

Abstract
Glucose uptake is the rate-limiting step in glucose utilization in mammalians and is tightly regulated by a family of specialized proteins, called the facilitated glucose transporters (GLUTs/SLC2). GLUT4, the major isoform in insulin-responsive tissue, translocates from an intracellular pool to the cell surface and as such determines insulin-stimulated glucose uptake. However, despite intensive research over 50 years, the insulin-dependent and -independent pathways that mediate GLUT4 translocation are not fully elucidated in any species. Insulin resistance (IR) is one of the hallmarks of equine metabolic syndrome and is the most common metabolic predisposition for laminitis in horses. IR is characterized by the impaired ability of insulin to stimulate glucose disposal into insulin-sensitive tissues. Similar to other species, the functional capability of the insulin-responsive GLUTs is impaired in muscle and adipose tissue during IR in horses. However, the molecular mechanisms of altered glucose transport remain elusive in all species, and there is still much to learn about the physiological and pathophysiological functions of the GLUT family members, especially in regard to class III. Since GLUTs are key regulators of whole-body glucose homeostasis, they have received considerable attention as potential therapeutic targets to treat metabolic disorders in human and equine patients.
1. Regulation of Glucose Transport in Healthy State
Glucose is of the most abundant and essential energy sources for both plants and animals, existing in various polymerized forms such as cellulose and glycogen [1]. Glucose uptake from the bloodstream into the cell is the rate-limiting step in glucose utilization primarily in insulin-sensitive tissue in all species. Striated (i.e., cardiac and skeletal) muscle is the main tissue to utilize glucose as an energy substrate, followed by adipose tissue. For instance, skeletal muscle, which makes up ~40% of the body mass in mammalian species, is the primary tissue responsible for the peripheral disposal of glucose, especially during exercise [2]. In addition, the energetic demands in the heart are extreme and as a result, the heart has the highest rate of oxygen consumption per gram of any tissue in the body [3]. In order to sustain this high energy demand, the rate of glucose utilization in the heart is greater than in skeletal muscle, adipose tissue, and lung, despite the ability of the myocardium to use other substrates (i.e., fatty acids, lactate, ketone bodies, and amino acids) [4]. Therefore, glucose transport and utilization by myocytes are critical for the maintenance of muscle function [5]. In addition, glucose absorbed from the gut stimulates the release of insulin from pancreatic β-cells, which both suppresses hepatic gluconeogenesis and promotes glucose uptake by muscle and adipose tissue [6]. Finally, glucose can regulate gene transcription, epigenetic processes, and enzyme activity, as well as glucose-sensitive neurons in the brain [1].
In all mammalian cells, blood glucose is maintained within a narrow range by homeostatic mechanisms. Since the lipid bilayers of cell membranes are impermeable to glucose, most cells take up glucose by a passive facilitative transport process, which is mediated by a family of integral membrane proteins collectively known as the glucose transporters (GLUTs) [7]. It is believed that only the epithelial cell brush border of the small intestine and the kidney proximal convoluted tubules absorb glucose by an active mechanism that uses Na+/glucose cotransporters to transport glucose against its electrochemical gradient [8]. The equine intestinal Na+/glucose cotransporter, SGLT1, has 85–89% homology at the nucleotide level and 84–87% at the amino acid level with SGLT1 of other species [9]. Similarly to other herbivorous and omnivorous species, there is an enhancement in the expression of the intestinal Na+/glucose cotransporter, SGLT1, and the capacity to absorb monosaccharides in response to increased dietary carbohydrate levels in horses [10].
This paper reviews the current knowledge on the physiological and pathophysiological functions of the GLUT/SLC2 family member, as they relate to the equine species focusing on insulin-responsive tissue (e.g., striated muscle and adipose tissue).
1.1. Function and Distribution of the Facilitative Glucose Transporters
The GLUT protein family are encoded by the SLC2 genes and are a member of the major facilitator superfamily of membrane transporters, which are ubiquitous proteins with >5,000 members identified in all three kingdoms [1, 11]. Since at least one GLUT isoform is present in every cell type of the body, GLUTs are widely recognized as key regulators of whole-body glucose homeostasis. The GLUT proteins, which are comprised of ~500 amino acid residues, have 12 membrane-spanning domains, an N-linked glycosylation site, and intracellular NH2 and COOH termini, as well as several conserved residues and motifs designated “sugar transporter signatures” [7, 12]. These facilitative GLUT proteins are expressed in a tissue-specific manner, with distinct regulatory and/or kinetic properties that reflect their specific roles in cellular and whole-body glucose homeostasis [1]. Currently, fourteen GLUT isoforms have been identified and divided into 3 different classes based on sequence similarity, as well as structural and functional characteristics: Class I (GLUTs 1–4, 14); Class II (GLUTs 5, 7, 9, and 11), and Class III (GLUTs 6, 8, 10, and 12 and HMIT). Although the major substrate(s) for several isoforms has not been identified, these facilitated transporters are capable of transporting glucose, fructose, myoinositol, and urate across the plasma membrane down-gradient. Tissue distribution and major function(s) are listed in Table 1. Since several reviews on the GLUT family have been published [1, 6–8, 13], this paper briefly summarizes key features of the major GLUTs, as they relate to the equine species. The function and distribution of two members of class I, GLUT-1 and -4, have been extensively studied [1, 14, 15]. GLUT1 is primarily located at the cell surface and thus is a basal transporter that does not require insulin stimulation for its activation. Therefore, GLUT1 is considered to be the primary transporter responsible for basal glucose uptake in many cell types. GLUT1 has a high expression in brain and erythrocytes [16], and as a consequence, ~1/3 of blood glucose is carried within the red blood cell cytoplasm [7]. Interestingly, GLUT1 is also highly abundant in the digital lamellae, indicating that this unique weight-bearing, dermoepidermal structure within the horse's hoof is a highly metabolic tissue [17, 18]. Similarly, articular chondrocytes, which express both GLUT-1 and -3, rely on glucose as the major energy substrate and as the main precursor for the synthesis of extracellular matrix glycosaminoglycans in cartilage [19, 20]. The equine small intestine is the major site of glucose absorption, with glucose transport being the highest in the duodenum, followed by the jejunum and ileum [9]. Glucose is transported across the brush-border membrane of the intestinal enterocytes by SGLT1 and then exits the cell across the basolateral membrane by the facilitated GLUT2, a low-affinity glucose and fructose transporter [10, 21]. Equine small intestine also expresses GLUT5, a fructose transporter, in the villus enterocytes with the highest levels in the duodenum and the lowest levels in the ileum [22].
Table 1.
| Protein | Major sites of expression | Proposed function/major substrate | Major species studied |
|---|---|---|---|
| GLUT1 | Many cell types (e.g., erythrocytes, brain); fetal tissues | Basal glucose uptake; transport across blood-tissue barriers | Rodents, humans, horses, cows, dogs, pigs |
| GLUT2 | Kidney, small intestine, liver, pancreatic islets, brain | High-capacity low-affinity facilitated glucose and fructose transporter | Rodents, humans, horses, cows |
| GLUT3 | Brain (neurons) and testis | High-affinity facilitated glucose transporter; neuronal transport | Rodents, humans, cows, dogs, pigs |
| GLUT4 | Striated muscle, fat, heart | High-affinity facilitated glucose transporter; insulin-regulated transport; linked to IR/diabetes | Rodents, humans, horses, cows, dogs, pigs |
| GLUT5 | Small intestine, kidney, striated muscle, fat, testis | Facilitated fructose transporter | Rodents, humans, horses, cows, dogs, pigs |
| GLUT6 | Leukocytes, brain, spleen | Facilitated glucose transporter | |
| GLUT7 | Small intestine, colon, testis | Transport of glucose and fructose | |
| GLUT8 | Testis, brain, blastocyst, striated muscle, fat, liver, spleen, lung | Facilitated glucose transporter widely expressed; neuronal transport; insulin-responsive transport in blastocyst | Rodents, humans, cows, horses |
| GLUT9 | Liver, kidney, small intestine | Facilitated urate (glucose) transporter | Rodents, humans |
| GLUT10 | Striated muscle, lung, liver, pancreas | ||
| GLUT11 | Striated muscle | Muscle-specific; facilitated glucose/fructose transporter | Rodents, humans |
| GLUT12 | Striated muscle, fat, prostate, mammary gland | A second insulin-responsive facilitated GLUT? | Rodents, humans, cows, horses |
| HMIT | Brain, fat | H+/myo-inositol cotransporter | |
| GLUT14 | Testis | Orphan transporter |
In contrast to basal GLUTs, GLUT4 translocates from an intracellular (nonactive) pool to the cell surface (active site) to enhance glucose uptake, a process called translocation. Therefore, GLUT4 is responsible for insulin-stimulated glucose uptake [23, 24]. Since GLUT4 is predominantly expressed in insulin-sensitive tissues, it plays a crucial role in whole-body glucose homeostasis [24] and as a result is one of the most studied GLUT isoforms, including horses. The equine GLUT4 gene has been characterized in equine skeletal muscle, which shares a high degree of homology with that of other mammalians [17, 25–27]. Similar to other species, GLUT4 protein is abundantly expressed in striated muscle and adipose tissue of horses [27, 28]. In addition, GLUT4 protein is expressed in equine muscle in a fiber-type specific manner with a higher expression in type 2B fibers, compared with that in type 1 fibers [27]. In contrast, immunostaining of GLUT4 in the equine digital lamellae has produced inconclusive results [29, 30], while western blot of crude membrane and PCR showed that GLUT4 is not a predominant isoform, suggesting that this insulin-responsive GLUT is unlikely to play a substantial role in the regulation of lamellar glucose transport [17, 18]. In addition, glucose uptake was not affected by insulin, further indicating the insulin-independent nature of the equine lamellae [17].
Although it has been believed that GLUT4 translocation is the rate-limiting step for glucose uptake and utilization in insulin-sensitive tissue, GLUT4 knockout mice do not develop hyperglycemia [31], suggesting that other GLUT isoforms could be involved in the regulation of whole-body glucose homeostasis [28]. Therefore, researchers have recently focused their attention on less understood GLUTs, particularly class III transporters, such as GLUT-8 and -12 [12, 32–35]. For instance, GLUT12 has been identified as a potential novel second insulin-sensitive GLUT [36], although its function is more likely to be tissue specific as for many other isoforms. In addition, its role is not well known in any species [37]. In horses, GLUT12 is expressed in insulin-sensitive tissues, with the highest protein expression in the omental adipose tissue, similar to the distribution pattern of GLUT4 protein across tissues [28]. In addition, it has been suggested that GLUT8, a dual-specific glucose and fructose transporter, is another insulin-regulated GLUT because it is highly expressed in striated muscle and adipose tissue, in addition to other tissues such as brain, testis, spermatozoa, liver, and kidney [38]. Furthermore, GLUT8 has been identified as an insulin-responsive GLUT in blastocysts [39]. Interestingly, GLUT8 protein has been recently characterized in striated muscle and digital lamellae of horses, with similar protein expression across tissue [18]. However, further studies are required to elucidate the physiological functions of the novel GLUT-8 and -12 isoforms.
1.2. Regulation of GLUT Translocation
Despite intensive research for over 50 years (primarily in rodents), the downstream signaling pathways that mediate the translocation of GLUT4 protein from an intracellular pool to the sarcolemma (active site) are not fully elucidated in any species. GLUT4 trafficking is largely regulated by insulin-dependent processes, although other factors including calcium/contraction, catecholamines, and hypoxia can alter glucose transport in skeletal and cardiac muscles (Figure 1) [40–47]. Since there are limited reports on the equine species, most of the information presented in this section is derived from studies on humans and rodents.
