

Abstract
The cryptococcal capsule is a critical virulence factor of an important pathogen, but little is known about how it is associated with the cell or released into the environment. Two mutants lacking PBX1 and PBX2 were found to shed reduced amounts of the capsule polysaccharide glucuronoxylomannan (GXM). Nuclear magnetic resonance, composition, and physical analyses showed that the shed material was of normal mass but was slightly enriched in xylose. In contrast to previous reports, this material contained no glucose. Notably, the capsule fibers of pbxΔ mutant cells grown under capsule-inducing conditions were present at a lower than usual density and were loosely attached to the cell wall. Mutant cell walls were also defective, as indicated by phenotypes including abnormal cell morphology, reduced mating filamentation, and altered cell integrity. All observed phenotypes were shared between the two mutants and exacerbated in a double mutant. Consistent with a role in surface glycan synthesis, the Pbx proteins localized to detergent-resistant membrane domains. These results, together with the sequence motifs in the Pbx proteins, suggest that Pbx1 and Pbx2 are redundant proteins that act in remodeling the cell wall to maintain normal cell morphology and precursor availability for other glycan synthetic processes. Their absence results in aberrant cell wall growth and metabolic imbalance, which together impact cell wall and capsule synthesis, cell morphology, and capsule association. The surface changes also lead to increased engulfment by host phagocytes, consistent with the lack of virulence of pbx mutants in animal models.
INTRODUCTION
The basidiomycetous yeast Cryptococcus neoformans is ubiquitous in the environment and is most commonly associated with bird excreta (1–3). In recent years, this yeast has emerged as an important opportunistic pathogen of humans. Cryptococcosis poses a particular challenge in the developing world, where large populations of individuals are immunocompromised due to HIV infection in the context of limited health care resources. These factors contribute to a global death toll due to cryptococcal infection that was recently estimated to exceed 625,000 per year (4).
Cryptococcosis is initiated by inhalation of airborne infectious particles, either basidiospores or desiccated yeast cells (5). In healthy individuals, C. neoformans is either cleared or causes a latent infection, but in the context of immune compromise, this facultative intracellular pathogen can multiply and disseminate throughout the body; its ability to cross the blood-brain barrier leads to a frequently fatal meningoencephalitis (6).
The virulence of C. neoformans is attributed to multiple factors that include its ability to grow at human body temperature and inhibit phagocytosis; the secretion of proteases, phospholipase, and urease; melanin synthesis; and the elaboration of a polysaccharide capsule (7–9). The capsule is a dynamic structure surrounding the cell wall that dramatically increases in thickness during infection, up to many times the cell radius. Capsule polysaccharide is also shed constitutively into the extracellular environment and impedes host responses to cryptococcal infection by inhibiting phagocytosis, altering antigen presentation, changing cytokine synthesis, and inhibiting leukocyte migration to the sites of inflammation (10).
The C. neoformans capsule is a complex structure composed of two high-molecular-mass polysaccharides, glucuronoxylomannan (GXM; ∼90% of the mass) and glucuronoxylomannogalactan (GXMGal; ∼7 to 8% of the mass), together with a small amount of mannoproteins (10–12). Purification of capsule polysaccharides shed from cryptococcal cells grown in vitro has enabled detailed structural studies of these components. GXM is a linear α-1,3-linked mannan of 1 to 7 MDa (13) whose mannose residues are variably O-acetylated and substituted with glucuronic acid and xylose (14). GXMGal is an α-1,6-galactan of roughly 100,000 Da. Alternate galactose residues of the linear backbone are branched with short side chains of galactose and mannose, which may be further substituted with xylose and glucuronic acid (15, 16); the branching backbone residues may be additionally substituted with galactofuranose (17) (other residues are all of the pyranose form). Both of the capsule polysaccharides demonstrate structural heterogeneity, depending on the strain and growth conditions (18, 19), and both have been implicated in cryptococcal pathogenesis (20, 21).
The synthesis of capsule polysaccharides and construction of the capsule involve multiple steps and significant metabolic investment (12, 22, 23). We previously reported that capsule polysaccharide synthesis occurs intracellularly, with the products subsequently being transported to the cell surface via elements of the protein secretory pathway (24). Once capsule polysaccharides exit the cell, they become associated with its surface (25). Little is known about the specific mechanisms of this interaction, although we have shown that it depends on cell wall α-1,3-glucan (26, 27). Consistent with this finding, deletion of the gene encoding α-1,3-glucan synthase (AGS1) yields acapsular cells that cannot bind GXM, although they do shed capsule polysaccharide like wild-type cells (27). Other investigators have suggested that chitin, another cell wall component, is also involved in capsule assembly (28, 29).
To identify additional factors involved in capsule assembly, we screened mutant cryptococci using a capsule transfer assay. This assay is based on the observation that exogenous capsule polysaccharide, such as the material shed from wild-type cells, is able to bind to the surfaces of acapsular mutant strains of C. neoformans (e.g., the cap59Δ mutant) (26, 30). We tested a library of cryptococcal deletion mutants (31) for their display of surface capsule and their ability to serve as capsule donors in this transfer assay. Strains lacking the genes PBX1 and PBX2 showed defects in both surface capsule and capsule transfer in our screen. It has previously been reported that these mutants have reduced virulence in mice (32), highlighting the importance of investigating the function of the encoded proteins. We found that these mutants additionally exhibit morphological defects, reduced capsule synthesis and association, and increased uptake by mammalian phagocytes. We hypothesize that the Pbx proteins act at the membrane in cell wall remodeling, such that their absence leads to this constellation of phenotypes.
MATERIALS AND METHODS
Reagents.
Difco medium components were from BD (Becton, Dickinson and Company, Sparks, MD), DNA polymerases (Taq and AccuPrime Pfx) and Escherichia coli DH5α competent cells were from Invitrogen (by Life Technologies Corporation), oligonucleotides were from Invitrogen or Sigma-Aldrich Co. LLC (St. Louis, MO), reagents for DNA isolation and biolistic transformation were from Bio-Rad Laboratories Inc., and restriction enzymes were from Invitrogen or New England BioLabs Inc. Kits for purification of DNA fragments and PCR products were from Qiagen, In-Fusion HD cloning kits were from Clontech Laboratories Inc., nourseothricin (ClonNat) was from Werner BioAgents (Germany), and G418 (Geneticin) was from Invitrogen. Specialty reagents for electron microscopy were from Polysciences Inc. or Ted Pella Inc., and colloidal gold-conjugated antibody was from Jackson ImmunoResearch Laboratories Inc., West Grove, PA. All other reagents or chemicals were from Sigma. Murine monoclonal anti-GXM antibodies 3C2 and 2H1 were generous gifts from Thomas Kozel (University of Nevada, Reno, NV) and Arturo Casadevall (Albert Einstein College of Medicine, Bronx, NY), respectively.
Culture conditions and strains.
C. neoformans strains were stored at −80°C in 25% glycerol and streaked onto yeast extract peptone dextrose (YPD) agar (1% [wt/vol] yeast extract, 2% [wt/vol] peptone, 2% [wt/vol] dextrose, 2% agar) plates for growth at 30°C. Liquid cultures were grown at 30°C with shaking at 230 rpm in YPD broth or in capsule induction medium (CIM; 0.17% [wt/vol] yeast nitrogen base without amino acids and ammonium sulfate, 0.15% [wt/vol] asparagine, 2% [wt/vol] glucose, 12 mM NaHCO3, 35 mM MOPS [morpholinepropanesulfonic acid], pH 7.1). As appropriate, media were supplemented with 100 μg/ml nourseothricin or G418. E. coli strains were grown at 37°C in Luria broth (LB; 1% [wt/vol] tryptone, 0.5% [wt/vol] yeast extract, 1% [wt/vol] NaCl) with shaking (230 rpm) or on LB agar plates (LB with 2% agar). As appropriate, the medium included 100 μg/ml ampicillin or 60 μg/ml kanamycin. The fungal strains used in this work are listed in Table 1. Wild-type (WT) KN99 MATα and MATa strains were provided by Joseph Heitman (Duke University Medical Center, Durham, NC) (33), an acapsular cap59Δ strain (in the KN99α background) was provided by Jennifer K. Lodge, Washington University Medical School (34), and the ags1Δ strain was from our previous work (27). Other strains used in this study (Table 1) were generated as described below.
TABLE 1.
Strains used in this study
| Strain descriptiona | Drug(s) used for selection | Reference or source | Strain |
|---|---|---|---|
| KN99 | 33 | WT KN99α | |
| KN99 MATa | 33 | WT KN99a | |
| KN99 cap59Δ | Hygromycin | 34 | cap59Δ mutant |
| JEC21 ags1Δ | Nourseothricin | 27 | ags1Δ mutant |
| KN99 pbx1Δ | G418 | This study | pbx1Δ mutant |
| KN99 pbx2Δ | Nourseothricin | This study | pbx2Δ mutant |
| KN99 pbx1Δ pbx2Δ | G418, nourseothricin | This study | pbx1Δ pbx2Δ mutant |
| KN99 pbx1Δ::PBX1 | Nourseothricin | This study | pbx1Δ::PBX1 mutant |
| KN99 pbx2Δ::PBX2 | G418 | This study | pbx2Δ::PBX2 mutant |
| KN99/pPBX1-HA | Nourseothricin | This study | Pbx1-HA |
| KN99/pPBX2-HA | Nourseothricin | This study | Pbx2-HA |
| KN99 cap59Δ pbx1Δ | Hygromycin, G418 | This study | cap59Δ pbx1Δ mutant |
| KN99 cap59Δ pbx2Δ | Hygromycin, nourseothricin | This study | cap59Δ pbx2Δ mutant |
All strains not otherwise designated were MATα.
Genetic manipulations.
The coding sequences of PBX1 (CNAG_01172.1) and PBX2 (CNAG_05562.1) were identified from the C. neoformans (serotype A strain H99) genome sequence available at http://www.broad.mit.edu/annotation/genome/cryptococcus_neoformans/Home.html. These were replaced by drug resistance markers according to the split marker strategy (35) shown in Fig. S1 in the supplemental material, using the primers listed in Table S1 in the supplemental material. Roughly 1-kb segments of the 5′ and 3′ untranslated regions (UTRs) of each gene were amplified from KN99α genomic DNA isolated as described previously (36). Drug resistance marker segments were amplified from plasmids pMH12-T (from Jennifer K. Lodge, Washington University Medical School) and pGMC200 (from Gary Cox, Duke University Medical Center) for G418 (37) and nourseothricin (38), respectively. PCR was used to fuse the upstream flank of each gene of interest with the 3′ part of the marker and the downstream flank with the 5′ part of the marker.
The fusion PCR products were biolistically transformed into KN99α as described in reference 39, with the expectation that homologous recombination events involving each flank and the overlapping region of the marker segments (∼500 bp) would yield replacement of the targeted gene by the marker (in reverse orientation; see Fig. S1 in the supplemental material). For biolistics, KN99α cells were grown in YPD broth to late log phase and harvested by centrifugation, and thick cell suspensions were plated as circular lawns at the center of YPD agar plates. The plates were incubated at 30°C for 3 h and then bombarded with 0.6-μm gold beads coated with DNA of the target construct (1.5 μg total) using a model PDS-1000/He biolistic particle delivery system (Bio-Rad Laboratories Inc.). After overnight growth, cells were transferred to selective YPD agar plates and individual colonies were screened by PCR to assess correct integration. Three independent strains were also confirmed to have each deletion by DNA blotting, using an AlkPhos direct labeling and detection system with a CDP-Star kit (GE Healthcare) according to the manufacturer's instructions. Ten-microgram aliquots of genomic DNA from WT KN99α and three independent pbx1Δ and pbx2Δ mutants were digested with HindIII and XhoI (which cuts within the PBX1-coding sequence but not that of the G418 resistance cassette) or BamHI and PvuII (which cuts within the PBX2-coding sequence but not that of the nourseothricin resistance cassette) and blotted using standard techniques (40) with probes corresponding to the 5′ or 3′ flanking regions of the PBX1 and PBX2 genes. These blots confirmed the gene replacement and that the marker cassette was present only once in the genome with no additional ectopic insertions (data not shown). The same approach was used to delete PBX1 and PBX2 in the cap59Δ mutant background for use in GXMGal isolation (see below). A pbx double mutant was also generated by deleting PBX2 (as above) in a pbx1Δ mutant strain.
