Gutterman D.D. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. American Journal of Physiology: Heart and Circulatory Physiology. 2007;292:H2023–H2031. doi: 10.1152/ajpheart.01283.2006. 17237240 [DOI] [PubMed] [Google Scholar]
- 125.Liu Y., Zhao H., Li H., Kalyanaraman B., Nicolosi A.C., Gutterman D.D. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circulation Research. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. 12919951 [DOI] [PubMed] [Google Scholar]
- 126.Zhang D.X., Borbouse L., Gebremedhin D., Mendoza S.A., Zinkevich N.S., Li R., Gutterman D.D. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circulation Research. 2012;110:471–480. doi: 10.1161/CIRCRESAHA.111.258871. 22158710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Barlow R.S., White R.E. Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity. American Journal of Physiology. 1998;275:H1283–H1289. doi: 10.1152/ajpheart.1998.275.4.H1283. 9746477 [DOI] [PubMed] [Google Scholar]
- 128.Brakemeier S., Eichler I., Knorr A., Fassheber T., Kohler R., Hoyer J. Modulation of Ca2+-activated K+ channel in renal artery endothelium in situ by nitric oxide and reactive oxygen species. Kidney International. 2003;64:199–207. doi: 10.1046/j.1523-1755.2003.00051.x. 12787410 [DOI] [PubMed] [Google Scholar]
- 129.Palen D.I., Ouhtit A., Belmadani S., Lucchesi P.A., Matrougui K. Hydrogen peroxide acts as relaxing factor in human vascular smooth muscle cells independent of map-kinase and nitric oxide. Frontiers in Bioscience: A Journal and Virtual Library. 2006;11:2526–2534. doi: 10.2741/1987. 16720330 [DOI] [PubMed] [Google Scholar]
- 130.Xi Q., Cheranov S.Y., Jaggar J.H. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circulation Research. 2005;97:354–362. doi: 10.1161/01.RES.0000177669.29525.78. 16020754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Saitoh S., Zhang C., Tune J.D., Potter B., Kiyooka T., Rogers P.A., Knudson J.D., Dick G.M., Swafford A., Chilian W.M. Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:2614–2621. doi: 10.1161/01.ATV.0000249408.55796.da. 17023676 [DOI] [PubMed] [Google Scholar]
- 132.Saitoh S., Kiyooka T., Rocic P., Rogers P.A., Zhang C., Swafford A., Dick G.M., Viswanathan C., Park Y., Chilian W.M. Redox-dependent coronary metabolic dilation. American Journal of Physiology: Heart and Circulatory Physiology. 2007;293:H3720–H3725. doi: 10.1152/ajpheart.00436.2007. 17965288 [DOI] [PubMed] [Google Scholar]
- 133.Rogers P.A., Chilian W.M., Bratz I.N., Bryan R.M., Jr., Dick G.M. H2O2 activates redox- and 4-aminopyridine-sensitive Kv channels in coronary vascular smooth muscle. American Journal of Physiology: Heart and Circulatory Physiology. 2007;292:H1404–H1411. doi: 10.1152/ajpheart.00696.2006. 17071731 [DOI] [PubMed] [Google Scholar]
- 134.Wang Q., Sun A.Y., Simonyi A., Kalogeris T.J., Miller D.K., Sun G.Y., Korthuis R.J. Ethanol preconditioning protects against ischemia/reperfusion-induced brain damage: role of NADPH oxidase-derived ROS. Free Radical Biology and Medicine. 2007;43:1048–1060. doi: 10.1016/j.freeradbiomed.2007.06.018. 17761301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang Q., Sun A.Y., Simonyi A., Miller D.K., Smith R.E., Luchtefeld R.G., Korthuis R.J., Sun G.Y. Oral administration of grape polyphenol extract ameliorates cerebral ischemia/reperfusion-induced neuronal damage and behavioral deficits in gerbils: comparison of pre- and post-ischemic administration. Journal of Nutritional Biochemistry. 2009;20:369–377. doi: 10.1016/j.jnutbio.2008.04.007. 18602816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang Q., Tompkins K.D., Simonyi A., Korthuis R.J., Sun A.Y., Sun G.Y. Apocynin protects against global cerebral ischemia–reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Research. 2006;1090:182–189. doi: 10.1016/j.brainres.2006.03.060. 16650838 [DOI] [PubMed] [Google Scholar]