Figure 1.
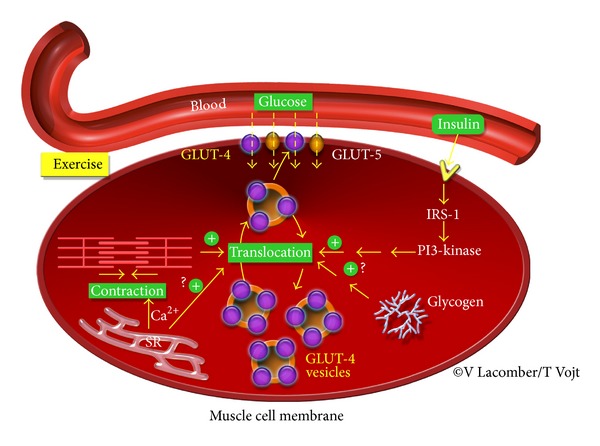
Regulation of glucose transport in striated muscle. GLUT4 translocation to the cell surface is mediated by both insulin-dependent and -independent pathways. GLUT: glucose transporter; PI 3-kinase: phosphatidylinositide 3-kinase; IRS: insulin receptor substrate; Ca2+: calcium; SR: sarcoplasmic reticulum.
GLUT4 is a continuously recycling protein with faster endocytosis than reexocytosis, resulting in a net, but dynamic, intracellular retention [48]. The concept that insulin mobilized GLUT4 from intracellular storage vesicles to the plasma membrane was proposed by Cushman and colleagues in 1980 [49]. It has been estimated that ~5% of total GLUT4 is located at the cell surface of myocytes and adipocytes during basal conditions and that this fraction increased to ~50% after insulin stimulation (Figure 2) [6]. However, there is still some considerable debate on the regulation of GLUT trafficking by the downstream insulin signaling pathways [6]. It is known that, upon insulin release, insulin binds to the insulin receptor, thereby triggering its intrinsic protein-tyrosine kinase activity. The subsequent autophosphorylation of several insulin receptor tyrosine residues promotes tyrosine phosphorylation of insulin receptor substrates [50]. That action is followed by the activation of phosphatidylinositide 3-kinase (PI 3-kinase) and the production of phosphatidylinositol 3,4,5 triphosphate (PIP3) in the membrane (Figure 1). Increased PIP3 levels activate PDK1, a serine/threonine kinase, which in turn recruits the pivotal serine/threonine protein kinase, namely, Akt. Since it is involved in many processes, including cellular growth, survival, and fuel metabolism, Akt has 3 isoforms that directly act on several substrates [7]. Importantly, Akt2 appears to regulate AS160 (namely, Akt substrate protein of 160 kDa), which is the most distal signaling protein that has been implicated in insulin-mediated GLUT4 translocation and as such has emerged as a key regulator of GLUT trafficking. Under basal conditions, AS160 retains GLUT4 vesicles intracellularly through activity of its GTPase activating protein (GAP) domain for members of the Rab G protein family. The Rab proteins, which are guanosine triphosphate (GTP)-binding proteins, are thought to promote vesicle traffic (formation, targeting, and fusion) and may act as molecular switches, catalyzing membrane trafficking events by conversion from an inactive guanosine diphosphate-bound form to an active GTP-bound form [48]. Phosphorylation of AS160, which occurs on at least 6 different phospho-Akt-substrate (PAS) sites, suppresses or alters GAP activity, thereby elevating the GTP form of Rab, so GLUT4 exocytosis is permitted [51–53]. Although AS160 is also expressed in insulin-sensitive tissue of horses, its role in regulating GLUT trafficking has not been established [28, 54]. Finally, it has been shown that insulin-dependent GLUT4 translocation also requires dynamic remodeling of filamentous actin [48, 55]. Recently, it has also been suggested that α3(V) collagen is critical for glucose homeostasis, as mice with null alleles of the α3(V) gene Col5a3 have defective GLUT4 translocation to the plasma membrane in response to insulin [56].
Figure 2.
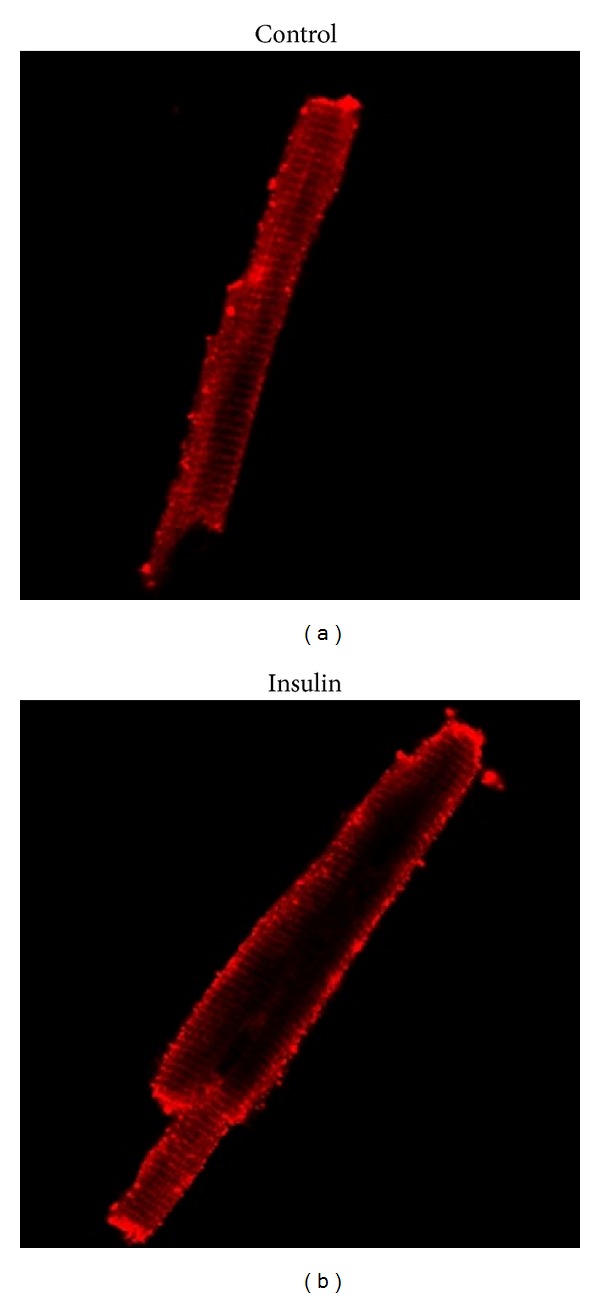
Insulin translocates GLUT4 from an intracellular pool (inactive site) to the cell surface (active site) in muscle, as visualized by confocal laser scanning microscopy. Adult rat cardiac myocytes were incubated without (a) or with insulin ((b), 100 μU/mL) for 30 min prior to immunofluorescent staining. Modified from [37].
Since exercise is vital for the maintenance of glucose homeostasis in diabetic patients, it has been proposed that insulin-independent pathways could be a major mechanism to regulate glucose uptake into striated muscles. Indeed, GLUT4 translocation to the cell surface can be regulated by contraction/Ca2+-dependent processes in striated muscle (Figure 1). For instance, contractile activity during exercise increases glucose uptake and the number of GLUTs at the cell surface of the skeletal muscle, in the absence of insulin [40–42, 57, 58]. Holloszy and Narahara, in 1967, suggested the possibility that exercise-induced contraction and the resultant increase in cytosolic Ca2+ provide the signal to activate glucose transport in frog skeletal muscle [59]. In addition, exposing skeletal muscle to caffeine, which induces Ca2+ release from the sarcoplasmic reticulum at a concentration too low to induce contractions, results in GLUT4 translocation, suggesting that intracellular Ca2+ spikes are sufficient to stimulate glucose transport [60]. However, since the seminal discovery in 1967, the molecular signaling mechanisms by which contraction/Ca2+ regulates muscle glucose transport have remained elusive in all species. It has been hypothesized that activated PI3-kinase directly or indirectly opens Ca2+ channels. In striated muscle, Ca2+ entry subsequently initiates contraction via Ca2+-induced Ca2+ release through the ryanodine receptors, which releases Ca2+ from the sarcoplasmic reticulum and subsequently activates intracellular calmodulin and calmodulin-dependent protein kinase II (CAMPKII). However, the mechanisms by which calmodulin and CAMPKII regulate glucose transport are not well known [58, 61]. Alternatively, it has been suggested that Ca2+ plays a direct role in the insertion of GLUT4 at the sarcolemma. Another potential candidate for contraction-stimulated glucose transport in skeletal muscle is the protein kinase C family [62]. However, the role of these potential candidates has not been well established because of the lack of specificity of most inhibitors studied [62]. Finally, during exercise, ATP is hydrolyzed to sustain the cross bridge cycle and the activity of ion channels and pumps involved in the excitation-contraction process, such as the sarcoplasmic reticulum calcium ATPase (SERCA) pump. The subsequent increase in AMP/ATP ratio has been implicated as a mediator of contraction-induced GLUT4 translocation in skeletal muscle, through activation of AMP-activated protein kinase (AMPK) [62]. There is recent evidence that activation of AMPK is involved in the regulation of AS160 through a contraction/Ca2+ dependent process in skeletal muscle. Indeed, AMPK phosphorylates AS160 at a site (namely, S711), which is distinct from phosphorylation sites that are modulated by insulin downstream signaling pathway [52, 63]. Although it has been shown that high-intensity exercise increases GLUT4 content in skeletal muscle of horses [64], further exploration of the molecular regulations of GLUT4 trafficking by insulin-independent pathways has not been established in this species. Only one study reported an increase in plasma AICAR (5-amino-4-imidazolecarboxamide riboside) after exercise in race horses [65]. Since AICAR activates AMPK, one could speculate that, similar to others species, AMPK-mediated metabolic regulation could be an important process for exercise-induced increased glucose transport in horses. Therefore, additional studies are needed to further understand the mechanisms of insulin-independent glucose transport.