To reconstitute confirmed pbx1Δ and pbx2Δ mutants, wild-type PBX sequences, including ∼1-kb flanking regions at both ends, were amplified. PCR was used to incorporate sequences for nourseothricin and G418 resistance after the 3′ ends of the PBX1 and PBX2 amplicons, respectively, followed by another ∼1 kb of appropriate 3′ flanking sequence (details are available on request). Biolistic transformation was then carried out using a gene replacement strategy (similar to that described above) to incorporate the PBX sequences at their original loci.
Epitope tagging of Pbx1 and Pbx2.
To episomally express tagged Pbx proteins, we constructed plasmids in which the PBX1 and PBX2 genes were modified by the addition of a sequence encoding a triple hemagglutinin (HA) epitope tag at the 3′ end. Briefly, an empty expression cassette was first assembled by PCR amplifying the ACT1 promoter and TRP1 terminator regions from C. neoformans KN99α genomic DNA using primers MSPC114 and MSPC115 and primers MSPC116 and MSPC117, respectively (primer sequences are provided in Table S1 in the supplemental material). The resulting amplicons were then fused by overlap PCR, using 5′ phosphorylated primers, to a synthetic spacer consisting of PmlI and MreI restriction sites, three copies of the HA sequence, and a stop codon. For drug selection, a fragment encoding the cryptococcal ACT1 promoter driving the NAT-coding sequence was released from plasmid pGMC200 (38) using PmeI and EcoRI and ligated to the PsiI and EcoRI restriction sites of plasmid pIBB103 (41). The resulting vector was digested with HpaI and ligated to the empty expression cassette containing the C. neoformans ACT1 promoter and TRP1 terminator. This expression plasmid was termed pMSC-043. The PBX1- and PBX2-coding regions were then PCR amplified from C. neoformans KN99α genomic DNA using primers PK201 and PK202 and primers PK203 and PK204, respectively, thereby incorporating PmlI and MreI restriction sites. The products were digested with PmlI and MreI and ligated to the corresponding sites of plasmid pMSC-043 to incorporate the three copies of the HA tag in frame with the 3′ end of each coding sequence before the stop codon. The resulting plasmids, pPBX1-HA and pPBX2-HA, respectively, were transformed into DH5α cells for sequence confirmation, and plasmids with correct sequences were used to transform C. neoformans KN99α by electroporation for episomal expression. To do this, 3 × 108 cells in early log phase were transformed with 1 to 5 μg of DNA as described previously (42), with parameters set at 0.5 kV, 25 μF, and resistance of 1,000 Ω. Transformed cells were grown in liquid YPD for 2 h at 30°C and then plated for drug selection.
Capsule transfer.
Capsule transfer assays were performed as described in reference 26, using the cap59Δ acapsular strain as the capsule acceptor and conditioned medium (CM) from 5-day-old cultures of KN99α or other indicated strains as the capsule donor. Acceptor cells were grown overnight, harvested by centrifugation, and washed twice with phosphate-buffered saline (PBS). Cells (5 × 106) were diluted to 499 μl with PBS, mixed with 1 μl CM (or medium control), and rotated for 1 h at room temperature (RT). After transfer, the acceptor cells were sedimented by centrifugation and washed twice with PBS. To visualize capsule acquisition by the acceptor cells, or endogenous capsule on other strains, the cells were further incubated for 1 h at RT with a final concentration of 8 μg/ml of anti-GXM antibody 3C2 that had been conjugated to a fluorophore (Cy3 monoreactive dye from GE Healthcare, used according to the manufacturer's recommendations). Stained cells were washed twice with PBS, resuspended in PBS, and examined on a Zeiss Axioskop 2 MOT Plus wide-field fluorescence microscope; all samples from each experiment were imaged with identical acquisition settings.
Capsule induction.
Overnight cultures of C. neoformans in YPD were harvested by centrifugation, washed twice with Dulbecco's modified Eagle's medium (DMEM), and resuspended at 2 × 106 cells/ml in DMEM. One milliliter of the suspension was incubated at 37°C with 5% CO2 for 36 h in a 24-well tissue culture plate. Cells were harvested by centrifugation, washed once with deionized water (diH2O), and resuspended in 1 ml diH2O. Eight microliters of the cell suspension was mixed with 2 μl of India ink stain, and 6 μl of the mixture was spotted on a microscope slide for bright-field image acquisition on a Zeiss Axioskop 2 MOT Plus wide-field fluorescence microscope. In some experiments, cells were sonicated for 10 s using a Fisher sonic dismembrator at 35% output before India ink staining.
Phenotypic assays.
To measure growth, cells were grown in YPD or CIM at 30°C or 37°C with shaking as described above. Samples were collected at regular intervals, and the cell number was assessed using a Cellometer Auto M10 automated cell counter (Nexcelom Bioscience). For plate assays, C. neoformans strains were grown to mid-log phase in YPD broth and diluted to 107 cells/ml, and five 10-fold serial dilutions were made. Five microliters of each dilution was spotted onto solid medium plates, which were incubated at 30°C, 37°C, or 39°C. The solid media tested included YPD containing NaCl (0.1, 0.5, or 1.2 M), SDS (0.01, 0.03, or 0.06%), caffeine (1, 5, or 15 mM), Congo red dye (0.1 or 0.5% from a 4% stock solution prepared in 50% ethanol), or calcofluor white (0.5, 1.0, or 1.5 mg/ml). Growth on various carbon sources was checked on minimal medium (0.17% [wt/vol] yeast nitrogen base without amino acids and ammonium sulfate, 0.5% [wt/vol] ammonium sulfate, 2% [wt/vol] agar) containing 2% (wt/vol) of the desired carbon source (glucose, galactose, or xylose).
Melanin synthesis was assessed by growing cells on glucose-free asparagine agar plates (0.1% [wt/vol] l-asparagine, 0.05% [wt/vol] MgSO4, 0.3% [wt/vol] KH2PO4, 0.0001% [wt/vol] thiamine, 1 mM l-3,4-dihydroxyphenylalanine [l-DOPA], 2% [wt/vol] Bacto agar) in the dark at 30°C, 37°C, or 39°C and examining them on days 2 through 5 for melanization, seen as a brown colony color. To compare melanin retention in KN99 and mutant cell walls, cells grown overnight in the same medium without agar were collected, washed once in the same medium, and counted. Cells (108) were inoculated into 2 ml of fresh medium in 10-ml culture tubes in triplicate and shaken at 30°C for 24 h. Following incubation, the culture tubes were centrifuged at 3,000 × g for 10 min and photographed to visualize the cell wall-associated melanin in the cell pellet; the optical density at 400 nm of the supernatant fraction was also recorded to assess melanin release from the cells into the growth medium (34).
Electron microscopy.
Overnight cultures were diluted to 2 × 106 cells/ml in YPD, grown for 9 h at 30°C, collected by centrifugation, quickly resuspended in 1 ml of primary fixation mix (0.1 M sorbitol, 1 mM MgCl2, 1 mM CaCl2, 2% glutaraldehyde in 0.1 M PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] buffer, pH 6.8 [43]), and fixed overnight at 4°C. For ultrastructural analysis, cells were fixed in 2% glutaraldehyde (Polysciences Inc., Warrington, PA) in 100 mM phosphate buffer, pH 7.2, for 1 h at room temperature, washed in the same buffer, and postfixed in 1% osmium tetroxide (Polysciences Inc.) for 1 h. Samples were then rinsed in the same buffer, followed by dehydration in a graded series of ethanol and propylene oxide prior to embedding in Eponate 12 resin (Ted Pella Inc., Redding, CA). Sections of 90 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc., Bannockburn, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200EX transmission electron microscope (JEOL USA Inc., Peabody, MA).
For immunoelectron microscopy, cells were fixed in glutaraldehyde as described above, washed three times in 50 mM citrate buffer, pH 6.0, and resuspended in 1 ml of 40 mg/ml lysing enzymes (L1412; Sigma) in the same buffer to partially digest the cell wall. After 30 min of incubation at 30°C, cells were washed in 0.1 M phosphate buffer and postfixed, and blocks were prepared as described above. Sections were blocked for 30 min with a solution of 5% fetal bovine serum in 100 mM PIPES buffer (pH 7.0), labeled for 1 h with an anti-GXM monoclonal antibody (2H1), washed in blocking buffer, and incubated with 12-nm gold-conjugated goat anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Sections were then washed once in 100 mM PIPES buffer, pH 7.0, and twice in diH2O, stained with uranyl acetate and lead citrate, and viewed as described above.
Purification of GXM.
For GXM purification, cells were inoculated into 200 ml of CIM and grown for 5 days at 30°C with shaking. The cultures were autoclaved and centrifuged at 18,000 × g for 1 h at RT, and the supernatant fraction was filtered through a 0.22-μm-pore-size membrane filter and used for GXM purification. Purification, based on precipitation of the negatively charged GXM with the cationic detergent cetyltrimethylammonium bromide (CTAB), was performed as described previously (22, 44).
Purification of GXMGal.
GXMGal was purified following the procedure described for the serotype D cap67Δ strain (16, 22). YPD medium (200 ml) was inoculated with the cap59Δ, cap59Δ pbx1Δ, or cap59Δ pbx2Δ mutant strain and incubated at 30°C with shaking for 5 days. Cells were sedimented (8,000 × g, 10 min, RT), and the supernatant fraction was concentrated to ∼10 ml with a Pall Jumbosep spin concentrator containing a 10-kDa-molecular-weight-cutoff membrane. The concentrated supernatant was used to isolate GXMGal as described by Vaishnav et al. (16) and Klutts and Doering (22) using a combination of affinity, anion exchange, and size exclusion chromatography techniques.
NMR spectroscopy.
Nuclear magnetic resonance (NMR) studies of GXM and GXMGal samples were performed as previously described (15, 22). GXM samples were dissolved in water at 5 mg/ml, sonicated to reduce the molecular mass, and deacetylated by adjusting the pH to 11 with concentrated NH4OH before incubation for 24 h at 25°C. Samples were then dialyzed, lyophilized, deuterium exchanged by lyophilization from D2O, and dissolved in 0.7 ml D2O for analysis. Proton, gradient-enhanced correlated spectroscopy (gCOSY), total correlation spectroscopy (TOCSY), heteronuclear single quantum correlation (HSQC), gradient-enhanced heteronuclear multiple-bond correlation (gHMBC), and nuclear Overhauser effect spectroscopy (NOESY) NMR spectra were acquired on a Varian Inova 500-MHz spectrometer at 70°C using standard Varian pulse sequences. TOCSY and NOESY mixing times were 120 and 300 ms, respectively. GXMGal samples were deuterium exchanged by dissolving the samples in D2O and lyophilization, before dissolving the samples in 0.27 ml D2O. One-dimensional proton spectra and two-dimensional gCOSY, TOCSY, NOESY, gHSQC, and gHMBC spectra were obtained on a Varian Inova 500-MHz spectrometer at 70°C using standard Varian pulse sequences. Proton chemical shifts were measured relative to the location of internal acetone (δH = 2.218 ppm, δC = 33.0 ppm). The relative molar amounts of GXM structural motifs were determined from the anomeric protons of the mannose residues after Lorentz-Gauss resolution enhancement of the proton spectra and deconvolution of the anomeric signals by use of the line-fitting routine in Mestre-C NMR software. The areas of the three peaks belonging to the same triad were averaged.