- 137.Fornazari M., de Paula J.G., Castilho R.F., Kowaltowski A.J. Redox properties of the adenoside triphosphate-sensitive K+ channel in brain mitochondria. Journal of Neuroscience Research. 2008;86:1548–1556. doi: 10.1002/jnr.21614. 18189325 [DOI] [PubMed] [Google Scholar]
- 138.Gaspar T., Snipes J.A., Busija A.R., Kis B., Domoki F., Bari F., Busija D.W. ROS-independent preconditioning in neurons via activation of mitoK(ATP) channels by BMS-191095. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:1090–1103. doi: 10.1038/sj.jcbfm.9600611. 18212794 [DOI] [PubMed] [Google Scholar]
- 139.Drose S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochimica et Biophysica Acta. 2013;1827:578–587. doi: 10.1016/j.bbabio.2013.01.004. 23333272 [DOI] [PubMed] [Google Scholar]
- 140.Ertracht O., Malka A., Atar S., Binah O. The mitochondria as a target for cardioprotection in acute myocardial ischemia. Pharmacology and Therapeutics. 2014;142:33–40. doi: 10.1016/j.pharmthera.2013.11.003. 24275322 [DOI] [PubMed] [Google Scholar]
- 141.Kis B., Rajapakse N., Snipes J.A., Nagy K., Horiguchi T., Busija D.W. Diazoxide induces delayed pre-conditioning in cultured rat cortical neurons. Journal of Neurochemistry. 2003;87:969–980. doi: 10.1046/j.1471-4159.2003.02072.x. 14622127 [DOI] [PubMed] [Google Scholar]
- 142.Krenz M., Baines C., Kalogeris T., Korthuis R. 2013. [Google Scholar]
- 143.Tullio F., Angotti C., Perrelli M.G., Penna C., Pagliaro P. Redox balance and cardioprotection. Basic Res Cardiol. 2013;108:392. doi: 10.1007/s00395-013-0392-7. 24158692 [DOI] [PubMed] [Google Scholar]
- 144.Penna C., Mancardi D., Rastaldo R., Pagliaro P. Cardioprotection: a radical view free radicals in pre and postconditioning. Biochimica et Biophysica Acta. 2009;1787:781–793. doi: 10.1016/j.bbabio.2009.02.008. 19248760 [DOI] [PubMed] [Google Scholar]
- 145.Boengler K., Heusch G., Schulz R. Mitochondria in postconditioning. Antioxidants and Redox Signaling. 2011;14:863–880. doi: 10.1089/ars.2010.3309. 20578961 [DOI] [PubMed] [Google Scholar]
- 146.Penna C., Mancardi D., Rastaldo R., Losano G., Pagliaro P. Intermittent activation of bradykinin B2 receptors and mitochondrial KATP channels trigger cardiac postconditioning through redox signaling. Cardiovascular Research. 2007;75:168–177. doi: 10.1016/j.cardiores.2007.03.001. 17400201 [DOI] [PubMed] [Google Scholar]
- 147.Oldenburg O., Qin Q., Krieg T., Yang X.M., Philipp S., Critz S.D., Cohen M.V., Downey J.M. Bradykinin induces mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP channel opening and leads to cardioprotection. American Journal of Physiology: Heart and Circulatory Physiology. 2004;286:H468–H476. doi: 10.1152/ajpheart.00360.2003. 12958031 [DOI] [PubMed] [Google Scholar]
- 148.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochimica et Biophysica Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. 20122895 [DOI] [PubMed] [Google Scholar]