1.3. Contribution of Glucose Transport to Muscle Glycogen Synthesis and Whole-Body Insulin Sensitivity in Healthy Horses
Similar to humans, horses rely on carbohydrate metabolism during both high- and low-to-moderate intensity exercise, where glycogen and glucose are the obligate substrates to sustain anaerobic ATP production [66]. Therefore, during exercise, the majority of circulating glucose is utilized by muscle fibers to sustain contraction. At rest, the majority of the glucose entering the cell is converted to triglycerides in adipose tissue and to glycogen in muscle fibers and liver. Therefore, the capacity to maximize muscle glycogen replenishment after exercise is an important factor for optimizing subsequent performance in horses [47]. After exercise, the feeding of a high soluble carbohydrate diet (such as grains and sweet feeds) hastens glycogen synthesis by increasing glucose availability and insulin release. Similar to other species, insulin release activates protein phosphatase, which converts glycogen synthase from its inactive form (D) to its active form (I) and inhibits glycogenolytic enzymes such as phosphorylase a (Figure 3). Since glucose is the required substrate for glycogen synthesis, insulin release also activates GLUT4 translocation to the plasma membrane to enhance glucose uptake [47, 50]. However, in contrast to other species, in which complete synthesis of the muscle glycogen pool takes 24 h, it requires 48–72 h after exhaustive exercise in horses, even after feeding a high soluble carbohydrate diet [67]. It has been proposed that the mechanisms underlying the slow rate of glycogen replenishment after exercise in horses may include limited ability for the small intestine to digest starch and slower activity of the muscle glycogen synthase enzyme compared with values reported in humans and rodents [68, 69]. In addition, there is evidence that glucose transport across the sarcolemma is the rate-limiting step for glucose uptake and subsequent glycogen synthesis in equine skeletal muscle [47, 68]. For instance, intravenous glucose infusion after exercise enhanced muscle glycogen synthesis but failed to enhance protein expression of the insulin-responsive GLUT4 in muscle 24 hours after a strenuous exercise [64]. Similarly, starch-rich meals failed to enhance GLUT4 gene expression, GLUT4 protein content, and muscle glycogen concentrations within the first 24 h after strenuous exercise [67, 70]. Finally, it was reported that in vitro insulin stimulation minimally increased GLUT4 translocation in the equine skeletal muscle, with the increase only being ~15% even after supraphysiological insulin concentrations in healthy ponies [54, 71]. These changes are much less than the ones reported in skeletal muscle of healthy human after physiological (80%) and supra-physiological (400%) insulin stimulation [71, 72]. To further evaluate in vivo glucose transport in horses, researchers used the euglycemic hyperinsulinemic clamp technique since the induced supraphysiological plasma insulin concentrations produce a maximal response of glucose uptake in insulin-sensitive tissue while inhibiting endogenous hepatic glucose production [2]. Interestingly, both short-term (i.e., 6 h) and long-term (i.e., 48 h) exogenous hyperinsulinemia in healthy horses did not affect the expression of GLUT4 gene and protein in equine skeletal muscle [18, 73]. Therefore, the observed lack of substantial increase in GLUT4 expression and/or translocation in healthy skeletal muscle in response to both in vivo and in vitro insulin stimulation may be the result of the insulin-dependent signaling pathways regulating GLUTs already being at near maximal physiologic limits for the species [54]. Alternatively, it has also been suggested that the relatively high resting muscle glycogen concentrations typical of the equine species may partially prevent further GLUT4 translocation to the plasma membrane via a negative feedback pathway [64]. Taken together, these studies demonstrated that glucose uptake across the sarcolemma is the rate-limiting step in glucose utilization and could be a major mechanism for the slow rate of muscle glycogen replenishment observed after exercise in this species [50].
Figure 3.
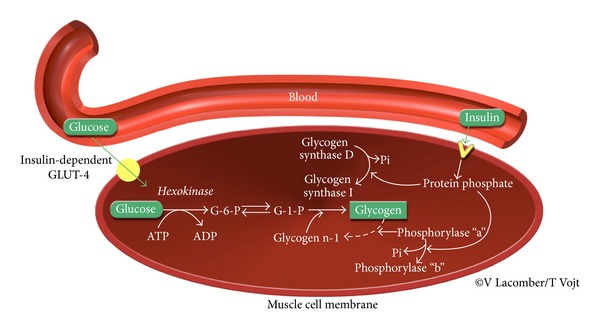
Biochemical pathways underlying glucose uptake and glycogen synthesis in the skeletal muscle after a high soluble carbohydrate diet. Insulin (1) activates GLUT4 translocation to enhance glucose uptake; (2) activates protein phosphatase, which converts glycogen synthase from its inactive form (D) to its active form (I); and (3) inhibits glycogenolytic enzymes such as phosphorylase a. G-6-P: glucose-6-phosphate; G-1-P: glucose-1-phosphate. Modified from [47].
The central role of adipose tissue glucose transport in regulating whole-body insulin sensitivity is becoming increasingly evident in mammalians, as adipose tissue-selective reduction of GLUT4 in mice results in impaired glucose tolerance and peripheral IR [74]. In addition, human omental adipocytes have higher GLUT4 expression and basal- and insulin-stimulated glucose uptake rate [75]. Similar to humans, GLUT4 protein expression is higher in visceral compared to subcutaneous adipose sites and skeletal muscle of insulin-sensitive horses, with the highest content present in the omental site [28]. Since GLUT4 is abundant in visceral adipose tissue, it is therefore likely to play a substantial role in the regulation of visceral glucose transport and in the maintenance of peripheral insulin sensitivity. Overall, the greater ability of insulin to promote glucose uptake in visceral compared with subcutaneous fat might be explained by higher levels of insulin receptors and downstream intermediates in the former fat depot, which could also explain the higher insulin-responsive GLUT content in this depot [76]. In support of these findings, GLUT12, a novel potential second insulin-responsive GLUT, has a higher protein content in visceral compared to subcutaneous adipose tissue and skeletal muscle in healthy horses [28]. Collectively, these findings suggest a central role for visceral GLUT-mediated glucose transport in the maintenance of peripheral insulin sensitivity in healthy horses.
2. Glucose Transporters: Implications for Metabolic Dysregulation
Although increased insulin sensitivity has been reported with equine polysaccharide storage myopathy and equine motor neuron disease, changes in GLUT expression and/or trafficking have not been reported [21, 77]. In contrast, impaired glucose uptake appears to play a key pathogenic role during insulin resistance (IR) and other related metabolic diseases in many species, including horses, as outlined below.
2.1. Dysregulation of Glucose Transport: Parallels between Equine Metabolic Syndrome and Type 2 Diabetes in Humans
There is an emerging and growing threat as well as considerable health issues associated with insulin resistance, diabetes, and obesity, which are primary health concerns for humans and affect a wide range of economically important domesticated and/or agricultural species [78–81]. In humans, the World Health Organization has declared diabetes an epidemic disease, with worldwide prevalence expected to rise to 438 million by 2030 and projects that the number of diabetes-related deaths will increase by more than 50% worldwide in the next 10 years [82]. Prior to the development of type 2 diabetes, patients almost always suffer from “prediabetes,” which is characterized by impaired glucose tolerance, increased fasting glucose (with values above the normal range but below the cutoff for the diagnosis of diabetes, 100 to 125 mg/dL), or both [83]. Development of diabetes from normal glucose tolerance is a continuous process, which may last for over a decade. Over 300 million people suffer from this preclinical stage of diabetes and this figure is expected to rise to 472 million worldwide by 2030 [83]. Insulin resistance is a core component of the preclinical stages of type 2 diabetes (i.e., “prediabetes”) and is characterized by an impaired ability of insulin to stimulate glucose uptake into muscle and adipose tissue in all species. Insulin resistance first leads to compensatory oversecretion of insulin by the pancreatic β-cells and, eventually, β-cell exhaustion and development of type 2 diabetes. In addition, IR in the liver results in failure to suppress glucose production and release into the blood, further worsening the hyperglycemia. Although the development of diabetes is rarely reported in horses [84], there have been increased clinical awareness and recognition of chronic IR without marked hyperglycemia [85–88]. In addition, equine metabolic syndrome phenotype has been defined as a general clustering of obesity, IR, hyperinsulinemia, hypertriglyceridemia, and an increased risk for laminitis [85–88]. Affected patients are adult horses of both genders, from a wide range of breeds, types, and performance levels, living in distinct parts of the country [85, 88–90]. With increasing awareness, there is increasing diagnosis. Although the true prevalence of IR is not known, it has been estimated to be 10–28% (based on hyperinsulinemia) when large populations of adult horses have been studied [91]. It is anticipated that the prevalence of animals at metabolic risk of developing IR is even greater. Although there is limited work on biomarkers or panels of risk factors, environmental and physical risk factors for equine IR have been identified. Diet appears to be one important factor, as significant parallels exist between the provision of refined, nutritionally dense rations to horses and the consumption of energy dense refined foods by people in modern society [92]. In addition, increasing age, pregnancy/maternal insulin sensitivity, and obesity are also associated with equine IR [92–96]. As in other domesticated animals (e.g., dogs and cats), obesity is prevalent in horses and reported to be 19% and 32% in two independent studies of at least 300 adult horses [86]. Similar to humans, obesity promotes IR in horses, although IR occurs in nonobese equines as well [95, 96].
Although there has been increased clinical recognition of IR and diabetes, a clear understanding of the pathophysiology of these insidious diseases is still missing in horses and in other species. Cellular defects associated with IR can include reduced insulin receptor number, reduced expression and activation of insulin signaling cascade proteins (e.g., PI3K), and an impaired glucose transport process [54, 97]. Importantly, alterations in GLUT4 translocation appear to be a key pathogenic factor underlying IR in multiple species, including in horses. For example, decreased insulin-stimulated GLUT4 translocation in skeletal muscle is a hallmark of IR associated with type 2 diabetes in humans and rodents [98, 99]. Although there have been limited studies in horses, it was recently demonstrated that naturally occurring IR decreases translocation of GLUT4 to the cell surface in equine skeletal muscle, without a change in its total protein expression [54]. It is worthy to note that these horses exhibited early stage IR (with normal baseline blood glucose concentrations), suggesting that the observed alteration in muscle glucose transport is an early pathogenic event [54].
In many species, it is becoming increasingly apparent that adipose tissue plays a key pathogenic role in the overall development of IR [74]. Chronic elevation of serum free fatty acid concentrations, as observed in many humans with obesity or diabetes and horses with Cushing's or metabolic syndrome, may also contribute to the decreased uptake of glucose into peripheral tissues. It is well accepted that preferential deposition of fat into visceral deposits instead of subcutaneous deposits increases the risk of IR and diabetes in humans [100]. The “Portal Theory” suggests that IR and many of its related features could arise from visceral adipose tissue delivering free fatty acids at a high rate to the liver via the portal vein into which visceral adipose tissue directly drains. This in turn increases hepatic glucose production, reduces hepatic insulin clearance, and ultimately leads to hyperinsulinemia and IR [50]. In addition, the development of obesity-induced IR may be caused by visceral fat being more sensitive to increase in adipocyte cell size during expansion of adiposity, which may result in a relative decrease in cellular GLUT4 content as cell size increases [28, 50]. In contrast, in horses, adiposity and expansion of fat deposits in the crest and tailhead regions are associated with IR [50]. However, the importance of these regional fat sites to the pathogenesis of IR and glucose transport in horses has not been well investigated. One study reported a decrease in GLUT4 translocation in both visceral and subcutaneous adipose tissue in insulin-resistant compared to insulin-sensitive horses, suggesting an impairment of glucose transport in both of these adipose tissues [28]. Similarly, GLUT4 protein expression was lower in visceral adipose tissue of type 2 diabetic humans [101] and to a lesser extent in subcutaneous adipose tissue [102].
Since AS160 is a downstream insulin signaling protein, it could be a potential mechanism for the observed alteration in GLUT4 translocation to the cell surface during metabolic diseases. However, IR did not induce alterations in total nor phosphorylated AS160 in skeletal and adipose tissue of horses, suggesting that alterations in glucose transport occurred despite normal AS160 activation [28, 54]. Similarly, the role of AS160 in the pathogenesis of IR has not been well established in other species [6].
Overall, the early pathophysiological mechanisms underlying the long latency period preceding overt diabetes remain largely undefined in humans. Interestingly, horses are frequently affected by chronic IR and rarely decompensate to diabetes. As such, this species could provide a unique naturally occurring animal model to elucidate the chronic, pathological changes accompanying the transition from normal to impaired glucose tolerance. However, it should be pointed out that, because of its evolution as a grazing, hindgut-fermenting herbivore, the horse presents some unique features in regard to its metabolism [103]. For example, the horse derives ~30% of its energy from volatile fatty acids produced in the cecum, and acetate (the main volatile fatty acid produced in the hindgut) could account for 30% of the energy utilization by the limb of healthy horses during periods of rest [104–106]. Nonetheless, horses with chronic IR present some similarities in regard to metabolic dysregulation with human patients affected by the preclinical stages of type 2 diabetes, both at the whole tissue and molecular levels.