GXMGal composition.
The glycosyl composition of GXMGal was analyzed by combined gas chromatography (GC)-mass spectrometry (MS) of the per-O-trimethylsilyl (TMS) derivatives of the monosaccharide methyl glycosides produced from the sample by acidic methanolysis as previously described (45, 46). GXMGal samples were dissolved in water, 20 μg of inositol was added as an internal standard, and the solutions were transferred to screw-top glass tubes and freeze-dried. Methyl glycosides were prepared by heating the samples in 1 M methanolic HCl at (80°C, 18 h), followed by re-N-acetylation with pyridine and acetic anhydride in methanol (for detection of amino sugars). The samples were then per-O-trimethylsilylated by treatment with Tri-Sil reagent (Pierce) at 80°C (0.5 h). GC-MS analysis of the TMS methyl glycosides was performed both on an AT 7890N GC interfaced to a 5975C MSD system (electron impact ionization mode) and on a Shimadzu GCMS QP 2010 plus apparatus (chemical ionization mode); separation was performed on a Grace EC-1 bonded-phase fused-silica capillary column.
GXM molecular mass determination.
The molecular masses of GXM were determined by multiangle light scattering (MALS) as described previously (13, 47). The dn/dc constant for GXM (change in the refractive index of the GXM solution with the change in the GXM concentration) was determined using an Optilab rEX refractive index detector (Wyatt Technology Corporation) in batch mode at 25°C. The instrument was preequilibrated in MALS buffer (150 mM sodium chloride, 20 mM HEPES [pH 7.4], 0.01% sodium azide), and solutions prepared from anhydrous NaCl (0 [blank], 0.1, 0.5, 1.0, 1.2, 2.0, 3.0, 4.0, and 5.0 mg/ml) were loaded as standards for dn/dc constant determination (0.172 ml/g for NaCl at 690 nm). GXM samples were then prepared in the same buffer, mixed overnight, and filtered through 0.8-μm-pore-size syringe filters. The carbohydrate concentration was determined by phenol sulfuric acid assay (48), and 50, 100, 200, 300, 400, or 500 μg/ml of GXM was loaded onto the differential refractometer. The Astra V macromolecular characterization software package (Wyatt Technology Corporation) was used to calculate the dn/dc constant.
GXM samples in MALS buffer were loaded onto a size exclusion chromatography column (pore size, 300 Å; WTC-030S5; Wyatt Technology Corporation) set up in series with a Dawn Helios II multiangle light-scattering detector (Wyatt), an Optilab rEX differential refractive index detector (Wyatt), and a 996 photodiode array detector (Waters). The data were recorded for light scattering, refractive index change, and UV absorbance over the elution profile of GXM, and results were analyzed with the Astra V macromolecular characterization software package (Wyatt) to calculate the molecular mass from the light scattering and refractive index change.
GXM immunoblotting.
Immunoblotting of GXM was performed essentially as we have previously reported (49). Briefly, 50 μl of CM from the indicated strains grown in YPD or CIM or 10 μg of GXM purified from CM of cells grown in CIM was mixed with 6× DNA loading dye, and the mixture was loaded on a 0.6% certified megabase agarose gel (Bio-Rad Laboratories Inc.) and subjected to electrophoresis for 15 h at 25 V in 0.5× TBE (44.5 mM Tris base, 44.5 mM boric acid, 1 mM EDTA, pH 8.3). The gel was transferred onto a positively charged nylon membrane and immunoblotted with 1 μg/ml anti-GXM antibody 3C2 as described in reference 49.
Protein purification and localization.
Protein purification was performed at 4°C using a previously described method (50) with some modifications. Cells were grown overnight in 50 ml of YPD to late log phase, harvested by centrifugation (3,000 × g, 10 min, RT), washed once with cold Tris-EDTA buffer (100 mM Tris-HCl, pH 7.5, 0.1 mM EDTA), and resuspended in an equal volume of the same buffer containing protease inhibitors (0.1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail [10 μl/ml; P8215; Sigma]). All subsequent steps were performed at 4°C. Aliquots (800 μl) were transferred to 2-ml screw-cap microcentrifuge tubes and mixed with an equal volume of 0.5-mm glass beads (BioSpec Products), and the samples were subjected to 1-min bursts on a Mini-BeadBeater (BioSpec Products) alternating with 2 min on ice. Eight to 10 cycles of bead beating were required to disrupt ∼75% of the cells (as assessed by microscopy). The lysate was then transferred to a 15-ml conical tube, the glass beads were rinsed twice with 800 μl of Tris-EDTA buffer, and both rinses were pooled with the lysate. The pooled material (total protein) was subjected to a low-speed clearing centrifugation step (1,000 × g, 25 min). The resulting cell-free supernatant fraction (S1; cytosol and membranes) was removed, and the pellet (P1) was stored at 4°C for later extraction. S1 was next fractionated by ultracentrifugation (60,000 × g, 45 min); the supernatant fraction from this step (S2; cytosol) was reserved, and the pellet (P2; total membranes) was resuspended in 200 μl of Tris buffer (100 mM Tris-HCl, pH 7.5) for detergent extraction of membrane proteins (see below). Protein concentrations were determined using a protein assay kit (catalog no. 500 0006; Bio-Rad Laboratories Inc.).
For Triton X-100 (TX-100) extraction of membrane proteins, P2 was extracted with 1% TX-100 on ice for 1 h with brief vortex mixing every 5 min. Particulate material was removed by ultracentrifugation (60,000 × g, 45 min), and both the pellet (TX-100-insoluble) and supernatant (TX-100-soluble) fractions were stored at 4°C. A parallel P2 sample was extracted with 1% SDS by boiling for 10 min, after which the sample was centrifuged (15,000 × g, 10 min, RT) and the supernatant and pellet were saved as SDS-soluble and SDS-insoluble fractions, respectively. Detergent was removed by using an SDS-PAGE cleanup kit (GE Healthcare) before protein gel electrophoresis. The cell wall-associated proteins were extracted as described in reference 51. Briefly, fraction P1 (see above) was resuspended in 1 ml of Tris buffer and disrupted by probe sonication (5 cycles of 10 s on and 10 s off; output, 35%). The sample was then centrifuged at 3,500 × g for 10 min, and the pellet was washed three times with Tris buffer, resuspended in 1 ml of lysing enzyme (20 mg/ml in diH2O with protease inhibitors, as described above; L1412; Sigma), and rotated at 37°C for 1 h. After digestion, the sample was centrifuged at 15,000 × g for 15 min and the supernatant was preserved as cell wall-extracted proteins.
Macrophage phagocytosis assay.
To measure uptake by macrophages, we used our previously published assay (52) with minor modifications. As host cells we used the human monocytic cell line THP-1, which was seeded onto 12-mm coverslips at a density of 2.5 × 105 cells/ml in medium containing 0.2 μg/ml of phorbol myristate acetate (PMA) to induce differentiation. After 48 h, the medium was replaced with fresh medium containing PMA to remove any nonadherent and dead cells, and 1 day later, uptake of serum-opsonized C. neoformans (multiplicity of infection, 5) was measured. Coverslips were fixed and stained as described previously (52), except that DAPI (4′,6-diamidino-2-phenylindole) staining was omitted and CellMask plasma membrane dye was used in place of CellMask HCS stain, and samples were imaged on a Zeiss LSM 510 confocal laser scanning system. At least 100 THP-1 cells were examined for each sample, and significance was evaluated by the Holm-Sidak method with alpha equal to 1%.
RESULTS
Screening for capsule mutants.
Our previous work showed that cell wall α-glucan is required for capsule association with C. neoformans cells (26, 27). We hypothesize that additional cellular elements are also required for the normal display of capsule on the cell surface. To identify such factors and further define the mechanism of capsule association in C. neoformans, we used a forward genetic approach, screening for mutants defective in either surface capsule display or the ability to donate capsule material to acapsular acceptor cells.
We tested surface capsule by labeling cryptococcal cells with an anti-GXM monoclonal antibody (3C2, kindly provided by Tom Kozel) and observing them by immunofluorescence microscopy (see Materials and Methods). As shown in Fig. 1 (left two columns), this antibody binds evenly to the cell surface of wild-type cells but does not label strains known to be acapsular (such as cap59Δ or ags1Δ mutants). We tested the ability of cryptococci to donate capsule (26) by collecting conditioned medium (CM) from cultures of the strains of interest and incubating it with cells of the acapsular cap59Δ mutant. We then washed the acceptor cells and used the 3C2 antibody to assess their acquisition of capsule material from the donor CM (see Materials and Methods). CM from wild-type cells was an effective donor of capsule material to the cap59Δ mutant in this assay, while CM from the cap59Δ mutant itself could not act as a donor (Fig. 1, right two columns). CM from cells of the ags1Δ mutant was transfer competent, although the mutant itself bore no capsule, showing that the defect in the ags1Δ mutant is one of capsule association with the cell surface, rather than a problem with capsule synthesis or release (Fig. 1) (27).
FIG 1.
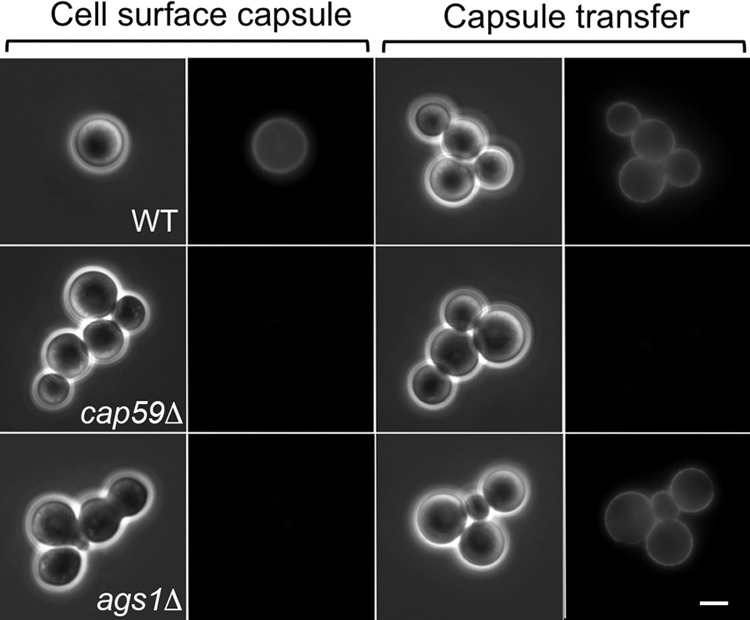
Detection of cell surface capsule and capsule transfer. (Left two columns) Endogenous surface capsule of the indicated strains was probed by staining with the anti-GXM monoclonal antibody 3C2. (Right two columns) The cap59Δ acapsular strain was used as the acceptor for exogenous capsule from CM of the indicated strains, washed, and stained similarly. The left panel of each pair is a differential interference contrast (DIC) image, and the right panel is an immunofluorescence (IF) image. All images are at the same magnification. Bar = 5 μm.