- 149.Costa A.D., Pierre S.V., Cohen M.V., Downey J.M., Garlid K.D. CGMP signalling in pre- and post-conditioning: the role of mitochondria. Cardiovascular Research. 2008;77:344–352. doi: 10.1093/cvr/cvm050. 18006449 [DOI] [PubMed] [Google Scholar]
- 150.Gidday J.M. Cerebral preconditioning and ischaemic tolerance. Nature Reviews: Neuroscience. 2006;7:437–448. doi: 10.1038/nrn1927. 16715053 [DOI] [PubMed] [Google Scholar]
- 151.Liu Y., Sato T., O’Rourke B., Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. 9641699 [DOI] [PubMed] [Google Scholar]
- 152.Liu B., Zhu X., Chen C.L., Hu K., Swartz H.M., Chen Y.R., He G. Opening of the mitoKATP channel and decoupling of mitochondrial complex II and III contribute to the suppression of myocardial reperfusion hyperoxygenation. Molecular and Cellular Biochemistry. 2010;337:25–38. doi: 10.1007/s11010-009-0283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Drose S., Hanley P.J., Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochimica et Biophysica Acta. 2009;1790:558–565. doi: 10.1016/j.bbagen.2009.01.011. 19364480 [DOI] [PubMed] [Google Scholar]
- 154.Wojtovich A.P., Brookes P.S. The complex II inhibitor atpenin A5 protects against cardiac ischemia–reperfusion injury via activation of mitochondrial KATP channels. Basic Research in Cardiology. 2009;104:121–129. doi: 10.1007/s00395-009-0001-y. 19242645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Heinzel F.R., Luo Y., Li X., Boengler K., Buechert A., Garcia-Dorado D., Di Lisa F., Schulz R., Heusch G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circulation Research. 2005;97:583–586. doi: 10.1161/01.RES.0000181171.65293.65. 16100048 [DOI] [PubMed] [Google Scholar]
- 156.Lacza Z., Snipes J.A., Kis B., Szabo C., Grover G., Busija D.W. Investigation of the subunit composition and the pharmacology of the mitochondrial ATP-dependent K+ channel in the brain. Brain Research. 2003;994:27–36. doi: 10.1016/j.brainres.2003.09.046. 14642445 [DOI] [PubMed] [Google Scholar]
- 157.Busija D.W., Katakam P., Rajapakse N.C., Kis B., Grover G., Domoki F., Bari F. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Research Bulletin. 2005;66:85–90. doi: 10.1016/j.brainresbull.2005.03.022. 15982523 [DOI] [PubMed] [Google Scholar]
- 158.Oldenburg O., Qin Q., Sharma A.R., Cohen M.V., Downey J.M., Benoit J.N. Acetylcholine leads to free radical production dependent on K(ATP) channels, G(i) proteins, phosphatidylinositol 3-kinase and tyrosine kinase. Cardiovascular Research. 2002;55:544–552. doi: 10.1016/s0008-6363(02)00332-2. 12160951 [DOI] [PubMed] [Google Scholar]
- 159.Krieg T., Landsberger M., Alexeyev M.F., Felix S.B., Cohen M.V., Downey J.M. Activation of Akt is essential for acetylcholine to trigger generation of oxygen free radicals. Cardiovascular Research. 2003;58:196–202. doi: 10.1016/s0008-6363(02)00861-1. 12667962 [DOI] [PubMed] [Google Scholar]
- 160.Rottlaender D., Boengler K., Wolny M., Michels G., Endres-Becker J., Motloch L.J., Schwaiger A., Buechert A., Schulz R., Heusch G., Hoppe U.C. Connexin 43 acts as a cytoprotective mediator of signal transduction by stimulating mitochondrial KATP channels in mouse cardiomyocytes. Journal of Clinical Investigation. 2010;120:1441–1453. doi: 10.1172/JCI40927. 20364086 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 161.Ishikawa S., Kuno A., Tanno M., Miki T., Kouzu H., Itoh T., Sato T., Sunaga D., Murase H., Miura T. Role of connexin-43 in protective PI3K-Akt-GSK-3b signaling in cardiomyocytes. AJP: Heart and Circulatory Physiology. 2012;302:H2536–H2544. doi: 10.1152/ajpheart.00940.2011. [DOI] [PubMed] [Google Scholar]