2.2. Dysregulated Glucose Transport and Inflammation: Potential Novel Link and Pathogenic Factor
Numerous studies in humans and animal models of metabolic diseases have demonstrated that obesity induces an inflammatory state, which plays a key pathogenic role during IR [107, 108]. With the development of obesity, adipose tissue is reported in humans and rodents to adopt a distinct inflammatory phenotype, characterized by increased gene expression and secretion of proinflammatory cytokines, such as tumor necrosis factor (TNF-α) and interleukins (IL)-1 and -6 [108]. In response to the increased proinflammatory cytokines (i.e., TNF-α), there is an increased production of suppressors of cytokine signaling (SOCS) proteins, which have been suggested to play a crucial link between inflammation and IR in the liver [109–111]. However, the feedback regulation of SOCS-3 is complex, and there are conflicting reports describing its relation with other components of the inflammatory signaling pathways, including toll-like receptors (TLR) [112], and ultimately the pathways linking inflammation to altered glucose transport remain elusive in all species [113].
It is well known that intravenous administration of endotoxin, which activates TNF-α and TLR, impairs insulin sensitivity in horses [114]. However, the role of inflammation during IR in horses has received scarce attention and findings remain controversial. It was reported that peripheral blood cells of obese hyperinsulinemic horses showed decreased endogenous proinflammatory cytokine gene expression [115]. In contrast, a correlative relationship was demonstrated between biomarkers of inflammation in the blood (i.e., TNF-α and IL-1) and obesity and IR in horses, as well as laminitis [116–118]. Dr. Burns and colleagues reported that subcutaneous depot (nuchal ligament) might be more likely to adopt an inflammatory phenotype than other adipose depots [119]. Furthermore, a significant increase in TNF-α protein expression in visceral but not subcutaneous tissue was reported in horses with compensated IR, suggesting a role for inflammation during IR [120].
Recent studies suggested that toll-like receptor (TLR) could be a key mechanism by which impaired fatty acid metabolism induces and propagates inflammation, and ultimately decreases insulin signaling and glucose transport [121]. TLRs recognize pathogen-associated molecular patterns and are of primary importance in the innate and adaptive immune system. TLR stimulation also leads to inflammation and cytokine production [121]. Although lipopolysaccharide is the key activator of TLR4 signaling, fatty acids have also been shown to be an endogenous ligand for TLR4 [122]. Activation of TLR4 signaling in adipocytes could activate JNK and NFkB signaling leading to increased production of proinflammatory cytokines, which in turn increases SOCS-3 production. Increased SOCS-3 in muscle and adipose tissue leads to decreased signal transduction through serine phosphorylation of IRS-1 and thus impairs the insulin signaling pathway, which ultimately results in dysregulation of glucose transport and homeostasis [122]. Therefore, TLR4, which is highly expressed in insulin-sensitive tissues, has been linked with the pathogenesis of IR [123, 124]. Recently, it has been shown that activation of TLR4, which is also highly expressed in striated muscle and adipose tissue of horses, was associated with an increased SOCS3 production during naturally occurring equine IR [120]. In addition, TLR4 signaling was activated in the digital lamellae of an experimental model of induced hyperinsulinemia, suggesting that inflammation contributes to the development of endocrinopathic laminitis [125]. Collectively, these findings suggest that TLR4 is a pathogenic marker during equine IR and/or metabolic dysfunction. However, further studies are required to elucidate the role of inflammation during dysregulated glucose metabolism in horses.
2.3. Complications Associated with Insulin Resistance: Potential Role of Dysregulated Glucose Transport
In humans, it is well accepted that IR even without overt diabetes can lead to multiorgan dysfunction and increased mortality [126, 127]. Evidence suggests that a similar state of inflammatory and multiple tissue complications occurs in insulin-resistant horses [85, 87]. For instance, IR is thought to be involved not only in the pathogenesis of equine metabolic syndrome and diabetes mellitus, but also in the pathogenesis of pars intermedia dysfunction, hyperlipemia, endotoxemia, osteoarthritis and osteochondrosis dissecans [19, 128]. Importantly, IR is well known to be a direct and independent risk factor for a painful, debilitating peripheral vascular limb disease in horses, namely, laminitis, with some similarities to peripheral vascular disease and ischemic necrosis of the foot in human diabetic patients [85, 87, 88]. The prognosis and outcomes for horses with laminitis are poor, varying from 8 to 20% dying or being euthanized after 1 year, and it has been suggested that hyperinsulinemic laminitis is ultimately not reversible [129]. Therefore, a better understanding of the early pathophysiological effects could improve our capacity to predict and prevent this debilitating disease. Although the proposed mechanisms involved in the pathogenesis of laminitis are numerous and interrelated, IR is at the forefront of current investigations [130–133]. To this end, the development of experimental models of hyperinsulinemia-induced laminitis in the horse has provided further insights into the pathogenesis of endocrinopathic laminitis, especially as it relates to the altered glucose metabolism [27, 131, 134]. This is particularly germane as the regulation of glucose transport in the lamellae in the healthy and laminitic horse remains scarce and controversial. Although GLUT1 has been consistently reported within lamellar keratinocytes, its expression during laminitis is variable [17, 18, 30]. An immunohistochemical study demonstrated loss of lamellar GLUT1 during chronic laminitis [29]. In contrast, lamellar GLUT1 gene and protein expression was unaffected in horses and ponies treated with a prolonged supraphysiologic infusion of insulin (which induces laminitis) suggesting that endocrinopathic laminitis is unlikely due to impaired glucose uptake and subsequent glucose deprivation in lamellae [17, 18]. Interestingly, it has been recently reported that both novel GLUT-8 and -12 (members of class III) proteins were upregulated in the equine lamellae following prolonged insulin infusion [18]. Interestingly, it has been recently suggested that GLUT8 mediates the deleterious metabolic effects associated with insulin resistance in mice fed a high-fructose diet [135]. However, it remains to be determined whether glucotoxicity contributes to the development of hyperinsulinemic laminitis in horses. In addition, it is not well established whether insulin resistance is required for the induction of endocrinopathic laminitis. Therefore, further studies are required to elucidate the pathogenesis of endocrinopathic laminitis, including additional potential mechanisms such as oxidative stress and vascular perturbations to the digit.
To further understand the impact of impaired glucose transport during diabetes, researchers have focused their attention on mechanisms by which chronic hyperglycemia induced micro- and macrovascular damages. Four main hypotheses have been proposed: (1) increased polyol pathway flux, (2) activation of protein kinase C isoforms, (3) increased hexosamine pathway flux, and (4) increased advanced glycation end products (AGEs) [136]. Importantly, chronic hyperglycemia accelerates the reaction between glucose and proteins and leads to the formation of advanced glycation end products, which form irreversible cross-links throughout the lifetime of many large proteins (such as collagen and hemoglobin), covalently modifying their structure and function [137–139]. In diabetes, and to a lesser extent during aging, AGEs accumulate at an accelerated rate in various cell types (in days to weeks) and cause multiple organ dysfunction [137, 139]. Once AGEs are formed, they are irreversible. At the organ level, AGEs stimulate collagen deposition, which ultimately leads to increased tissue fibrosis [137–141]. AGEs can also initiate alterations in cellular function by binding to a specific receptor known as the receptor for advanced glycation end products (RAGE), which is found on the surface of many cells, including macrophages, epithelial and endothelial cells, and smooth muscle cells. Although there is evidence that RAGE activation could occur early during the pathophysiological process, the role of RAGE and AGEs during metabolic dysfunction in horses has received scarce attention, in contrast to the extensive studies performed in human and rodents. Recently, one study reported a modest accumulation of AGE in the lamellar tissue of horses after 48 h of hyperinsulinemia-induced laminitis, suggesting a potential pathogenic role [142].
3. Glucose Transporters: Potential Therapeutic Targets
Since GLUTs are key regulators of whole-body glucose homeostasis during physiologic and pathophysiologic states, they have received considerable attention as potential therapeutic targets to treat metabolic disorders in humans, such as during diabetes and IR. However, despite the increasing clinical awareness, research on new pharmacologic approaches for equine IR is limited and effective insulin-sensitizing strategies are lacking.
As in humans, careful dietary management and increasing physical activity are suggested in horses to promote insulin sensitivity [86]. However, the difficulty of encouraging exercise and weight loss in a recurrently laminitic horse can make the management very challenging or even be contraindicated. Therefore, there is a need for pharmacological intervention in horses, and levothyroxine sodium (i.e., T4) has been prescribed to induce weight loss and to improve insulin sensitivity [143]. In other species, it has been shown that thyroid hormone increases glucose uptake in insulin-sensitive tissue, in part by the activation of GLUTs [144]. In addition, veterinarians have increasingly prescribed the off-label use of human medications, such as metformin, a biguanide [145]. Although the main mechanism of action of metformin is through the suppression of hepatic gluconeogenesis, its spectrum of action is wide and remains not completely understood [146]. Importantly, metformin activates AMPK, which induces GLUT4 translocation to the cell surface, and thereby increases insulin sensitivity by enhancing peripheral glucose uptake. However, no clinical effect was observed with oral doses of 11–16 mg/kg/day [147], and either no effect or only modest increases in insulin sensitivity were observed in insulin-resistant horses following 30 mg/kg/day oral metformin [145, 148]. In addition, long-term results appear to be variable, and possible adverse drug effects have been reported [147, 148]. This lack of prolonged therapeutic effect could be in part explained by the poor bioavailability of metformin in horses (36–52% lower than in humans) and thus, chronic dosing may not achieve therapeutic blood concentrations [146, 149]. Recently, pioglitazone, an FDA-approved thiazolidinedione to treat type 2 diabetes, has been evaluated in healthy horses. It is absorbed after oral administration without any clinically detectable adverse effects or any abnormalities in hematological and biochemical analyses [150]. Its mechanism of action relies on the activation of peroxisome proliferators-activated receptor gamma (i.e., PPAR-γ agonist). PPAR-γ, which is expressed in adipose tissue and muscle, is an intracellular transcription factor that controls the transcription of genes regulating glucose homeostasis [151, 152]. In particular, pioglitazone increases the efficiency of insulin-stimulated glucose uptake from blood into insulin-sensitive tissue by targeting GLUT proteins [153]. This increase in glucose uptake following pioglitazone treatment would in turn increase whole-body insulin sensitivity. In addition, pioglitazone possesses some anti-inflammatory properties including reduction of inflammatory markers (i.e., IL6, TNF-α), as well as prevention of SOCS-3 upregulation in the liver of insulin-resistant rodents fed a high fat/fructose diet [109], although similar effects have not been demonstrated in horses yet [154]. Pioglitazone could become an affordable therapeutic option for IR in horses since a generic preparation is going to be available soon [150]. However, only a few studies have investigated its therapeutic efficacy in horses. Although twelve days of pioglitazone treatment (1 mg/kg bodyweight PO) did not improve basal insulin-sensitivity, an increased skeletal muscle tissue transcript abundance of GLUT1 and insulin receptor was reported [155]. Therefore, further investigation of different dosing regimens of pioglitazone and metformin might be warranted in horses [146], as well as the investigation of alternative insulin-sensitizing strategies.