We screened the 1,201 strains in a C. neoformans deletion collection generated by the Madhani laboratory (31) and identified 13 with consistent differences in surface capsule or capsule transfer compared with wild-type C. neoformans (Table 2 and data not shown). Four known acapsular mutants, the CAP10, CAP59, CAP60, and CAP64 mutants, were negative for surface capsule, as shown previously (53–56), and for the ability to donate capsule material. A similar profile was seen in cells lacking an enzyme (Ugd1) required for the production of UDP-glucuronic acid and UDP-xylose; these molecules are important sugar donors for capsule polysaccharide synthesis (57–59). Several other mutants showed partial defects in the ability to donate capsule or in their surface capsule, but we were particularly interested in three strains that bore a surface capsule, as detected by antibody staining, but were not able to donate capsule in our transfer assay (Table 2, bold font). This phenotype was opposite that of the ags1Δ mutant cells, which do not display surface capsule but do shed capsule that can bind normally to acapsular cells (26). It suggested that these mutants (Table 2, bold font) made polysaccharide that could associate with the cells of origin but had some defect that precluded appreciable binding to acapsular cells. Findings for one of these mutants, the cpl1Δ mutant, will be presented elsewhere (Meng Yang and Tamara L. Doering, unpublished data). Below we present our findings on the other two mutants, the pbx1Δ and pbx2Δ mutants.
TABLE 2.
Capsule phenotypes of C. neoformans mutant strains
| Locus deleted (CNAGa no.) | Protein encoded | Cell surface capsule | Capsule donor |
|---|---|---|---|
| None | + | + | |
| 02628 | Cap10 | − | − |
| 00721 | Cap59 | − | − |
| 00600 | Cap60 | − | − |
| 02885 | Cap64 | − | − |
| 04969 | Ugd1 | − | − |
| 06813 | Cap1 | −/+ | −/+ |
| 00440 | Ssn801 | + | −/+ |
| 06889 | Na/H antiporter | + | −/+ |
| 06902 | Hypothetical | + | −/+ |
| 05258 | Hypothetical | + | −/+ |
| 02797 | Cpl1 | + | − |
| 01172 | Pbx1 | + | − |
| 05562 | Pbx2 | + | − |
| 03120 | Ags1 | − | + |
CNAG, Cryptococcus neoformans assigned gene number (see http://www.broadinstitute.org/annotation/genome/cryptococcus_neoformans).
In previous studies of the pbx1Δ and pbx2Δ mutants, the missing proteins were implicated in capsule synthesis (32), although capsule transfer was not assessed at that time (see Discussion). To further address their function, we first replaced PBX1 and PBX2 in the serotype A strain KN99α with sequences that confer resistance to G418 and nourseothricin, respectively (37, 38). KN99α is closely related to the strain that was used to generate the mutant library (H99) but is more genetically tractable (33). We confirmed the new mutant strains (Table 1) by PCR and DNA blotting (data not shown) and tested them for surface capsule: as expected from our screen results, anti-GXM antibody staining of both mutants was similar to that of wild-type cells (Fig. 2A). Also consistent with our earlier results, CM harvested from cultures of the pbx1Δ or pbx2Δ mutant failed to donate capsule to the cap59Δ mutant; the same result was obtained with a double mutant (the pbx1Δ pbx2Δ mutant) that we generated by deleting the PBX2 gene in a pbx1Δ mutant background (Fig. 2B, left two columns). Complementation of the mutants by reintegration of the wild-type sequences (pbx1Δ::PBX1 and pbx2Δ::PBX2) restored normal capsule transfer (Fig. 2B, right two columns).
FIG 2.
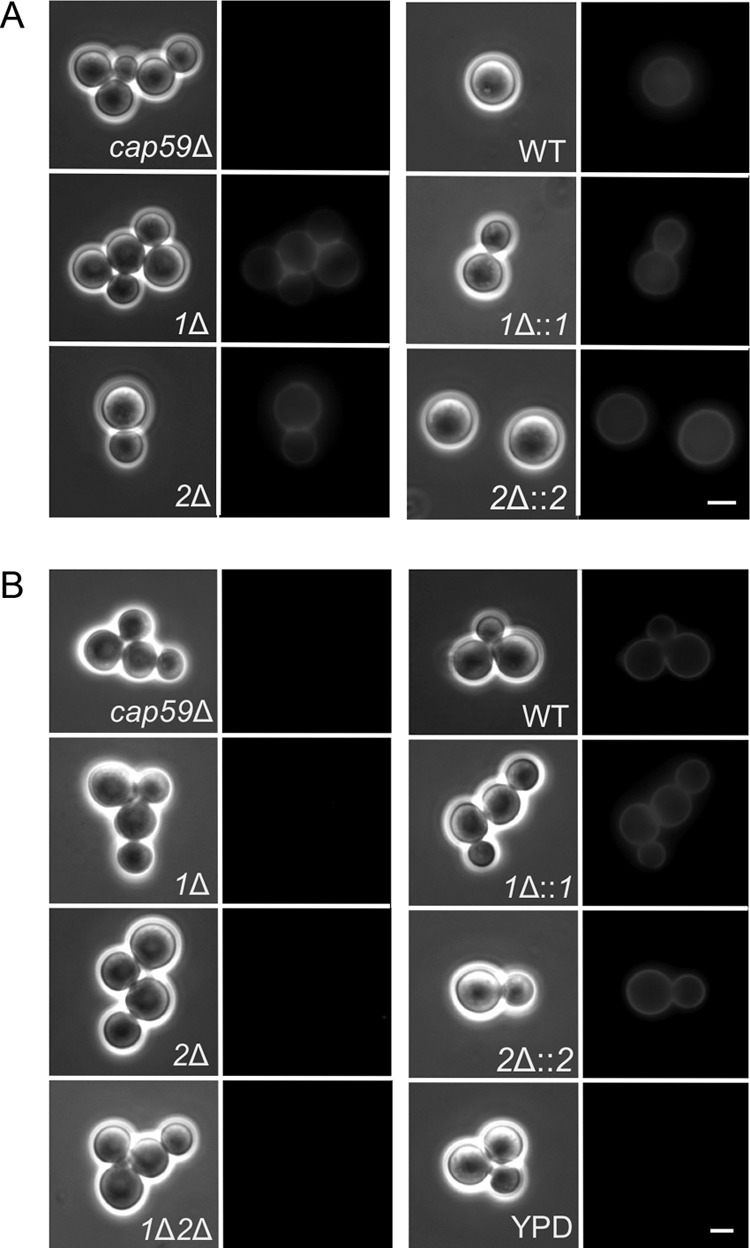
pbxΔ strains display cell surface capsule but cannot donate capsule to acapsular acceptor cells in standard assays. (A) Surface capsule. Cells of the indicated strains were labeled with antibody 3C2 and imaged as described in the legend to Fig. 1. (B) Capsule transfer. CM from the indicated strains (or YPD alone as a control) was used as the capsule donor in transfer assays, with the cap59Δ mutant as the acceptor strain. Imaging is as described in the legend to Fig. 1. 1Δ, pbx1Δ mutant; 2Δ, pbx2Δ mutant; 1Δ::1, pbx1Δ mutant complemented with PBX1; 2Δ::2, pbx2Δ mutant complemented with PBX2; 1Δ2Δ, pbx1Δ pbx2Δ mutant. All images in both panels are at the same magnification. Bar = 5 μm.
GXM release.
We wondered whether the inability of cells lacking Pbx1 and Pbx2 to act as capsule donors was due to the nature or amount of shed GXM. To test this, we performed standard capsule transfer assays in the presence of increasing concentrations of CM. At the standard CM dilution of 1:500, we observed no transfer of polysaccharide from mutant CM to acapsular acceptor cells, although wild-type CM showed a robust signal at this dilution (Fig. 3); similar results were obtained with a 1:20 dilution of CM. Notably, use of undiluted CM did result in transfer to cap59Δ mutant cells, although the capsule staining of the acceptor cells was not as complete as that with wild-type CM, demonstrated by an uneven and patchy signal (Fig. 3). These studies showed that mutant capsule could bind to acapsular cells, albeit poorly, but did not establish whether the underlying defect was a reduced amount of shed GXM in the CM or a reduced affinity of the polysaccharide material for the acapsular acceptor cells due to some fundamental change.
FIG 3.
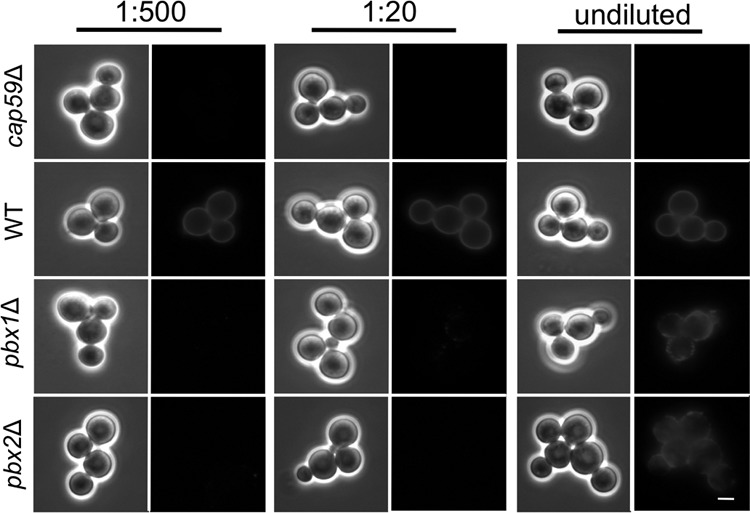
pbxΔ mutants are poor capsule donors in transfer experiments. Capsule transfer assays were performed as described in the legend to Fig. 2, using the indicated dilutions of CM from the strains noted at the left (the 1:500 dilution is that used in the standard assay; see Materials and Methods). Bar = 5 μm.
To directly examine the GXM that was shed from pbx mutant cells, we compared the polysaccharide-containing CM from each single mutant to that from the wild type by resolving the material on agarose gels and immunoblotting with anti-GXM 3C2 antibody (49). CM from the pbx mutant strains contained minimal antibody-reactive material compared to that from equal volumes of wild-type CM (Fig. 4, left); this material could be detected only on long exposures of the immunoblots (see Fig. S2 in the supplemental material). Similar analysis of CM from cultures grown in capsule-inducing medium (see Materials and Methods) showed slightly more GXM in the mutant CM, although this amount was still significantly less than that seen in wild-type CM (Fig. 4, center). (The growth rates of all strains were comparable in both media; see Fig. S3 in the supplemental material.) Because this analysis was based on antibody detection, we considered the possibility that the reduced signal might reflect altered interactions of mutant GXM with the 3C2 antibody. To test this, we purified GXM from each strain and compared equivalent mass amounts in a parallel blotting experiment. In this study, the GXM from all strains appeared to be identical in both signal intensity and gel migration behavior (Fig. 4, right). Furthermore, the purified GXM from each mutant showed the same efficacy as a capsule donor as wild-type purified GXM when comparable masses were included in transfer assays (not shown). Together, these experiments suggest that the poor function of pbx mutants as capsule donors does not reflect a difference in their GXM size (as assessed by gel electrophoresis) or structure (as assessed by antibody reactivity) but, rather, reflects a reduction in the amount of polysaccharide that is shed from the mutant cells during growth in culture.
FIG 4.