- 162.Cao C.M., Xia Q., Gao Q., Chen M., Wong T.M. Calcium-activated potassium channel triggers cardioprotection of ischemic preconditioning. Journal of Pharmacology and Experimental Therapeutics. 2005;312:644–650. doi: 10.1124/jpet.104.074476. 15345753 [DOI] [PubMed] [Google Scholar]
- 163.Liu Y., Kalogeris T., Wang M., Zuidema M.Y., Wang Q., Dai H., Davis M.J., Hill M.A., Korthuis R.J. Hydrogen sulfide preconditioning or neutrophil depletion attenuates ischemia-reperfusion-induced mitochondrial dysfunction in rat small intestine. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2012;302:G44–G54. doi: 10.1152/ajpgi.00413.2010. 21921289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shintani Y., Node K., Asanuma H., Sanada S., Takashima S., Asano Y., Liao Y., Fujita M., Hirata A., Shinozaki Y., Fukushima T., Nagamachi Y., Okuda H., Kim J., Tomoike H., Hori M., Kitakaze M. Opening of Ca2+-activated K+ channels is involved in ischemic preconditioning in canine hearts. Journal of Molecular and Cellular Cardiology. 2004;37:1213–1218. doi: 10.1016/j.yjmcc.2004.09.012. 15572051 [DOI] [PubMed] [Google Scholar]
- 165.Wang X., Yin C., Xi L., Kukreja R.C. Opening of Ca2+-activated K+ channels triggers early and delayed preconditioning against I/R injury independent of NOS in mice. American Journal of Physiology: Heart and Circulatory Physiology. 2004;287:H2070–H2077. doi: 10.1152/ajpheart.00431.2004. 15217801 [DOI] [PubMed] [Google Scholar]
- 166.Xu W., Liu Y., Wang S., McDonald T., Van Eyk J.E., Sidor A., O’Rourke B. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science (New York, N.Y.) 2002;298:1029–1033. doi: 10.1126/science.1074360. 12411707 [DOI] [PubMed] [Google Scholar]
- 167.Stowe D.F., Aldakkak M., Camara A.K., Riess M.L., Heinen A., Varadarajan S.G., Jiang M.T. Cardiac mitochondrial preconditioning by big Ca2+-sensitive K+ channel opening requires superoxide radical generation. American Journal of Physiology: Heart and Circulatory Physiology. 2006;290:H434–H440. doi: 10.1152/ajpheart.00763.2005. 16126810 [DOI] [PubMed] [Google Scholar]
- 168.Wang Q., Kalogeris T.J., Wang M., Jones A.W., Korthuis R.J. Antecedent ethanol attenuates cerebral ischemia/reperfusion-induced leukocyte–endothelial adhesive interactions and delayed neuronal death: role of large conductance, Ca2+-activated K+ channels. Microcirculation (New York, N.Y.: 1994) 2010;17:427–438. doi: 10.1111/j.1549-8719.2010.00041.x. 20690981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zuidema M.Y., Yang Y., Wang M., Kalogeris T., Liu Y., Meininger C.J., Hill M.A., Davis M.J., Korthuis R.J. Antecedent hydrogen sulfide elicits an anti-inflammatory phenotype in postischemic murine small intestine: role of BK channels. AJP: Heart and Circulatory Physiology. 2010;299:H1554–H1567. doi: 10.1152/ajpheart.01229.2009. 20833953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Heinen A., Camara A.K., Aldakkak M., Rhodes S.S., Riess M.L., Stowe D.F. Mitochondrial Ca2+-induced K+ influx increases respiration and enhances ROS production while maintaining membrane potential. American Journal of Physiology. Cell Physiology. 2007;292:C148–C156. doi: 10.1152/ajpcell.00215.2006. 16870831 [DOI] [PubMed] [Google Scholar]