4. Conclusions
Glucose uptake is the rate-limiting step in whole-body glucose utilization and is tightly regulated by a family of specialized proteins, the glucose transporters. Insulin resistance is characterized by an impaired ability of insulin to stimulate glucose uptake into insulin sensitive tissues (i.e., muscle and adipose tissue) in all species. As for other species, the functional capability of the insulin-responsive glucose transporter GLUT4 is impaired in skeletal muscle and adipose tissue during naturally occurring IR in horses. Therefore, these insulin-responsive GLUTs provide molecular tool to further understand the pathophysiology of metabolic diseases across species and constitute potential therapeutic targets for equine patients affected by IR. However, despite intensive research for over 50 years, the molecular mechanisms of altered glucose transport remain elusive, and further research is required to characterize impairments of the downstream signaling pathways regulating GLUT4 translocation. Importantly, there is still much to learn about the physiological and pathophysiological functions of the GLUT family member, especially in regard to class III, and such knowledge will be essential in understanding the pathogenesis of IR and metabolic syndrome. Finally, future studies could also investigate novel physical, nutritional, and/or therapeutic strategies to activate GLUTs, which may be beneficial for both healthy equine athletes and horses with IR.
Acknowledgments
The author is grateful to Tim Vojt for his excellent assistance with the medical illustrations, as well as to members of her laboratory, in particular Dr. Amanda Waller and Dr. Melody de Laat, for valuable discussions. This work was supported by grants from the United States Equestrian Federation Equine Health Research Fund, the Animal Health Foundation, and the National Institutes of Health (K01RR023083).
Abbreviations
- GLUT:
Glucose transporter
- IR:
Insulin resistance
- PI 3-kinase:
Phosphatidylinositide 3-kinase
- PIP3:
Phosphatidylinositol 3,4,5 trisphosphate
- Ca2+:
Calcium
- AMPK:
AMP-activated protein kinase
- IRS:
Insulin receptor substrate
- AGE:
Advanced glycation end products
- TNF:
Tumor necrosis factor
- IL:
Interleukins
- TLR:
Toll-like receptor
- SOCS:
Suppressors of cytokine signaling proteins
- PPAR-γ:
Peroxisome proliferators-activated receptor gamma
- G-6-P:
Glucose-6-phosphate
- G-1-P:
Glucose-1-phosphate
- PDK1:
Phosphoinositide-dependent kinase 1.
Conflict of Interests
The author declares that there is no conflict of interests regarding the publication of this paper.
References
- 1.Thorens B, Mueckler M. Glucose transporters in the 21st Century. The American Journal of Physiology—Endocrinology and Metabolism. 2010;298(2):E141–E145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Kolk JH, Rijnen KEPM, Rey F, de Graaf-Roelfsema E, Grinwis GCM, Wijnberg ID. Evaluation of glucose metabolism in three horses with lower motor neuron degeneration. American Journal of Veterinary Research. 2005;66(2):271–276. doi: 10.2460/ajvr.2005.66.271. [DOI] [PubMed] [Google Scholar]
- 3.Grueter CE, Van Rooij E, Johnson BA, et al. A cardiac MicroRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149(3):671–683. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiological Reviews. 2005;85(3):1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 5.Schwenk RW, Luiken JJFP, Bonen A, Glatz JFC. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovascular Research. 2008;79(2):249–258. doi: 10.1093/cvr/cvn116. [DOI] [PubMed] [Google Scholar]
- 6.Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annual Review of Biochemistry. 2012;81:507–532. doi: 10.1146/annurev-biochem-060109-094246. [DOI] [PubMed] [Google Scholar]
- 7.Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Molecular Aspects of Medicine. 2013;34(2-3):121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao F-Q, Keating AF. Expression and regulation of glucose transporters in the bovine mammary gland. Journal of Dairy Science. 2007;90:E76–E86. doi: 10.3168/jds.2006-470. [DOI] [PubMed] [Google Scholar]
- 9.Dyer J, Fernandez-Castaño Merediz E, Salmon KSH, Proudman CJ, Edwards GB, Shirazi-Beechey SP. Molecular characterisation of carbohydrate digestion and absorption in equine small intestine. Equine Veterinary Journal. 2002;34(4):349–358. doi: 10.2746/042516402776249209. [DOI] [PubMed] [Google Scholar]
- 10.Dyer J, Al-Rammahi M, Waterfall L, et al. Adaptive response of equine intestinal Na+/glucose co-transporter (SGLT1) to an increase in dietary soluble carbohydrate. Pflugers Archiv European Journal of Physiology. 2009;458(2):419–430. doi: 10.1007/s00424-008-0620-4. [DOI] [PubMed] [Google Scholar]
- 11.Pao SS, Paulsen IT, Saier MH., Jr. Major facilitator superfamily. Microbiology and Molecular Biology Reviews. 1998;62(1):1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt S, Joost H-G, Schürmann A. GLUT8, the enigmatic intracellular hexose transporter. The American Journal of Physiology—Endocrinology and Metabolism. 2009;296(4):E614–E618. doi: 10.1152/ajpendo.91019.2008. [DOI] [PubMed] [Google Scholar]
- 13.Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. The American Journal of Physiology—Endocrinology and Metabolism. 2008;295(2):E227–E237. doi: 10.1152/ajpendo.90245.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joost H-G, Thorens B. The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members. Molecular Membrane Biology. 2001;18(4):247–256. doi: 10.1080/09687680110090456. [DOI] [PubMed] [Google Scholar]
- 15.Karnieli E, Armoni M. Transcriptional regulation of the insulin-responsive glucose transporter GLUT4 gene: from physiology to pathology. The American Journal of Physiology—Endocrinology and Metabolism. 2008;295(1):E38–E45. doi: 10.1152/ajpendo.90306.2008. [DOI] [PubMed] [Google Scholar]
- 16.Arai T, Washizu T, Sagara M, et al. D-glucose transport and glycolytic enzyme activities in erythrocytes of dogs, pigs, cats, horses, cattle and sheep. Research in Veterinary Science. 1995;58(2):195–196. doi: 10.1016/0034-5288(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 17.Asplin KE, Curlewis JD, Mcgowan CM, Pollitt CC, Sillence MN. Glucose transport in the equine hoof. Equine Veterinary Journal. 2011;43(2):196–201. doi: 10.1111/j.2042-3306.2010.00127.x. [DOI] [PubMed] [Google Scholar]
- 18.de Laat CKC MA, Pollitt CC, McGowan CM, Sillence MN, Lacombe VA. Regulation of glucose transport during equine hyperinsulinaemia. Proceeding of the American College of Veterinary Internal Medecine Forum; 2013. [Google Scholar]
- 19.Mobasheri A, Vannucci SJ, Bondy CA, et al. Glucose transport and metabolism in chondrocytes: a key to understanding chondrogenesis, skeletal development and cartilage degradation in osteoarthritis. Histology and Histopathology. 2002;17(4):1239–1267. doi: 10.14670/HH-17.1239. [DOI] [PubMed] [Google Scholar]
- 20.Phillips T, Ferraz I, Bell S, Clegg PD, Carter SD, Mobasheri A. Differential regulation of the GLUT1 and GLUT3 glucose transporters by growth factors and pro-inflammatory cytokines in equine articular chondrocytes. Veterinary Journal. 2005;169(2):216–222. doi: 10.1016/j.tvjl.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 21.Benders NA, Dyer J, Wijnberg ID, Shirazi-Beechey SP, van der Kolk JH. Evaluation of glucose tolerance and intestinal luminal membrane glucose transporter function in horses with equine motor neuron disease. American Journal of Veterinary Research. 2005;66(1):93–99. doi: 10.2460/ajvr.2005.66.93. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez-Castaño Merediz E, Dyer J, Salmon KSH, Shirazi-Beechey SP. Molecular characterisation of fructose transport in equine small intestine. Equine Veterinary Journal. 2004;36(6):532–538. doi: 10.2746/0425164044877378. [DOI] [PubMed] [Google Scholar]
- 23.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nature Reviews Molecular Cell Biology. 2002;3(4):267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 24.Zisman A, Peroni OD, Abel ED, et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nature Medicine. 2000;6(8):924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]
- 25.Nout YS, Hinchcliff KW, Jose-Cunilleras E, Dearth LR, Sivko GS, DeWille JW. Effect of moderate exercise immediately followed by induced hyperglycemia on gene expression and content of the glucose transporter-4 protein in skeletal muscles of horses. American Journal of Veterinary Research. 2003;64(11):1401–1408. doi: 10.2460/ajvr.2003.64.1401. [DOI] [PubMed] [Google Scholar]
- 26.Jose-Cunilleras E, Hayes KA, Toribio RE, Mathes LE, Hinchcliff KW. Expression of equine glucose transporter type 4 in skeletal muscle after glycogen-depleting exercise. American Journal of Veterinary Research. 2005;66(3):379–385. doi: 10.2460/ajvr.2005.66.379. [DOI] [PubMed] [Google Scholar]
- 27.van Dam KG, van Breda E, Schaart G, et al. Investigation of the expression and localization of glucose transporter 4 and fatty acid translocase/CD36 in equine skeletel muscle. American Journal of Veterinary Research. 2004;65(7):951–956. doi: 10.2460/ajvr.2004.65.951. [DOI] [PubMed] [Google Scholar]
- 28.Waller AP, Kohler K, Burns TA, Mudge MC, Belknap JK, Lacombe VA. Naturally occurring compensated insulin resistance selectively alters glucose transporters in visceral and subcutaneous adipose tissues without change in AS160 activation. Biochimica et Biophysica Acta. 2011;1812(9):1098–1103. doi: 10.1016/j.bbadis.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mobasheri A, Critchlow K, Clegg PD, Carter SD, Canessa CM. Chronic equine laminitis is characterised by loss of GLUT1, GLUT4 and ENaC positive laminar keratinocytes. Equine Veterinary Journal. 2004;36(3):248–254. doi: 10.2746/0425164044877224. [DOI] [PubMed] [Google Scholar]
- 30.Wattle O, Pollitt CC. Lamellar metabolism. Clinical Techniques in Equine Practice. 2004;3(1):22–33. [Google Scholar]
- 31.Katz EB, Stenbit AE, Hatton K, DePinho R, Charron MJ. Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature. 1995;377(6545):151–155. doi: 10.1038/377151a0. [DOI] [PubMed] [Google Scholar]
- 32.D’Cruz SC, Jubendradass R, Mathur PP. Bisphenol a induces oxidative stress and decreases levels of insulin receptor substrate 2 and glucose transporter 8 in rat testis. Reproductive Sciences. 2012;19(2):163–172. doi: 10.1177/1933719111415547. [DOI] [PubMed] [Google Scholar]
- 33.Dawson PA, Mychaleckyj JC, Fossey SC, Mihic SJ, Craddock AL, Bowden DW. Sequence and functional analysis of GLUT10: a glucose transporter in the type 2 diabetes-linked region of chromosome 20q12–13.1. Molecular Genetics and Metabolism. 2001;74(1-2):186–199. doi: 10.1006/mgme.2001.3212. [DOI] [PubMed] [Google Scholar]