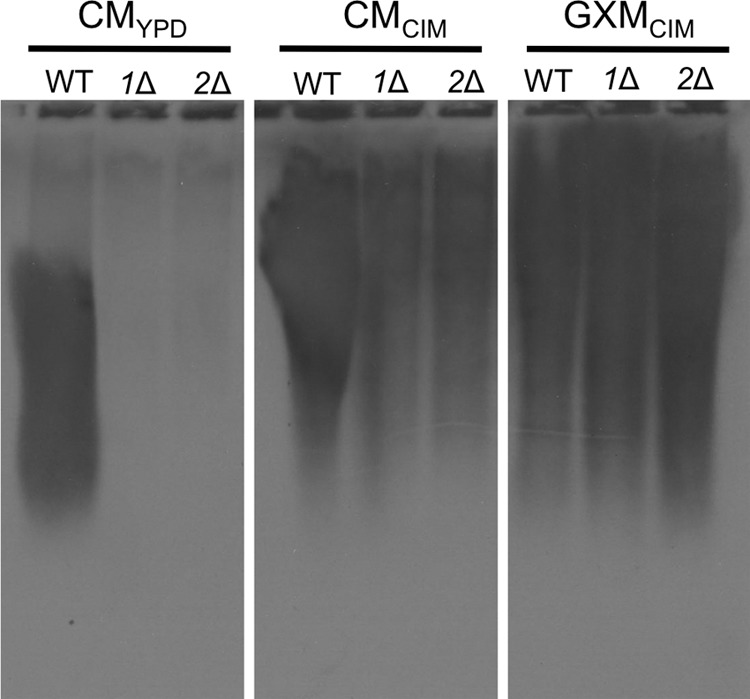
pbxΔ mutants are deficient in capsule shedding. Shown are the results of immunoblot analysis of CM or purified GXM from the indicated strains grown in rich medium (YPD) or CIM. Samples (50 μl of CM or 10 μg of GXM) were resolved by agarose gel electrophoresis and immunoblotted with antibody 3C2. The exposure time was 2 s (images after a longer exposure are shown in Fig. S2 in the supplemental material). The reduced migration of GXM from cells grown in CIM reflects the increased polymer size that results from capsule induction (49).
Intracellular GXM.
The observed reduced shedding of GXM could indicate either a defect in the production of this polysaccharide or a failure to release it into the medium. To distinguish between these possibilities, we probed thin sections of cryptococcal cells with a colloidal gold-tagged monoclonal antibody to GXM (2H1). The resulting immunoelectron micrographs (Fig. 5) showed abundant labeling of wild-type cell surfaces with occasional intracellular signal, especially on vesicular structures. In contrast, the pbx1Δ, pbx2Δ, and pbx1Δ pbx2Δ mutant strains showed less surface staining and no intracellular signal, consistent with a general reduction in GXM synthesis. Although reduced surface staining together with reduced shedding could also occur if the cells had a defect in secretion of capsule polysaccharide, we would expect such cells to demonstrate a corresponding increase in intracellular signal (24); the absence of any internal signal in the mutants, let alone such an increase (Fig. 5 and data not shown), further supports a defect in capsule polysaccharide synthesis.
FIG 5.
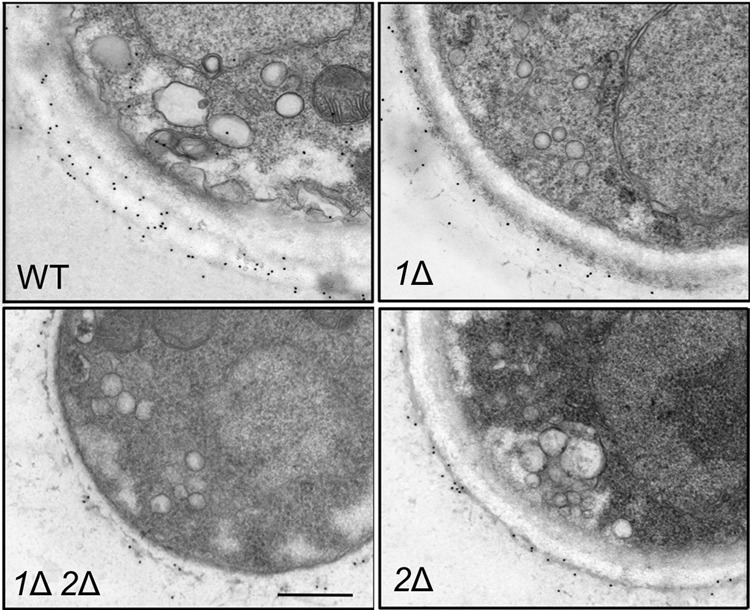
pbxΔ mutants show reduced intracellular and surface capsule polysaccharide. Cryptococcal cells grown in YPD medium were examined by immunoelectron microscopy using colloidal gold-labeled anti-GXM monoclonal antibody 2H1. Each panel shows a portion of a cell of the indicated strain. All images are at the same scale. Bar = 500 nm.
Surface capsule.
Our analysis of shed GXM (Fig. 4) suggested that the release of polysaccharides from pbx mutants increased in capsule induction medium. To examine the induction of cell-associated capsule polysaccharides, we grew cells under inducing conditions (in DMEM with 5% CO2 at 37°C; see Materials and Methods) and assessed capsule size by negative staining with India ink. Upon induction, all of the pbx mutants produced capsules with a radius similar to that of the wild type (see the top row of Fig. S4 in the supplemental material), although the mutant capsules were more heterogeneous in thickness (data not shown).
We further examined the capsule structure of cells grown under inducing conditions using quick-freeze deep-etch imaging. As we showed previously (25), the capsule fibers of wild-type cells grown in this way form a dense meshwork extending from the cell wall. In contrast, the pbx mutant capsules formed a much looser network (Fig. 6). The association between the capsule and wall was also weaker, as brief sonication was sufficient to release almost all of the capsule material from pbx1Δ, pbx2Δ, and pbx1Δ pbx2Δ mutant cells, even though wild-type capsules were not perturbed by this treatment (see the bottom row in Fig. S4 in the supplemental material). This phenotype, which is consistent with results previously reported by Liu et al. for pbx1Δ and pbx2Δ mutants (32), was reversed upon complementation with the corresponding wild-type genes (see Fig. S4 in the supplemental material).
FIG 6.

The capsules of pbxΔ mutants have reduced density. Quick-freeze, deep-etch electron micrographs for the indicated strains are shown. Each image shows a portion of the cell edge, with the plasma membrane at the top and capsule fibers at the bottom (extending downwards) separated by the cell wall. All strains were grown under the same conditions, and all images are at the same magnification. Bar = 100 nm.
Capsule polysaccharide analysis.
The loose association of the capsule with the cell surface of pbxΔ mutants could be due to differences in either the capsule itself or the underlying cell wall. Previous studies of cells lacking PBX1 and PBX2 reported that GXM in these mutants contained glucose (which the wild type does not), but the chemical structure of the mutant polysaccharide was not determined (32). To pursue this observation, we purified GXM from pbx1Δ mutant, pbx2Δ mutant, and wild-type cells for NMR analysis (see Tables S2 and S3 in the supplemental material). All three samples were composed of structures corresponding to mannosyl triads M2 and M3 (Fig. 7), although the fraction of M3 was higher in the mutant strains than in the wild type (Table 3). This difference would correspond to an additional 25% of GXM reporter groups containing xylose β-1,4 linked to the backbone mannose residue that also bears glucuronic acid (ManA in Fig. 7), translating to an overall increase in GXM xylose of ∼10%. We did not detect any glucose in the GXM structure (see Discussion).
FIG 7.
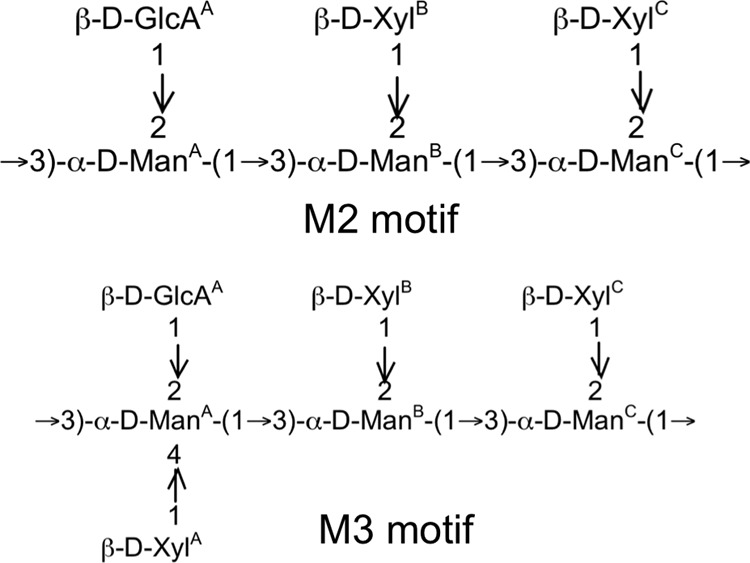
GXM structural motifs. The two structural reporter groups that were detected in GXM of wild-type strain KN99 and mutant strains are shown; GXM is composed of multiple repeats of these motifs. Superscripts denote specific residues for reference to the structural information provided in Tables S2 and S3 in the supplemental material.
TABLE 3.
Molar ratios of the mannosyl triads detected in GXM from WT and mutant strains
| Triad | Molar ratio |
||
|---|---|---|---|
| WT KN99 | pbx1Δ mutant | pbx2Δ mutant | |
| M2 | 85 | 63 | 60 |
| M3 | 15 | 37 | 40 |
To further characterize GXM purified from mutant and wild-type strains, we determined its molecular mass using multiangle light scattering (MALS). The masses of all three strains were very similar to each other (Table 4) and to the masses typical for serotype A strains (13). The slightly higher masses of the mutant GXM may reflect the presence of the modestly increased amount of xylose detected by NMR.
TABLE 4.
Molecular masses for purified GXM derived from MALS analysis
| GXM source | dn/dc constant | Molecular mass (Da) |
|---|---|---|
| WT KN99 | 0.173 | 1.578 × 106 |
| pbx1Δ mutant | 0.157 | 1.635 × 106 |
| pbx2Δ mutant | 0.167 | 1.636 × 106 |
We also purified the second capsule polysaccharide, GXMGal, from wild-type and mutant strains of C. neoformans. For this study, we deleted PBX1 and PBX2 in a cap59Δ mutant background to avoid contamination of the GXMGal preparations with the more abundant GXM produced by wild-type cells. The compositions of mutant and wild-type GXMGal were very similar (Table 5), suggesting no alterations that could be attributed to the lack of Pbx proteins. We also used these samples for NMR determination of the GXMGal structure, as this has not been reported for acapsular serotype A strains. While the basic structure is similar to that reported for serotype D strains, the serotype A polymer contains more galactofuranose (data not shown) and exhibits more 2-acetylation of mannose residues (see Table S4 in the supplemental material). Overall, these studies suggested that the structures of both capsule polysaccharides in the pbx mutant strains are close to those of the wild type, although the amount of shed GXM is significantly reduced. These results are consistent with our observation that GXM purified from mutant strains binds normally to acapsular acceptor cells (not shown).
TABLE 5.
GXMGal glycosyl composition
| Monosaccharide | Composition |
|||||
|---|---|---|---|---|---|---|
|
cap59Δ mutant |
cap59Δ pbx1Δ mutant |
cap59Δ pbx2Δ mutant |
||||
| Mass (μg) | mol% | Mass (μg) | mol% | Mass (μg) | mol% | |
| Xylose | 72.6 | 15.5 | 278.5 | 12.9 | 116.4 | 14.1 |
| Glucuronic acid | 10.3 | 1.7 | 101.5 | 3.6 | 15.7 | 1.5 |
| Mannose | 122.0 | 21.7 | 576.8 | 22.3 | 235.1 | 23.7 |
| Galactose | 342.4 | 61.0 | 1,580.3 | 61.1 | 603.9 | 60.8 |
| Total | 547 | 100 | 2,537.1 | 100 | 971.2 | 100 |
Cell wall analysis.