- 171.Bodyak N., Rigor D.L., Chen Y.S., Han Y., Bisping E., Pu W.T., Kang P.M. Uncoupling protein 2 modulates cell viability in adult rat cardiomyocytes. American Journal of Physiology: Heart and Circulatory Physiology. 2007;293:H829–H835. doi: 10.1152/ajpheart.01409.2006. 17468330 [DOI] [PubMed] [Google Scholar]
- 172.Hoerter J., Gonzalez-Barroso M.D., Couplan E., Mateo P., Gelly C., Cassard-Doulcier A.M., Diolez P., Bouillaud F. Mitochondrial uncoupling protein 1 expressed in the heart of transgenic mice protects against ischemic–reperfusion damage. Circulation. 2004;110:528–533. doi: 10.1161/01.CIR.0000137824.30476.0E. 15262832 [DOI] [PubMed] [Google Scholar]
- 173.Sack M.N. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovascular Research. 2006;72:210–219. doi: 10.1016/j.cardiores.2006.07.010. 16914124 [DOI] [PubMed] [Google Scholar]
- 174.Nadtochiy S.M., Baker P.R., Freeman B.A., Brookes P.S. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: implications for cardioprotection. Cardiovascular Research. 2009;82:333–340. doi: 10.1093/cvr/cvn323. 19050010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Skulachev V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Quarterly Reviews of Biophysics. 1996;29:169–202. doi: 10.1017/s0033583500005795. 8870073 [DOI] [PubMed] [Google Scholar]
- 176.Aldakkak M., Stowe D.F., Chen Q., Lesnefsky E.J., Camara A.K. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovascular Research. 2008;77:406–415. doi: 10.1016/j.cardiores.2007.08.008. 17900548 [DOI] [PubMed] [Google Scholar]
- 177.Shiva S., Sack M.N., Greer J.J., Duranski M., Ringwood L.A., Burwell L., Wang X., MacArthur P.H., Shoja A., Raghavachari N., Calvert J.W., Brookes P.S., Lefer D.J., Gladwin M.T. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. Journal of Experimental Medicine. 2007;204:2089–2102. doi: 10.1084/jem.20070198. 17682069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vinten-Johansen J., Shi W. Perconditioning and postconditioning: current knowledge, knowledge gaps, barriers to adoption, and future directions. Journal of Cardiovascular Pharmacology and Therapeutics. 2011;16:260–266. doi: 10.1177/1074248411415270. 21821526 [DOI] [PubMed] [Google Scholar]
- 179.Cohen M.V., Yang X.M., Downey J.M. Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning's success. Basic Research in Cardiology. 2008;103:464–471. doi: 10.1007/s00395-008-0737-9. 18626679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Penna C., Rastaldo R., Mancardi D., Raimondo S., Cappello S., Gattullo D., Losano G., Pagliaro P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Research in Cardiology. 2006;101:180–189. doi: 10.1007/s00395-006-0584-5. 16450075 [DOI] [PubMed] [Google Scholar]
- 181.Hausenloy D., Wynne A., Duchen M., Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–1717. doi: 10.1161/01.CIR.0000126294.81407.7D. 15066952 [DOI] [PubMed] [Google Scholar]
- 182.Hausenloy D.J., Ong S.B., Yellon D.M. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Research in Cardiology. 2009;104:189–202. doi: 10.1007/s00395-009-0010-x. 19242644 [DOI] [PubMed] [Google Scholar]