- 34.Gómez O, Ballester-Lurbe B, Mesonero JE, Terrado J. Glucose transporters GLUT4 and GLUT8 are upregulated after facial nerve axotomy in adult mice. Journal of Anatomy. 2011;219(4):525–530. doi: 10.1111/j.1469-7580.2011.01410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheepers A, Joost H-G, Schürmann A. The glucose transporter families SGLT and GLUT: molecular basis of normal and aberrant function. Journal of Parenteral and Enteral Nutrition. 2004;28(5):364–371. doi: 10.1177/0148607104028005364. [DOI] [PubMed] [Google Scholar]
- 36.Purcell SH, Aerni-Flessner LB, Willcockson AR, Diggs-Andrews KA, Fisher SJ, Moley KH. Improved insulin sensitivity by GLUT12 overexpression in mice. Diabetes. 2011;60(5):1478–1482. doi: 10.2337/db11-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waller AP, George M, Kalyanasundaram A, et al. GLUT12 functions as a basal and insulin-independent glucose transporter in the heart. Biochimica et Biophysica Acta-Molecular Basis of Disease. 2013;1832(1):121–127. doi: 10.1016/j.bbadis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 38.Aerni-Flessner L, Abi-Jaoude M, Koenig A, Payne M, Hruz PW. GLUT4, GLUT1, and GLUT8 are the dominant GLUT transcripts expressed in the murine left ventricle. Cardiovascular Diabetology. 2012;11(article 63) doi: 10.1186/1475-2840-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carayannopoulos MO, Chi MM-Y, Cui Y, et al. GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(13):7313–7318. doi: 10.1073/pnas.97.13.7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brozinick JT, Jr., Etgen GJ, Jr., Yaspelkis BB, III, Ivy JL. The effects of muscle contraction and insulin on glucose-transporter translocation in rat skeletal muscle. Biochemical Journal. 1994;297(3):539–545. doi: 10.1042/bj2970539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodyear LJ, Hirshman MF, Horton ES. Exercise-induced translocation of skeletal muscle glucose transporters. The American Journal of Physiology—Endocrinology and Metabolism. 1991;261(6):E795–E799. doi: 10.1152/ajpendo.1991.261.6.E795. [DOI] [PubMed] [Google Scholar]
- 42.Hayashi T, Wojtaszewski JFP, Goodyear LJ. Exercise regulation of glucose transport in skeletal muscle. The American Journal of Physiology—Endocrinology and Metabolism. 1997;273(6):E1039–E1051. doi: 10.1152/ajpendo.1997.273.6.E1039. [DOI] [PubMed] [Google Scholar]
- 43.Kolter T, Uphues I, Wichelhaus A, Reinauer H, Eckel J. Contraction-induced translocation of the glucose transporter Glut4 in isolated ventricular cardiomyocytes. Biochemical and Biophysical Research Communications. 1992;189(2):1207–1214. doi: 10.1016/0006-291x(92)92333-s. [DOI] [PubMed] [Google Scholar]
- 44.Till M, Kolter T, Eckel J. Molecular mechanisms of contraction-induced translocation of GLUT4 in isolated cardiomyocytes. American Journal of Cardiology. 1997;80(3):85A–89A. doi: 10.1016/s0002-9149(97)00461-x. [DOI] [PubMed] [Google Scholar]
- 45.Till M, Ouwens DM, Kessler A, Eckel J. Molecular mechanisms of contraction-regulated cardiac glucose transport. Biochemical Journal. 2000;346(3):841–847. [PMC free article] [PubMed] [Google Scholar]
- 46.Zorzano A, Sevilla L, Camps M, et al. Regulation of glucose transport, and glucose transporters expression and trafficking in the heart: studies in cardiac myocytes. American Journal of Cardiology. 1997;80(3, Supplement 1):65A–76A. doi: 10.1016/s0002-9149(97)00459-1. [DOI] [PubMed] [Google Scholar]
- 47.Lacombe VA, Hinchcliff KW, Taylor LE. Interactions of substrate availability, exercise performance, and nutrition with muscle glycogen metabolism in horses. Journal of the American Veterinary Medical Association. 2003;223(11):1576–1585. doi: 10.2460/javma.2003.223.1576. [DOI] [PubMed] [Google Scholar]
- 48.Klip A. The many ways to regulate glucose transporter 4. Applied Physiology, Nutrition and Metabolism. 2009;34(3):481–487. doi: 10.1139/H09-047. [DOI] [PubMed] [Google Scholar]
- 49.Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. Journal of Biological Chemistry. 1980;255(10):4758–4762. [PubMed] [Google Scholar]
- 50.Lacombe VA. Glucose metabolism in insulin-sensitive tissue: from health to disease (vol 31, pg 578, 2011) Journal of Equine Veterinary Science. 2012;32(2):116–116. [Google Scholar]
- 51.Larance M, Ramm G, Stöckli J, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. Journal of Biological Chemistry. 2005;280(45):37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 52.Kramer HF, Witczak CA, Fujii N, et al. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006;55(7):2067–2076. doi: 10.2337/db06-0150. [DOI] [PubMed] [Google Scholar]
- 53.Kramer HF, Taylor EB, Witczak CA, Fujii N, Hirshman MF, Goodyear LJ. Calmodulin-binding domain of AS160 regulates contraction- but not insulin-stimulated glucose uptake in skeletal muscle. Diabetes. 2007;56(12):2854–2862. doi: 10.2337/db07-0681. [DOI] [PubMed] [Google Scholar]
- 54.Waller AP, Burns TA, Mudge MC, Belknap JK, Lacombe VA. Insulin resistance selectively alters cell-surface glucose transporters but not their total protein expression in equine skeletal muscle. Journal of Veterinary Internal Medicine. 2011;25(2):315–321. doi: 10.1111/j.1939-1676.2010.0674.x. [DOI] [PubMed] [Google Scholar]
- 55.Talior-Volodarsky I, Randhawa VK, Zaid H, Klip A. α-actinin-4 is selectively required for insulin-induced GLUT4 translocation. Journal of Biological Chemistry. 2008;283(37):25115–25123. doi: 10.1074/jbc.M801750200. [DOI] [PubMed] [Google Scholar]
- 56.Huang G, Ge G, Wang D, et al. α3(V) Collagen is critical for glucose homeostasis in mice due to effects in pancreatic islets and peripheral tissues. Journal of Clinical Investigation. 2011;121(2):769–783. doi: 10.1172/JCI45096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Douen AG, Ramlal T, Cartee GD, Klip A. Exercise modulates the insulin-induced translocation of glucose transporters in rat skeletal muscle. The FEBS Letters. 1990;261(2):256–260. doi: 10.1016/0014-5793(90)80566-2. [DOI] [PubMed] [Google Scholar]
- 58.Wright DC, Geiger PC, Holloszy JO, Han D-H. Contraction- and hypoxia-stimulated glucose transport is mediated by a Ca2+-dependent mechanism in slow-twitch rat soleus muscle. The American Journal of Physiology—Endocrinology and Metabolism. 2005;288(6):E1062–E1066. doi: 10.1152/ajpendo.00561.2004. [DOI] [PubMed] [Google Scholar]
- 59.Holloszy JO, Narahara HT. Enhanced permeability to sugar associated with muscle contraction. Studies of the role of Ca++ . Journal of General Physiology. 1967;50(3):551–562. doi: 10.1085/jgp.50.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Youn JH, Gulve EA, Holloszy JO. Calcium stimulates glucose transport in skeletal muscle by a pathway independent of contraction. The American Journal of Physiology—Cell Physiology. 1991;260(3):C555–C561. doi: 10.1152/ajpcell.1991.260.3.C555. [DOI] [PubMed] [Google Scholar]
- 61.Wright DC, Fick CA, Olesen JB, Lim K, Barnes BR, Craig BW. A role for calcium/calmodulin kinase in insulin stimulated glucose transport. Life Sciences. 2004;74(7):815–825. doi: 10.1016/j.lfs.2003.06.041. [DOI] [PubMed] [Google Scholar]
- 62.Jessen N, Goodyear LJ. Invited review: contraction signaling to glucose transport in skeletal muscle. Journal of Applied Physiology. 2005;99(1):330–337. doi: 10.1152/japplphysiol.00175.2005. [DOI] [PubMed] [Google Scholar]
- 63.Treebak JT, Taylor EB, Witczak CA, et al. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. The American Journal of Physiology—Cell Physiology. 2010;298(2):C377–C385. doi: 10.1152/ajpcell.00297.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lacombe VA, Hinchcliff KW, Devor ST. Effects of exercise and glucose administration on content of insulin-sensitive glucose transporter in equine skeletal muscle. American Journal of Veterinary Research. 2003;64(12):1500–1506. doi: 10.2460/ajvr.2003.64.1500. [DOI] [PubMed] [Google Scholar]
- 65.Robinson MA, Liu Y, Uboh CE, Soma LR. Exercise increases plasma AICAR (5-amino-4-imidazolecarboxamide riboside) in the horse. The FASEB Journal. 2012;26 [Google Scholar]
- 66.Lacombe VA, Hinchcliff KW, Geor RJ, Baskin CR. Muscle glycogen depletion and subsequent replenishment affect anaerobic capacity of horses. Journal of Applied Physiology. 2001;91(4):1782–1790. doi: 10.1152/jappl.2001.91.4.1782. [DOI] [PubMed] [Google Scholar]
- 67.Lacombe VA, Hinchcliff KW, Kohn CW, Devor ST, Taylor LE. Effects of feeding meals with various soluble-carbohydrate content on muscle glycogen synthesis after exercise in horses. American Journal of Veterinary Research. 2004;65(7):916–923. doi: 10.2460/ajvr.2004.65.916. [DOI] [PubMed] [Google Scholar]
- 68.Votion DM, Navet R, Lacombe VA, et al. Muscle energetics in exercising horses. Equine and Comparative Exercise Physiology. 2007;4(3/4):105–118. [Google Scholar]
- 69.Ivy JL, Kuo C-H. Regulation of GLUT4 protein and glycogen synthase during muscle glycogen synthesis after exercise. Acta Physiologica Scandinavica. 1998;162(3):295–304. doi: 10.1046/j.1365-201X.1998.0302e.x. [DOI] [PubMed] [Google Scholar]
- 70.Jose-Cunilleras E, Hinchcliff KW, Lacombe VA, et al. Ingestion of starch-rich meals after exercise increases glucose kinetics but fails to enhance muscle glycogen replenishment in horses. Veterinary Journal. 2006;171(3):468–477. doi: 10.1016/j.tvjl.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 71.Duehlmeier R, Hacker A, Widdel-Bigdely A, Engelhardt WV, Sallmann H-P. Insulin stimulates GLUT4 translocation in the semitendinosus muscle of Shetland ponies. Veterinary Journal. 2010;184(2):176–181. doi: 10.1016/j.tvjl.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 72.Ryder JW, Yang J, Galuska D, et al. Use of a novel impermeable biotinylated photolabeling reagent to assess insulin- and hypoxia-stimulated cell surface GLUT4 content in skeletal muscle from type 2 diabetic patients. Diabetes. 2000;49(4):647–654. doi: 10.2337/diabetes.49.4.647. [DOI] [PubMed] [Google Scholar]
- 73.Suagee JK, Corl BA, Hulver MW, McCutcheon LJ, Geor RJ. Effects of hyperinsulinemia on glucose and lipid transporter expression in insulin-sensitive horses. Domestic Animal Endocrinology. 2011;40(3):173–181. doi: 10.1016/j.domaniend.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 74.Abel ED, Peroni O, Kim JK, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409(6821):729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 75.Virtanen KA, Lönnroth P, Parkkola R, et al. Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. Journal of Clinical Endocrinology and Metabolism. 2002;87(8):3902–3910. doi: 10.1210/jcem.87.8.8761. [DOI] [PubMed] [Google Scholar]