Because the relatively modest differences in the capsule polysaccharide structure of the pbx mutants may not be enough to explain the defects in capsule association with the cell wall (revealed by sonication), we examined the latter structure more closely. Cell wall-perturbing agents such as SDS, calcofluor white, and Congo red did not alter the growth of the mutant compared to that of the wild type (data not shown), suggesting normal overall cell integrity. We also compared the morphology of mutant cells to that of wild-type cells. Although the mutants exhibited no alterations in the thickness or general appearance of the cell wall when assessed at high magnification by thin-section electron microscopy (not shown), lower-magnification views showed that the cells were less regular in shape than wild-type or complemented mutant cells. While most wild-type or complemented mutant cells were round and smooth surfaced, the mutants showed asymmetric and uneven boundaries (Fig. 8). In other phenotypic changes that are potentially related to the cell wall, the pbx1Δ, pbx2Δ, and pbx1Δ pbx2Δ mutants showed modest defects in the production of cell wall melanin, and the pbx1Δ pbx2Δ double mutant showed reduced production of mating filaments (see Fig. S5 and S6 in the supplemental material). These cells also displayed heightened sensitivity to oxidative stress (Fig. 9), which has been related to cell walls in yeasts (60, 61).
FIG 8.
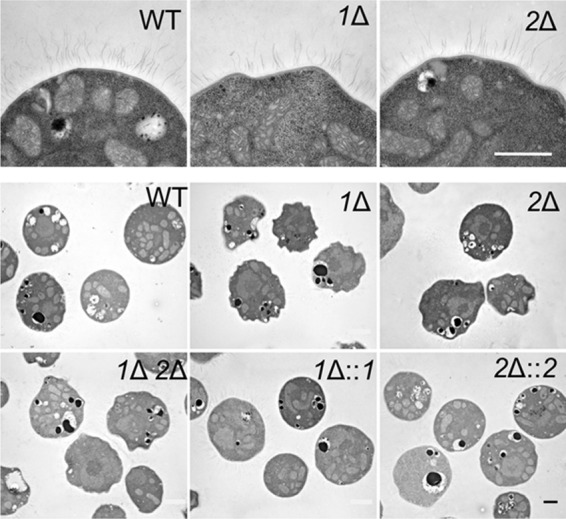
pbxΔ mutants show irregular cell morphology. Thin-section electron micrographs in the top row are all at the same magnification, and images in the bottom two rows are all at the same (lower) magnification. Bars = 1 μm.
FIG 9.
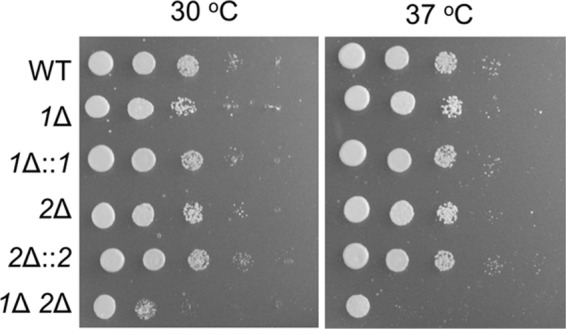
pbx1Δ pbx2Δ mutant cells show increased sensitivity to oxidative stress. Serial 10-fold dilutions were spotted on yeast nitrogen base agar containing 1 mM H2O2 and grown at the indicated temperatures for 4 days.
Pbx protein localization.
The Pbx proteins are predicted to have transmembrane domains and localize to membranes (Fig. S8 in the supplemental material). Consistent with this, immunoblotting analysis of HA-tagged versions of Pbx1 and Pbx2 expressed in C. neoformans showed that both proteins were completely absent from the cytosolic fraction (Fig. 10). The proteins were not extracted from membranes by 1% Triton X-100 (4°C, 1 h), although they could be extracted with 1% SDS (100°C, 10 min), and they were not associated with cell wall fractions (Fig. 10). These characteristics suggest that they are located in detergent-resistant membranes (see Discussion).
FIG 10.
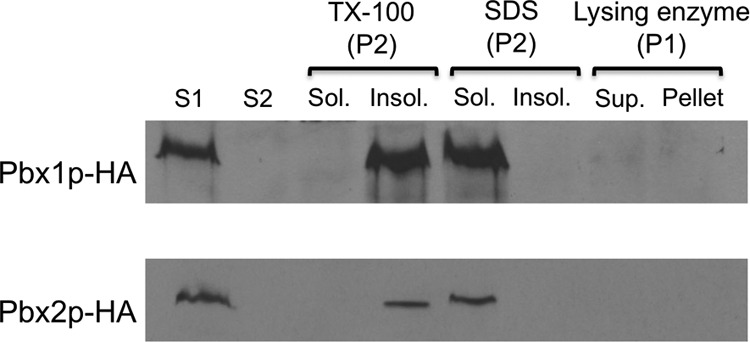
Pbx proteins occur in detergent-resistant membranes. Total cell lysate cleared of cell debris (S1) was subjected to ultracentrifugation, and the supernatant fraction (S2) was removed. The pellet (P2) was extracted with TX-100 or SDS and fractionated into detergent-insoluble (Insol.) and detergent-soluble (Sol.) fractions. The cell wall-containing fraction (P1) was treated with lysing enzyme and fractionated into supernatant (Sup.) and pellet fractions. The same number of cell equivalents of each fraction of the indicated strains was resolved by SDS-PAGE and immunoblotted with anti-HA antibody. See Materials and Methods for details.
Host cell interaction.
Finally, we investigated the interactions of the pbx mutant strains with host phagocytes, which have been implicated in fungal latency and dissemination, in addition to host defense. We found that phagocyte engulfment of these strains was increased by about 50% compared to the level of internalization of wild-type cells (Fig. 11; P = 2.2 × 10−10). This may contribute to the reduced virulence of single pbx mutants that has previously been reported (32) (see Discussion).
FIG 11.
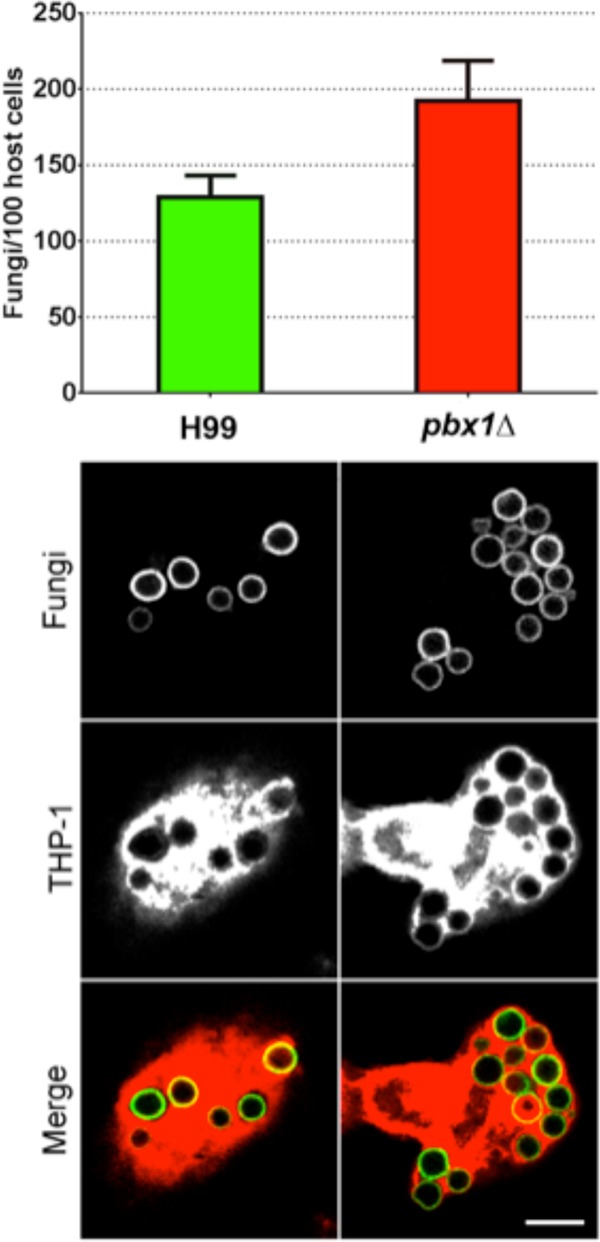
C. neoformans lacking Pbx1 is more efficiently engulfed by human phagocytes than wild-type fungi. (Top) Plot showing the 1.5-fold increase in uptake of the pbx1Δ mutant by THP-1 cells compared to that of wild-type fungi (mean values, 192 and 129 fungal cells/100 host cells, respectively). (Bottom) Confocal images of cryptococci (cell walls stained green) engulfed by THP-1 cells (the cytosol stained red). Left column, wild type; right column, pbx1Δ mutant. In some instances, fungi appear outlined in yellow in the merged image; this is because of pixel overlap with the intense cytosolic stain. Results are representative of those from two independent experiments. All images are at the same scale. Bar = 10 μm.
DISCUSSION
The cell wall of C. neoformans is required for fungal integrity, and its polysaccharide capsule is critical for pathogenesis (12, 23, 62, 63). Although these two key structures are physically linked, the mechanism of their association is only poorly understood. In previous work, we developed a capsule transfer assay to assess the ability of capsule polysaccharides shed from donor cells to bind to the cell walls of acapsular mutant acceptor cells. This enabled us to identify cell wall α-glucan as an important attribute of the acceptor cell walls which was needed for cell surface display of capsule polysaccharides (26, 27). We hypothesized that characteristics of the donor cell would also contribute to this process. To identify such features, we screened a deletion collection generated by the Madhani group (31) for mutants that could not effectively function as capsule donors in our transfer assay. In parallel, we assessed the surface capsule of these strains.
Our screen identified several strains with an impaired ability to serve as capsule donors and two groups of mutants that were completely unable to do so. One of these groups included four Cap mutants that have previously been characterized to be acapsular (53–56), along with another strain that we previously showed has an acapsular phenotype (ugd1) (58). Our identification of these strains, which, as expected from the literature, also lacked surface capsule (Table 2), validated our screening approach.
A second group of interesting mutants included strains that were unable to serve as capsule donors, even though they did display surface capsule (Table 2). This report describes our findings on two of these strains, the pbx1Δ and pbx2Δ mutants, which have indistinguishable phenotypes. Compared to wild-type cells, both mutants have less dense arrays of capsule fibers and capsules that are more loosely associated with the cell wall. The pbx mutants also shed greatly reduced amounts of capsule compared to the wild type, which accounts for their transfer defect. This most likely reflects a global defect in synthesis, because capsule material within the cell and at the cell surface is also reduced (Fig. 5). Both pbx mutants also displayed an aberrant cell wall morphology and exhibited some mild phenotypes associated with cell wall dysfunction that were more severe in the double mutant. pbx mutant cells were also engulfed more avidly than wild-type cells by human phagocytic cells, a process that has been implicated in host defense as well as fungal latency and dissemination. In some scenarios, residence within host cells may benefit C. neoformans. However, in the context of mutant cells that have deformed cell walls and increased sensitivity to oxidative stress, this engulfment is likely a dead end, and the increased phagocytosis may contribute to the observed virulence defect of cells lacking Pbx proteins (32).
Pbx1 and Pbx2 have parallel beta-helix domains, like cell wall-degrading enzymes such as pectin lyase, homogalacturonase, and xyloglucanase. Despite significant effort, we were unable to directly demonstrate glucanase activity for these proteins (see the text in the supplemental material). One possibility is that the Pbx proteins act to remodel the cell wall, liberating sugars for other synthetic processes. According to this model, the cell wall would overgrow in the absence of Pbx1 and Pbx2 at the expense of capsule synthesis; the abnormal wall would also mediate looser associations with the capsule. This model is consistent with the phenotypes that we observed in the pbx mutants, including their aberrant morphology (Fig. 8), less dense capsules (Fig. 5) that are easily released from the cell surface (32) (see Fig. S4 in the supplemental material), and a reduction in capsule shedding (Fig. 4) and the resultant transfer (Fig. 2 and 3). Based on the identical phenotypes of the two mutants and defects that were generally more severe in the double mutant, we believe that Pbx1 and Pbx2 serve redundant functions, although confirmation of this would require additional investigation.