- 183.Lim S.Y., Davidson S.M., Hausenloy D.J., Yellon D.M. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovascular Research. 2007;75:530–535. doi: 10.1016/j.cardiores.2007.04.022. 17512507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Petronilli V., Miotto G., Canton M., Brini M., Colonna R., Bernardi P., Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophysical Journal. 1999;76:725–734. doi: 10.1016/S0006-3495(99)77239-5. 9929477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tsutsumi Y.M., Yokoyama T., Horikawa Y., Roth D.M., Patel H.H. Reactive oxygen species trigger ischemic and pharmacological postconditioning: in vivo and in vitro characterization. Life Sciences. 2007;81:1223–1227. doi: 10.1016/j.lfs.2007.08.031. 17915258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yao Y.T., Li L.H., Chen L., Wang W.P., Li L.B., Gao C.Q. Sevoflurane postconditioning protects isolated rat hearts against ischemia-reperfusion injury: the role of radical oxygen species, extracellular signal-related kinases 1/2 and mitochondrial permeability transition pore. Molecular Biology Reports. 2010;37:2439–2446. doi: 10.1007/s11033-009-9755-4. 19693689 [DOI] [PubMed] [Google Scholar]
- 187.Penna C., Perrelli M.G., Pagliaro P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxidants and Redox Signaling. 2013;18:556–599. doi: 10.1089/ars.2011.4459. 22668069 [DOI] [PubMed] [Google Scholar]
- 188.Martindale J.J., Metzger J.M. Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. Journal of Molecular and Cellular Cardiology. 2014;67:26–37. doi: 10.1016/j.yjmcc.2013.12.008. 24362314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dudley R.W., Khairallah M., Mohammed S., Lands L., Des Rosiers C., Petrof B.J. Dynamic responses of the glutathione system to acute oxidative stress in dystrophic mouse (mdx) muscles. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2006;291:R704–R710. doi: 10.1152/ajpregu.00031.2006. 16614063 [DOI] [PubMed] [Google Scholar]
- 190.Kyoi S., Otani H., Sumida T., Okada T., Osako M., Imamura H., Kamihata H., Matsubara H., Iwasaka T. Loss of intracellular dystrophin: a potential mechanism for myocardial reperfusion injury. Circulation Journal: Official Journal of the Japanese Circulation Society. 2003;67:725–727. doi: 10.1253/circj.67.725. 12890920 [DOI] [PubMed] [Google Scholar]
- 191.Rodriguez M., Cai W.J., Kostin S., Lucchesi B.R., Schaper J. Ischemia depletes dystrophin and inhibits protein synthesis in the canine heart: mechanisms of myocardial ischemic injury. Journal of Molecular and Cellular Cardiology. 2005;38:723–733. doi: 10.1016/j.yjmcc.2005.02.019. 15850566 [DOI] [PubMed] [Google Scholar]
- 192.Armstrong S.C., Latham C.A., Shivell C.L., Ganote C.E. Ischemic loss of sarcolemmal dystrophin and spectrin: correlation with myocardial injury. Journal of Molecular and Cellular Cardiology. 2001;33:1165–1179. doi: 10.1006/jmcc.2001.1380. 11444921 [DOI] [PubMed] [Google Scholar]
- 193.Kido M., Otani H., Kyoi S., Sumida T., Fujiwara H., Okada T., Imamura H. Ischemic preconditioning-mediated restoration of membrane dystrophin during reperfusion correlates with protection against contraction-induced myocardial injury. American Journal of Physiology: Heart and Circulatory Physiology. 2004;287:H81–H90. doi: 10.1152/ajpheart.01140.2003. 15001448 [DOI] [PubMed] [Google Scholar]
- 194.Kyoi S., Otani H., Hamano A., Matsuhisa S., Akita Y., Fujiwara H., Hattori R., Imamura H., Kamihata H., Iwasaka T. Dystrophin is a possible end-target of ischemic preconditioning against cardiomyocyte oncosis during the early phase of reperfusion. Cardiovascular Research. 2006;70:354–363. doi: 10.1016/j.cardiores.2006.01.004. 16466703 [DOI] [PubMed] [Google Scholar]