- 76.MacLaren R, Cui W, Simard S, Cianflone K. Influence of obesity and insulin sensitivity on insulin signaling genes in human omental and subcutaneous adipose tissue. Journal of Lipid Research. 2008;49(2):308–323. doi: 10.1194/jlr.M700199-JLR200. [DOI] [PubMed] [Google Scholar]
- 77.Annandale EJ, Valberg SJ, Mickelson JR, Seaquist ER. Insulin sensitivity and skeletal muscle glucose transport in horses with equine polysaccharide storage myopathy. Neuromuscular Disorders. 2004;14(10):666–674. doi: 10.1016/j.nmd.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 78.Chagas LM, Lucy MC, Back PJ, et al. Insulin resistance in divergent strains of Holstein-Friesian dairy cows offered fresh pasture and increasing amounts of concentrate in early lactation. Journal of Dairy Science. 2009;92(1):216–222. doi: 10.3168/jds.2008-1329. [DOI] [PubMed] [Google Scholar]
- 79.Ohtsuka H, Koiwa M, Hatsugaya A, et al. Relationship between serum TNF activity and insulin resistance in dairy cows affected with naturally occurring fatty liver. Journal of Veterinary Medical Science. 2001;63(9):1021–1025. doi: 10.1292/jvms.63.1021. [DOI] [PubMed] [Google Scholar]
- 80.Feldhahn JR, Rand JS, Martin G. Insulin sensitivity in normal and diabetic cats. Journal of Feline Medicine and Surgery. 1999;1(2):107–115. doi: 10.1016/S1098-612X(99)90067-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.German AJ, Hervera M, Hunter L, et al. Improvement in insulin resistance and reduction in plasma inflammatory adipokines after weight loss in obese dogs. Domestic Animal Endocrinology. 2009;37(4):214–226. doi: 10.1016/j.domaniend.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 82.Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus—present and future perspectives. Nature Reviews Endocrinology. 2012;8(4):228–236. doi: 10.1038/nrendo.2011.183. [DOI] [PubMed] [Google Scholar]
- 83.Tabak AG, Herder C, Rathmann W, Brunner EJ, Kivimaki M. Prediabetes: a high-risk state for diabetes development. The Lancet. 2012;379(9833):2279–2290. doi: 10.1016/S0140-6736(12)60283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Durham AE, Hughes KJ, Cottle HJ, Rendle DI, Boston RC. Type 2 diabetes mellitus with pancreatic beta cell dysfunction in 3 horses confirmed with minimal model analysis. Equine Veterinary Journal. 2009;41(9):924–929. doi: 10.2746/042516409x452152. [DOI] [PubMed] [Google Scholar]
- 85.Frank N, Geor RJ, Bailey SR, Durham AE, Johnson PJ. Equine metabolic syndrome. Journal of Veterinary Internal Medicine. 2010;24(3):467–475. doi: 10.1111/j.1939-1676.2010.0503.x. [DOI] [PubMed] [Google Scholar]
- 86.Geor RJ, Harris P. Dietary management of obesity and insulin resistance: countering risk for laminitis. Veterinary Clinics of North America. 2009;25(1):51–65. doi: 10.1016/j.cveq.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 87.Geor R, Frank N. Metabolic syndrome-From human organ disease to laminar failure in equids. Veterinary Immunology and Immunopathology. 2009;129(3-4):151–154. doi: 10.1016/j.vetimm.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 88.Johnson PJ, Wiedmeyer CE, LaCarrubba A, Seshu Ganjam VK, Messer NT. Laminitis and the equine metabolic syndrome. Veterinary Clinics of North America. 2010;26(2):239–255. doi: 10.1016/j.cveq.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 89.Frank N, Elliott SB, Brandt LE, Keisler DH. Physical characteristics, blood hormone concentrations, and plasma lipid concentrations in obese horses with insulin resistance. Journal of the American Veterinary Medical Association. 2006;228(9):1383–1390. doi: 10.2460/javma.228.9.1383. [DOI] [PubMed] [Google Scholar]
- 90.Treiber KH, Kronfeld DS, Hess TM, Byrd BM, Splan RK, Staniar WB. Evaluation of genetic and metabolic predispositions and nutritional risk factors for pasture-associated laminitis in ponies. Journal of the American Veterinary Medical Association. 2006;228(10):1538–1545. doi: 10.2460/javma.228.10.1538. [DOI] [PubMed] [Google Scholar]
- 91.Geor RJ, Thatcher CD, Pleasant RS, Elvinger F, Gay L, Werre SR. Prevalence of hyperinsulinemia in mature horses: relationship to adiposity. Journal of Veterinary Internal Medicine. 2007;21(3):p. 601. [Google Scholar]
- 92.Hoffman RM, Boston RC, Stefanovski D, Kronfeld DS, Harris PA. Obesity and diet affect glucose dynamics and insulin sensitivity in Thoroughbred geldings. Journal of Animal Science. 2003;81(9):2333–2342. doi: 10.2527/2003.8192333x. [DOI] [PubMed] [Google Scholar]
- 93.George LA, Staniar WB, Treiber KH, Harris PA, Geor RJ. Insulin sensitivity and glucose dynamics during pre-weaning foal development and in response to maternal diet composition. Domestic Animal Endocrinology. 2009;37(1):23–29. doi: 10.1016/j.domaniend.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 94.Ralston SL, Nockels CF, Squires EL. Differences in diagnostic test results and hematologic data between aged and young horses. American Journal of Veterinary Research. 1988;49(8):1387–1392. [PubMed] [Google Scholar]
- 95.Bailey SR, Habershon-Butcher JL, Ransom KJ, Elliott J, Menzies-Gow NJ. Hypertension and insulin resistance in a mixed-breed population of ponies predisposed to laminitis. American Journal of Veterinary Research. 2008;69(1):122–129. doi: 10.2460/ajvr.69.1.122. [DOI] [PubMed] [Google Scholar]
- 96.Bailey SR, Menzies-Gow NJ, Harris PA, et al. Effect of dietary fructans and dexamethasone administration on the insulin response of ponies predisposed to laminitis. Journal of the American Veterinary Medical Association. 2007;231(9):1365–1373. doi: 10.2460/javma.231.9.1365. [DOI] [PubMed] [Google Scholar]
- 97.Kitamura Y, Accili D. New insights into the integrated physiology of insulin action. Reviews in Endocrine and Metabolic Disorders. 2004;5(2):143–149. doi: 10.1023/B:REMD.0000021436.91347.93. [DOI] [PubMed] [Google Scholar]
- 98.Handberg A, Vaag A, Damsbo P, Beck-Nielsen H, Vinten J. Expression of insulin regulatable glucose transporters in skeletal muscle from Type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1990;33(10):625–627. doi: 10.1007/BF00400207. [DOI] [PubMed] [Google Scholar]
- 99.Zierath JR, Houseknecht KL, Gnudi L, Kahn BB. High-fat feeding impairs insulin-stimulated GLUT4 recruitment via an early insulin-signaling defect. Diabetes. 1997;46(2):215–223. doi: 10.2337/diab.46.2.215. [DOI] [PubMed] [Google Scholar]
- 100.Cartwright MJ, Tchkonia T, Kirkland JL. Aging in adipocytes: potential impact of inherent, depot-specific mechanisms. Experimental Gerontology. 2007;42(6):463–471. doi: 10.1016/j.exger.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garvey WT, Maianu L, Zhu J-H, Hancock JA, Golichowski AM. Multiple defects in the adipocyte glucose transport system cause cellular insulin resistance in gestational diabetes: heterogeneity in the number and a novel abnormality in subcellular localization of GLUT4 glucose transporters. Diabetes. 1993;42(12):1773–1785. doi: 10.2337/diab.42.12.1773. [DOI] [PubMed] [Google Scholar]
- 102.Maianu L, Keller SR, Garvey WT. Adipocytes exhibit abnormal subcellular distribution and translocation of vesicles containing glucose transporter 4 and insulin-regulated aminopeptidase in type 2 diabetes mellitus: implications regarding defects in vesicle trafficking. Journal of Clinical Endocrinology and Metabolism. 2001;86(11):5450–5456. doi: 10.1210/jcem.86.11.8053. [DOI] [PubMed] [Google Scholar]
- 103.Glinsky MJ, Smith RM, Spires HR, Davis CL. Measurement of volatile fatty acid production rates in the cecum of the pony. Journal of Animal Science. 1976;42(6):1465–1470. doi: 10.2527/jas1976.4261465x. [DOI] [PubMed] [Google Scholar]
- 104.Pethick DW, Rose RJ, Bryden WL, Gooden JM. Nutrient utilisation by the hindlimb of thoroughbred horses at rest. Equine Veterinary Journal. 1993;25(1):41–44. doi: 10.1111/j.2042-3306.1993.tb02899.x. [DOI] [PubMed] [Google Scholar]
- 105.Moore-Colyer MJS, Hyslop JJ, Longland AC, Cuddeford D. Intra-caecal fermentation parameters in ponies fed botanically diverse fibre-based diets. Animal Feed Science and Technology. 2000;84(3-4):183–197. [Google Scholar]
- 106.Hintz HF, Argenzio RA, Schryver HF. Digestion coefficients, blood glucose levels and molar percentage of volatile acids in intestinal fluid of ponies fed varying forage-grain ratios. Journal of Animal Science. 1971;33(5):992–995. doi: 10.2527/jas1971.335992x. [DOI] [PubMed] [Google Scholar]
- 107.Clément K, Viguerie N, Poitou C, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. The FASEB Journal. 2004;18(14):1657–1669. doi: 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- 108.Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. Journal of Clinical Endocrinology and Metabolism. 2008;93(11, supplement 1):S64–S73. doi: 10.1210/jc.2008-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Collino M, Aragno M, Castiglia S, et al. Pioglitazone improves lipid and insulin levels in overweight rats on a high cholesterol and fructose diet by decreasing hepatic inflammation. British Journal of Pharmacology. 2010;160(8):1892–1902. doi: 10.1111/j.1476-5381.2010.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Farrell GC. Signalling links in the liver: knitting SOCS with fat and inflammation. Journal of Hepatology. 2005;43(1):193–196. doi: 10.1016/j.jhep.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 111.Ueki K, Kondo T, Tseng Y-H, Kahn CR. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(28):10422–10427. doi: 10.1073/pnas.0402511101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu L, Gu H, Zhao Y, Yu F. Study on SOCS-3, PPARgamma and ACO mRNA expression of adipose tissues in obese rats with leptin resistance. Wei Sheng Yan Jiu. 2008;37(1):40–42. [PubMed] [Google Scholar]
- 113.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nature Reviews Immunology. 2007;7(6):454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 114.Tóth F, Frank N, Elliott SB, Geor RJ, Boston RC. Effects of an intravenous endotoxin challenge on glucose and insulin dynamics in horses. American Journal of Veterinary Research. 2008;69(1):82–88. doi: 10.2460/ajvr.69.1.82. [DOI] [PubMed] [Google Scholar]
- 115.Holbrook TC, Tipton T, McFarlane D. Neutrophil and cytokine dysregulation in hyperinsulinemic obese horses. Veterinary Immunology and Immunopathology. 2012;145(1-2):283–289. doi: 10.1016/j.vetimm.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 116.Vick MM, Adams AA, Murphy BA, et al. Relationships among inflammatory cytokines, obesity, and insulin sensitivity in the horse. Journal of Animal Science. 2007;85(5):1144–1155. doi: 10.2527/jas.2006-673. [DOI] [PubMed] [Google Scholar]
- 117.Carter RA, Treiber KH, Geor RJ, Douglass L, Harris PA. Prediction of incipient pasture-associated laminitis from hyperinsulinaemia, hyperleptinaemia and generalised and localised obesity in a cohort of ponies. Equine Veterinary Journal. 2009;41(2):171–178. doi: 10.2746/042516408x342975. [DOI] [PubMed] [Google Scholar]