In addition to the gross morphological changes in pbx mutant cell walls (Fig. 8), we observed several more subtle alterations suggesting cell wall abnormalities. The pbx1Δ and pbx2Δ mutants showed decreased production of melanin (see Fig. S5 in the supplemental material), which occurs at the cell wall, and heightened binding of the cell wall stain lucifer yellow (data not shown). The double mutant was further defective in generating mating structures (see Fig. S6 in the supplemental material). Interestingly, none of the mutants differed from the wild type in sensitivity to calcofluor white and Congo red (not shown), two dyes typically used to assess cell wall integrity (64). Since these compounds bind with the highest affinity to chitin (65), this suggests that cell wall chitin is normal in the pbx mutants. Supporting this suggestion, these compounds and several other reagents that bind to related cellular components, including wheat germ agglutinin (N-acetylglucosamine), pontamine (chitin and chitosan), and eosin Y (chitosan), bind similarly to mutant and wild-type cells (not shown).
Additional support for our hypothesis that Pbx proteins act in cell wall remodeling comes from their localization in detergent-resistant membranes (Fig. 10), also termed lipid rafts (66). There is significant precedent in fungi for membrane proteins playing critical roles in cell wall assembly and morphogenesis; examples include Saccharomyces cerevisiae, Neurospora crassa, and Aspergillus fumigatus (67–69).
Our model suggests that the absence of Pbx proteins leads to reduced cell wall remodeling, which has consequences for cell morphology and capsule association. This would also alter the metabolic balance of the cells and the substrates available for synthesis of glycans, potentially explaining the reduced capsule polysaccharide synthesis of the pbx mutants. Metabolic imbalance might also explain the slightly increased xylosylation of the mutant capsule polysaccharide. Although xylose is incorporated into cryptococcal glycoproteins (70, 71), it is a relatively minor component of the cell wall overall, whereas it plays a major role in capsule polysaccharides. Reduced wall turnover might therefore have a lesser effect on this capsule moiety than on mannose, for example, which is a major component of both structures, leading to the altered ratio of these components that we observed in GXM.
We tried hard to demonstrate a specific biochemical activity for the Pbx proteins by growth on various substrates, in vitro assays using cell lysates or purified proteins, and several physical methods for assessing protein-glycan binding (see the text in the supplemental material). Our inability to do this may have several explanations. The cell wall of C. neoformans differs from that of other model yeast, and there is precedent for this organism having unique glycan structures; cryptococcal glycans have also not been fully characterized. We may therefore not have tested the appropriate substrates. It is also possible that a lipid association of these membrane proteins is required for their function. Finally, our glycan array binding studies were based on the currently available array (version 5.0; http://www.functionalglycomics.org/static/consortium/resources/resourcecoreh16.shtml), which is primarily composed of mammalian glycans, so we may not have had access to the appropriate binding partner. As we learn more about cryptococcal glycobiology and the community develops additional fungal glycan reagents, we will be able to productively revisit these experiments.
On the basis of compositional analysis of GXM, it was previously proposed that Pbx proteins act to remove glucose residues that are aberrantly incorporated into capsule polysaccharides (32). Our analysis of the composition of GXM and GXMGal from wild-type and mutant cells and detailed determination of their structures by NMR do not indicate the presence of any glucose and therefore do not support this model. GXM preparations may be contaminated with cell wall glucans, particularly when the strain being analyzed has low levels of shed capsule polysaccharides (unpublished observations), as is the case for the pbx mutants. The presence of cell wall glucans may have led to the previous observation of glucose in capsule polysaccharide samples from these strains.
We conclude that Pbx proteins act in C. neoformans cell wall remodeling. Their absence leads to an abnormal cell wall structure, which impairs capsule association, and altered metabolic balance, which reduces capsule polysaccharide synthesis. These changes yield cells that are less fit to cause disease, as demonstrated by their lack of virulence (32). Future work will be needed to demonstrate a specific biochemical function for these intriguing enzymes; this may require lipid reconstitution and further definition of cryptococcal glycans to suggest potential substrates. Nonetheless, our work has demonstrated the interplay between cell wall and capsule synthesis and elucidated how perturbations in these processes critically affect the virulence of this deadly pathogen.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Meng Yang and Mike Skowyra for helpful discussions, Mike Skowyra for plasmid pMSC-043, Tom Kozel and Arturo Casadevall for monoclonal antibodies, Jennifer Lodge and Gary Cox for plasmids, Joe Heitman and Jennifer Lodge for strains, and Zhuo Wang and Lucy Li for comments on the manuscript. We also thank Barbara Kunkel and Andrew Mutka for help in establishing C. neoformans infections in Arabidopsis thaliana, Daved Fremont for use of MALS equipment, Christopher Nelson for advice on GXM molecular weight determination, Wandy Beatty for performing thin-section microscopy, David Smith and Jamie Heimburg-Molinaro for glycan array analysis, Marcellene Hollingsworth and Tom Kozel for surface plasmon resonance studies of GXM binding, and Robyn Roth and John Heuser for quick-freeze deep-etch micrographs.
Work on cryptococcal glycan synthesis in the T. L. Doering lab was supported by NIH grant R01 GM071007, and polysaccharide analysis performed at the Complex Carbohydrate Research Center was supported by DOE grant DE-FG02-93ER20097. The Consortium for Functional Glycomics, which supported the glycan array studies at the Protein-Glycan Interaction Core at the Emory University School of Medicine and related compound synthesis at the Glycan Array Synthesis Core at the Scripps Research Institute, was funded by NIH grants GM062116 and GM098791. F.H.S.-T. was partially supported by NIH grant R01 AI78795 and a postdoctoral enrichment award for URM scientists from the Burroughs Wellcome Foundation.
Footnotes
Published ahead of print 28 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00290-13.
REFERENCES
- 1.Casadevall A, Perfect JR. (ed). 1998. Cryptococcus neoformans. ASM Press, Washington, DC [Google Scholar]
- 2.Ellis DH, Pfeiffer TJ. 1990. Natural habitat of Cryptococcus neoformans var. gattii. J. Clin. Microbiol. 28:1642–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emmons CW. 1951. Isolation of Cryptococcus neoformans from soil. J. Bacteriol. 62:685–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530. 10.1097/QAD.0b013e328322ffac [DOI] [PubMed] [Google Scholar]
- 5.Heitman J, Kozel TR, Kwon-Chung KJ, Perfect JR, Casadevall A. (ed). 2011. Cryptococcus. From human pathogen to model yeast. ASM Press, Washington, DC [Google Scholar]
- 6.Sabiiti W, May RC. 2012. Mechanisms of infection by the human fungal pathogen Cryptococcus neoformans. Future Microbiol. 7:1297–1313. 10.2217/fmb.12.102 [DOI] [PubMed] [Google Scholar]
- 7.Buchanan KL, Murphy JW. 1998. What makes Cryptococcus neoformans a pathogen? Emerg. Infect. Dis. 4:71–83. 10.3201/eid0401.980109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigues ML, Alviano CS, Travassos LR. 1999. Pathogenicity of Cryptococcus neoformans: virulence factors and immunological mechanisms. Microbes Infect. 1:293–301. 10.1016/S1286-4579(99)80025-2 [DOI] [PubMed] [Google Scholar]
- 9.Chun CD, Brown JC, Madhani HD. 2011. A major role for capsule-independent phagocytosis-inhibitory mechanisms in mammalian infection by Cryptococcus neoformans. Cell Host Microbe 9:243–251. 10.1016/j.chom.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Meara TR, Alspaugh JA. 2012. The Cryptococcus neoformans capsule: a sword and a shield. Clin. Microbiol. Rev. 25:387–408. 10.1128/CMR.00001-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherniak R. 1988. Soluble polysaccharides of Cryptococcus neoformans. Curr. Top. Med. Mycol. 2:40–54. 10.1007/978-1-4612-3730-3_2 [DOI] [PubMed] [Google Scholar]
- 12.Doering TL. 2009. How sweet it is! Cell wall biogenesis and polysaccharide capsule formation in Cryptococcus neoformans. Annu. Rev. Microbiol. 63:223–247. 10.1146/annurev.micro.62.081307.162753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McFadden DC, De Jesus M, Casadevall A. 2006. The physical properties of the capsular polysaccharides from Cryptococcus neoformans suggest features for capsule construction. J. Biol. Chem. 281:1868–1875. 10.1074/jbc.M509465200 [DOI] [PubMed] [Google Scholar]
- 14.Cherniak R, Valafar H, Morris LC, Valafar F. 1998. Cryptococcus neoformans chemotyping by quantitative analysis of 1H nuclear magnetic resonance spectra of glucuronoxylomannans with a computer-simulated artificial neural network. Clin. Diagn. Lab. Immunol. 5:146–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heiss C, Klutts JS, Wang Z, Doering TL, Azadi P. 2009. The structure of Cryptococcus neoformans galactoxylomannan contains beta-d-glucuronic acid. Carbohydr. Res. 344:915–920. 10.1016/j.carres.2009.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaishnav VV, Bacon BE, O'Neill M, Cherniak R. 1998. Structural characterization of the galactoxylomannan of Cryptococcus neoformans Cap67. Carbohydr. Res. 306:315–330. 10.1016/S0008-6215(97)10058-1 [DOI] [PubMed] [Google Scholar]
- 17.Heiss C, Skowyra ML, Liu H, Klutts JS, Wang Z, Williams M, Srikanta D, Beverley SM, Azadi P, Doering TL. 2013. Unusual galactofuranose modification of a capsule polysaccharide in the pathogenic yeast Cryptococcus neoformans. J. Biol. Chem. 288:10994–11003. 10.1074/jbc.M112.441998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Jesus M, Chow SK, Cordero RJ, Frases S, Casadevall A. 2010. Galactoxylomannans from Cryptococcus neoformans varieties neoformans and grubii are structurally and antigenically variable. Eukaryot. Cell 9:1018–1028. 10.1128/EC.00268-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McFadden DC, Fries BC, Wang F, Casadevall A. 2007. Capsule structural heterogeneity and antigenic variation in Cryptococcus neoformans. Eukaryot. Cell 6:1464–1473. 10.1128/EC.00162-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vecchiarelli A, Monari C. 2010. Microbial polysaccharide: new insights for treating autoimmune diseases. Front. Biosci. (Schol. Ed.) 2:256–267 [DOI] [PubMed] [Google Scholar]
- 21.Vecchiarelli A, Pericolini E, Gabrielli E, Chow SK, Bistoni F, Cenci E, Casadevall A. 2011. Cryptococcus neoformans galactoxylomannan is a potent negative immunomodulator, inspiring new approaches in anti-inflammatory immunotherapy. Immunotherapy 3:997–1005. 10.2217/imt.11.86 [DOI] [PubMed] [Google Scholar]