- 118.Treiber K, Carter R, Gay L, Williams C, Geor R. Inflammatory and redox status of ponies with a history of pasture-associated laminitis. Veterinary Immunology and Immunopathology. 2009;129(3-4):216–220. doi: 10.1016/j.vetimm.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 119.Burns TA, Geor RJ, Mudge MC, McCutcheon LJ, Hinchcliff KW, Belknap JK. Proinflammatory cytokine and chemokine gene expression profiles in subcutaneous and visceral adipose tissue depots of insulin-resistant and insulin-sensitive light breed horses. Journal of Veterinary Internal Medicine. 2010;24(4):932–939. doi: 10.1111/j.1939-1676.2010.0551.x. [DOI] [PubMed] [Google Scholar]
- 120.Waller AP, Huettner L, Kohler K, Lacombe VA. Novel link between inflammation and impaired glucose transport during equine insulin resistance. Veterinary Immunology and Immunopathology. 2012;149(3-4):208–215. doi: 10.1016/j.vetimm.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 121.Könner AC, Brüning JC. Toll-like receptors: linking inflammation to metabolism. Trends in Endocrinology and Metabolism. 2011;22(1):16–23. doi: 10.1016/j.tem.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 122.Kim JK. Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metabolism. 2006;4(6):417–419. doi: 10.1016/j.cmet.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 123.Suganami T, Mieda T, Itoh M, Shimoda Y, Kamei Y, Ogawa Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochemical and Biophysical Research Communications. 2007;354(1):45–49. doi: 10.1016/j.bbrc.2006.12.190. [DOI] [PubMed] [Google Scholar]
- 124.Tsukumo DML, Carvalho-Filho MA, Carvalheira JBC, et al. Loss-of-function mutation in toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56(8):1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 125.de Laat MA, Clement CK, McGowan CM, Sillence MN, Pollitt CC, Lacombe VA. Toll-like receptor and pro-inflammatory cytokine expression during prolonged hyperinsulinaemia in horses: implications for laminitis. Veterinary Immunology and Immunopathology. 2014;157(1-2):78–86. doi: 10.1016/j.vetimm.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 126.Meigs JB, Nathan DM, D’Agostino RB, Sr., Wilson PWF. Fasting and postchallenge glycemia and cardiovascular disease risk: the framingham offspring study. Diabetes Care. 2002;25(10):1845–1850. doi: 10.2337/diacare.25.10.1845. [DOI] [PubMed] [Google Scholar]
- 127.Sorkin JD, Muller DC, Fleg JL, Andres R. The relation of fasting and 2-h postchallenge plasma glucose concentrations to mortality: data from the Baltimore Longitudinal Study of Aging with a critical review of the literature. Diabetes Care. 2005;28(11):2626–2632. doi: 10.2337/diacare.28.11.2626. [DOI] [PubMed] [Google Scholar]
- 128.Firshman AM, Valberg SJ. Factors affecting clinical assessment of insulin sensitivity in horses. Equine Veterinary Journal. 2007;39(6):567–575. doi: 10.2746/042516407X238512. [DOI] [PubMed] [Google Scholar]
- 129.Cripps PJ, Eustace RA. Factors involved in the prognosis of equine laminitis in the UK. Equine Veterinary Journal. 1999;31(5):433–442. doi: 10.1111/j.2042-3306.1999.tb03845.x. [DOI] [PubMed] [Google Scholar]
- 130.Venugopal CS, Eades S, Holmes EP, Beadle RE. Insulin resistance in equine digital vessel rings: an in vitro model to study vascular dysfunction in equine laminitis. Equine Veterinary Journal. 2011;43(6):744–749. doi: 10.1111/j.2042-3306.2010.00351.x. [DOI] [PubMed] [Google Scholar]
- 131.de Laat MA, McGowan CM, Sillence MN, Pollitt CC. Equine laminitis: induced by 48 h hyperinsulinaemia in Standardbred horses. Equine Veterinary Journal. 2010;42(2):129–135. doi: 10.2746/042516409X475779. [DOI] [PubMed] [Google Scholar]
- 132.Walsh DM. Field treatment and management of endocrinopathic laminitis in horses and ponies. Veterinary Clinics of North America. 2010;26(2):379–390. doi: 10.1016/j.cveq.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 133.Asplin KE, Sillence MN, Pollitt CC, McGowan CM. Induction of laminitis by prolonged hyperinsulinaemia in clinically normal ponies. Veterinary Journal. 2007;174(3):530–535. doi: 10.1016/j.tvjl.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 134.de Laat MA, Kyaw-Tanner MT, Nourian AR, McGowan CM, Sillence MN, Pollitt CC. The developmental and acute phases of insulin-induced laminitis involve minimal metalloproteinase activity. Veterinary Immunology and Immunopathology. 2011;140(3-4):275–281. doi: 10.1016/j.vetimm.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 135.Debosch BJ, Chen Z, Finck BN, Chi M, Moley KH. Glucose transporter-8 (GLUT8) mediates glucose intolerance and dyslipidemia in high-fructose diet-fed male mice. Molecular Endocrinology. 2013;27(11) doi: 10.1210/me.2013-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115(25):3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 137.Cooper ME. Importance of advanced glycation end products in diabetes-associated cardiovascular and renal disease. American Journal of Hypertension. 2004;17(12):31S–38S. doi: 10.1016/j.amjhyper.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 138.Liu J, Masurekar MR, Vatner DE, et al. Glycation end-product cross-link breaker reduces collagen and improves cardiac function in aging diabetic heart. The American Journal of Physiology—Heart and Circulatory Physiology. 2003;285(6):H2587–H2591. doi: 10.1152/ajpheart.00516.2003. [DOI] [PubMed] [Google Scholar]
- 139.Kranstuber AL, Del Rio C, Biesiadecki BJ, et al. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Frontiers in Physiology. 2012;3(article 292) doi: 10.3389/fphys.2012.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Asif M, Egan J, Vasan S, et al. An advanced glycation endproduct cross-link breaker can reverse age-related increases in myocardial stiffness. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(6):2809–2813. doi: 10.1073/pnas.040558497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vaitkevicius PV, Lane M, Spurgeon H, et al. A cross-link breaker has sustained effects on arterial and ventricular properties in older rhesus monkeys. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(3):1171–1175. doi: 10.1073/pnas.98.3.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.de Laat MA, Kyaw-Tanner MT, Sillence MN, McGowan CM, Pollitt CC. Advanced glycation endproducts in horses with insulin-induced laminitis. Veterinary Immunology and Immunopathology. 2012;145(1-2):395–401. doi: 10.1016/j.vetimm.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 143.Frank N, Sommardahl CS, Eiler H, Webb LL, Denhart JW, Boston RC. Effects of oral administration of levothyroxine sodium on concentrations of plasma lipids, concentration and composition of very-low-density lipoproteins, and glucose dynamics in healthy adult mares. American Journal of Veterinary Research. 2005;66(6):1032–1038. doi: 10.2460/ajvr.2005.66.1032. [DOI] [PubMed] [Google Scholar]
- 144.Teixeira SS, Tamrakar AK, Goulart-Silva F, Serrano-Nascimento C, Klip A, Nunes MT. Triiodothyronine acutely stimulates glucose transport into l6 muscle cells without increasing surface GLUT4, GLUT1, or GLUT3. Thyroid. 2012;22(7):747–754. doi: 10.1089/thy.2011.0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tinworth KD, Edwards S, Harris PA, Sillence MN, Hackett LP, Noble GK. Pharmacokinetics of metformin after enteral administration in insulin-resistant ponies. American Journal of Veterinary Research. 2010;71(10):1201–1206. doi: 10.2460/ajvr.71.10.1201. [DOI] [PubMed] [Google Scholar]
- 146.Durham AE. Metformin in equine metabolic syndrome: an enigma or a dead duck? Veterinary Journal. 2012;191(1):17–18. doi: 10.1016/j.tvjl.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 147.Vick MM, Sessions DR, Murphy BA, Kennedy EL, Reedy SE, Fitzgerald BP. Obesity is associated with altered metabolic and reproductive activity in the mare: effects of metformin on insulin sensitivity and reproductive cyclicity. Reproduction, Fertility and Development. 2006;18(6):609–617. doi: 10.1071/rd06016. [DOI] [PubMed] [Google Scholar]
- 148.Durham AE, Rendle DI, Newton JR. The effect of metformin on measurements of insulin sensitivity and β cell response in 18 horses and ponies with insulin resistance. Equine Veterinary Journal. 2008;40(5):493–500. doi: 10.2746/042516408X273648. [DOI] [PubMed] [Google Scholar]
- 149.Hustace JL, Firshman AM, Mata JE. Pharmacokinetics and bioavailability of metformin in horses. American Journal of Veterinary Research. 2009;70(5):665–668. doi: 10.2460/ajvr.70.5.665. [DOI] [PubMed] [Google Scholar]
- 150.Wearn JMG, Crisman MV, Davis JL, et al. Pharmacokinetics of pioglitazone after multiple oral dose administration in horses. Journal of Veterinary Pharmacology and Therapeutics. 2011;34(3):252–258. doi: 10.1111/j.1365-2885.2010.01217.x. [DOI] [PubMed] [Google Scholar]
- 151.Ding S-Y, Shen Z-F, Chen Y-T, Sun S-J, Liu Q, Xie M-Z. Pioglitazone can ameliorate insulin resistance in low-dose streptozotocin and high sucrose-fat diet induced obese rats. Acta Pharmacologica Sinica. 2005;26(5):575–580. doi: 10.1111/j.1745-7254.2005.00090.x. [DOI] [PubMed] [Google Scholar]
- 152.Hammarstedt A, Rotter Sopasakis V, Gogg S, Jansson P-A, Smith U. Improved insulin sensitivity and adipose tissue dysregulation after short-term treatment with pioglitazone in non-diabetic, insulin-resistant subjects. Diabetologia. 2005;48(1):96–104. doi: 10.1007/s00125-004-1612-3. [DOI] [PubMed] [Google Scholar]
- 153.El-Kebbi IM, Roser S, Pollet RJ. Regulation of glucose transport by pioglitazone in cultured muscle cells. Metabolism. 1994;43(8):953–958. doi: 10.1016/0026-0495(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 154.Wearn JG, Suagee JK, Crisman MV, et al. Effects of the insulin sensitizing drug, pioglitazone, and lipopolysaccharide administration on markers of systemic inflammation and clinical parameters in horses. Veterinary Immunology and Immunopathology. 2012;145(1-2):42–49. doi: 10.1016/j.vetimm.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 155.Suagee JK, Corl BA, Wearn JG, et al. Effects of the insulin-sensitizing drug pioglitazone and lipopolysaccharide administration on insulin sensitivity in horses. Journal of Veterinary Internal Medicine. 2011;25(2):356–364. doi: 10.1111/j.1939-1676.2011.0681.x. [DOI] [PubMed] [Google Scholar]
- 156.Bucci D, Isani G, Spinaci M, et al. Comparative immunolocalization of GLUTs 1, 2, 3 and 5 in boar, stallion and dog spermatozoa. Reproduction in Domestic Animals. 2010;45(2):315–322. doi: 10.1111/j.1439-0531.2008.01307.x. [DOI] [PubMed] [Google Scholar]