- 22.Klutts JS, Doering TL. 2008. Cryptococcal xylosyltransferase 1 (Cxt1p) from Cryptococcus neoformans plays a direct role in the synthesis of capsule polysaccharides. J. Biol. Chem. 283:14327–14334. 10.1074/jbc.M708927200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, Yang M, Haynes BC, Skowyra ML, Doering TL. 2011. Emerging themes in cryptococcal capsule synthesis. Curr. Opin. Struct. Biol. 21:597–602. 10.1016/j.sbi.2011.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoneda A, Doering TL. 2006. A eukaryotic capsular polysaccharide is synthesized intracellularly and secreted via exocytosis. Mol. Biol. Cell 17:5131–5140. 10.1091/mbc.E06-08-0701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pierini LM, Doering TL. 2001. Spatial and temporal sequence of capsule construction in Cryptococcus neoformans. Mol. Microbiol. 41:105–115. 10.1046/j.1365-2958.2001.02504.x [DOI] [PubMed] [Google Scholar]
- 26.Reese AJ, Doering TL. 2003. Cell wall alpha-1,3-glucan is required to anchor the Cryptococcus neoformans capsule. Mol. Microbiol. 50:1401–1409. 10.1046/j.1365-2958.2003.03780.x [DOI] [PubMed] [Google Scholar]
- 27.Reese AJ, Yoneda A, Breger JA, Beauvais A, Liu H, Griffith CL, Bose I, Kim MJ, Skau C, Yang S, Sefko JA, Osumi M, Latge JP, Mylonakis E, Doering TL. 2007. Loss of cell wall alpha(1-3) glucan affects Cryptococcus neoformans from ultrastructure to virulence. Mol. Microbiol. 63:1385–1398. 10.1111/j.1365-2958.2006.05551.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fonseca FL, Nimrichter L, Cordero RJ, Frases S, Rodrigues J, Goldman DL, Andruszkiewicz R, Milewski S, Travassos LR, Casadevall A, Rodrigues ML. 2009. Role for chitin and chitooligomers in the capsular architecture of Cryptococcus neoformans. Eukaryot. Cell 8:1543–1553. 10.1128/EC.00142-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodrigues ML, Alvarez M, Fonseca FL, Casadevall A. 2008. Binding of the wheat germ lectin to Cryptococcus neoformans suggests an association of chitinlike structures with yeast budding and capsular glucuronoxylomannan. Eukaryot. Cell 7:602–609. 10.1128/EC.00307-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozel TR, Hermerath CA. 1984. Binding of cryptococcal polysaccharide to Cryptococcus neoformans. Infect. Immun. 43:879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu OW, Chun CD, Chow ED, Chen C, Madhani HD, Noble SM. 2008. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 135:174–188. 10.1016/j.cell.2008.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu OW, Kelly MJ, Chow ED, Madhani HD. 2007. Parallel beta-helix proteins required for accurate capsule polysaccharide synthesis and virulence in the yeast Cryptococcus neoformans. Eukaryot. Cell 6:630–640. 10.1128/EC.00398-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J. 2003. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and alpha isolates. Infect. Immun. 71:4831–4841. 10.1128/IAI.71.9.4831-4841.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker LG, Specht CA, Donlin MJ, Lodge JK. 2007. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot. Cell 6:855–867. 10.1128/EC.00399-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu J, Hettler E, Wickes BL. 2006. Split marker transformation increases homologous integration frequency in Cryptococcus neoformans. Fungal Genet. Biol. 43:200–212. 10.1016/j.fgb.2005.09.007 [DOI] [PubMed] [Google Scholar]
- 36.Nelson RT, Hua J, Pryor B, Lodge JK. 2001. Identification of virulence mutants of the fungal pathogen Cryptococcus neoformans using signature-tagged mutagenesis. Genetics 157:935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hua J, Meyer JD, Lodge JK. 2000. Development of positive selectable markers for the fungal pathogen Cryptococcus neoformans. Clin. Diagn. Lab. Immunol. 7:125–128. 10.1128/CDLI.7.1.125-128.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDade HC, Cox GM. 2001. A new dominant selectable marker for use in Cryptococcus neoformans. Med. Mycol. 39:151–154. 10.1080/714030997 [DOI] [PubMed] [Google Scholar]
- 39.Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. 1993. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J. Bacteriol. 175:1405–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown T. 2001. Southern blotting. Current protocols in molecular biology, unit 2.9A. John Wiley & Sons, Hoboken, NJ. 10.1002/0471142727.mb0209as21 [DOI] [PubMed] [Google Scholar]
- 41.Bose I, Doering TL. 2011. Efficient implementation of RNA interference in the pathogenic yeast Cryptococcus neoformans. J. Microbiol. Methods 86:156–159. 10.1016/j.mimet.2011.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edman JC, Kwon-Chung KJ. 1990. Isolation of the URA5 gene from Cryptococcus neoformans var. neoformans and its use as a selective marker for transformation. Mol. Cell. Biol. 10:4538–4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright R. 2000. Transmission electron microscopy of yeast. Microsc. Res. Tech. 51:496–510. [DOI] [PubMed] [Google Scholar]
- 44.Sheng S, Cherniak R. 1997. Structure of the 13C-enriched O-deacetylated glucuronoxylomannan of Cryptococcus neoformans serotype A determined by NMR spectroscopy. Carbohydr, Res. 301:33–40. 10.1016/S0008-6215(97)00084-0 [DOI] [PubMed] [Google Scholar]
- 45.Merkle RK, Poppe I. 1994. Carbohydrate composition analysis of glycoconjugates by gas-liquid chromatography/mass spectrometry. Methods Enzymol. 230:1–15. 10.1016/0076-6879(94)30003-8 [DOI] [PubMed] [Google Scholar]
- 46.York WS, Darvill AG, McNeil M, Stevenson TT, Albersheim P. 1986. Isolation and characterization of plant cell walls and cell wall components. Methods Enzymol. 118:3–40. 10.1016/0076-6879(86)18062-1 [DOI] [Google Scholar]
- 47.Frases S, Nimrichter L, Viana NB, Nakouzi A, Casadevall A. 2008. Cryptococcus neoformans capsular polysaccharide and exopolysaccharide fractions manifest physical, chemical, and antigenic differences. Eukaryot. Cell 7:319–327. 10.1128/EC.00378-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ashwell G. 1957. Colorimetric analysis of sugars. Methods Enzymol. 3:73–105. 10.1016/S0076-6879(57)03350-9 [DOI] [Google Scholar]
- 49.Yoneda A, Doering TL. 2008. Regulation of Cryptococcus neoformans capsule size is mediated at the polymer level. Eukaryot. Cell 7:546–549. 10.1128/EC.00437-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reilly MC, Levery SB, Castle SA, Klutts JS, Doering TL. 2009. A novel xylosylphosphotransferase activity discovered in Cryptococcus neoformans. J. Biol. Chem. 284:36118–36127. 10.1074/jbc.M109.056226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilbert NM, Baker LG, Specht CA, Lodge JK. 2012. A glycosylphosphatidylinositol anchor is required for membrane localization but dispensable for cell wall association of chitin deacetylase 2 in Cryptococcus neoformans. mBio 3(1):e00007–12. 10.1128/mBio.00007-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Srikanta D, Yang M, Williams M, Doering TL. 2011. A sensitive high-throughput assay for evaluating host-pathogen interactions in Cryptococcus neoformans infection. PLoS One 6:e22773. 10.1371/journal.pone.0022773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang YC, Kwon-Chung KJ. 1994. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol. Cell. Biol. 14:4912–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang YC, Kwon-Chung KJ. 1998. Isolation of the third capsule-associated gene, CAP60, required for virulence in Cryptococcus neoformans. Infect. Immun. 66:2230–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang YC, Kwon-Chung KJ. 1999. Isolation, characterization, and localization of a capsule-associated gene, CAP10, of Cryptococcus neoformans. J. Bacteriol. 181:5636–5643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang YC, Penoyer LA, Kwon-Chung KJ. 1996. The second capsule gene of Cryptococcus neoformans, CAP64, is essential for virulence. Infect. Immun. 64:1977–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bar-Peled M, Griffith CL, Ory JJ, Doering TL. 2004. Biosynthesis of UDP-GlcA, a key metabolite for capsular polysaccharide synthesis in the pathogenic fungus Cryptococcus neoformans. Biochem. J. 381:131–136. 10.1042/BJ20031075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griffith CL, Klutts JS, Zhang L, Levery SB, Doering TL. 2004. UDP-glucose dehydrogenase plays multiple roles in the biology of the pathogenic fungus Cryptococcus neoformans. J. Biol. Chem. 279:51669–51676. 10.1074/jbc.M408889200 [DOI] [PubMed] [Google Scholar]
- 59.Moyrand F, Janbon G. 2004. UGD1, encoding the Cryptococcus neoformans UDP-glucose dehydrogenase, is essential for growth at 37 degrees C and for capsule biosynthesis. Eukaryot. Cell 3:1601–1608. 10.1128/EC.3.6.1601-1608.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Souza Pereira R, Geibel J. 1999. Direct observation of oxidative stress on the cell wall of Saccharomyces cerevisiae strains with atomic force microscopy. Mol. Cell. Biochem. 201:17–24. 10.1023/A:1007007704657 [DOI] [PubMed] [Google Scholar]
- 61.Gerik KJ, Bhimireddy SR, Ryerse JS, Specht CA, Lodge JK. 2008. PKC1 is essential for protection against both oxidative and nitrosative stresses, cell integrity, and normal manifestation of virulence factors in the pathogenic fungus Cryptococcus neoformans. Eukaryot. Cell 7:1685–1698. 10.1128/EC.00146-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baker LG, Specht CA, Lodge JK. 2011. Cell wall chitosan is necessary for virulence in the opportunistic pathogen Cryptococcus neoformans. Eukaryot. Cell 10:1264–1268. 10.1128/EC.05138-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gilbert NM, Donlin MJ, Gerik KJ, Specht CA, Djordjevic JT, Wilson CF, Sorrell TC, Lodge JK. 2010. KRE genes are required for beta-1,6-glucan synthesis, maintenance of capsule architecture and cell wall protein anchoring in Cryptococcus neoformans. Mol. Microbiol. 76:517–534. 10.1111/j.1365-2958.2010.07119.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ram AF, Klis FM. 2006. Identification of fungal cell wall mutants using susceptibility assays based on calcofluor white and Congo red. Nat. Protoc. 1:2253–2256. 10.1038/nprot.2006.397 [DOI] [PubMed] [Google Scholar]
- 65.Roncero C, Duran A. 1985. Effect of calcofluor white and Congo red on fungal cell wall morphogenesis: in vivo activation of chitin polymerization. J. Bacteriol. 163:1180–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malinsky J, Opekarova M, Grossmann G, Tanner W. 2013. Membrane microdomains, rafts, and detergent-resistant membranes in plants and fungi. Annu. Rev. Plant Biol. 64:501–529. 10.1146/annurev-arplant-050312-120103 [DOI] [PubMed] [Google Scholar]
- 67.Alvarez FJ, Douglas LM, Konopka JB. 2009. The Sur7 protein resides in punctate membrane subdomains and mediates spatial regulation of cell wall synthesis in Candida albicans. Commun. Integr. Biol. 2:76–77. 10.1091/mbc.E08-05-0479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Free SJ. 2013. Fungal cell wall organization and biosynthesis. Adv. Genet. 81:33–82. 10.1016/B978-0-12-407677-8.00002-6 [DOI] [PubMed] [Google Scholar]
- 69.Wang HX, Douglas LM, Aimanianda V, Latge JP, Konopka JB. 2011. The Candida albicans Sur7 protein is needed for proper synthesis of the fibrillar component of the cell wall that confers strength. Eukaryot. Cell 10:72–80. 10.1128/EC.00167-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park JN, Lee DJ, Kwon O, Oh DB, Bahn YS, Kang HA. 2012. Unraveling unique structure and biosynthesis pathway of N-linked glycans in human fungal pathogen Cryptococcus neoformans by glycomics analysis. J. Biol. Chem. 287:19501–19515. 10.1074/jbc.M112.354209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reilly MC, Aoki K, Wang ZA, Skowyra ML, Williams M, Tiemeyer M, Doering TL. 2011. A xylosylphosphotransferase of Cryptococcus neoformans acts in protein O-glycan synthesis. J. Biol. Chem. 286:26888–26899. 10.1074/jbc.M111.262162 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.