

Abstract
One of the most challenging aspects of RT-qPCR data analysis is the identification of reliable reference genes. Ideally, they should be neither induced nor repressed under different experimental conditions. To date, few reference genes have been adequately studied for sugarcane (Saccharum spp.) using statistical approaches. In this work, six candidate genes (αTUB, GAPDH, H1, SAMDC, UBQ, and 25S rRNA) were tested for gene expression normalization of sugarcane root tissues from drought-tolerant and -sensitive accessions after continuous dehydration (24 h). By undergoing different approaches (GeNorm, NormFinder, and BestKeeper), it was shown that most of them could be used in combinations for normalization purposes, with the exception of SAMDC. Nevertheless three of them (H1, αTUB, and GAPDH) were considered the most reliable reference genes. Their suitability as reference genes validated the expression profiles of two targets (AS and PFPα1), related to SuperSAGE unitags, in agreement with results revealed by previous in silico analysis. The other two sugarcane unitags (ACC oxidase and PIP1-1), after salt stress (100 mM NaCl), presented their expressions validated in the same way. In conclusion, these reference genes will be useful for dissecting gene expression in sugarcane roots under abiotic stress, especially in transcriptomic studies using SuperSAGE or RNAseq approaches.
1. Introduction
Sugarcane (Saccharum spp.) is a major crop with vegetative propagation and capacity to accumulate high levels of sucrose in the culms [1–3]. World production of this crop in 2011 resulted in about two billion tons of raw material, which corresponded to a gross income of more than US$ 52 billion [4]. Despite the economic importance of sugarcane, the knowledge of relevant genetic mechanisms remains challenging, due to the fact that this crop presents one of the largest and most intricate genomes of the plant kingdom, with diploid numbers ranging from 100 to 130 chromosomes, indicating a high ploidy level, as well as regular aneuploidy events [5–7]. Because of this complexity, the use of molecular tools represents an attractive approach to the improvement of sugarcane breeding programs. Moreover, transcriptomic studies have been prioritized, allowing identification of candidate genes involved in developmental processes and plant responses to environmental cues, which have eventually led to the discovery of functional molecular markers [8]. Reverse transcription quantitative real-time PCR (RT-qPCR) is based on a high specific polymerase chain reaction associated with sensitive fluorescence, allowing the detection of variations in gene expression, including discreetly transcribed genes [9, 10]. This technology has been used as a diagnostic tool for identification of plant pathogens, transgene expression [9], and human diseases [11, 12] and confirmation of transcriptional profiles generated by different methodologies, such as EST libraries [13], Microarray [14], HT-SuperSAGE [15], and RNAseq [16]. The reliability of RT-qPCR data based on relative quantification is depending upon comparative transcription of target genes to stable reference genes [17, 18]. The use of reference genes that undergo changes in transcription across experimental groups can dramatically alter the conclusions on targeted gene expression [18]. In order to test for normalization of reference gene expression, several statistical algorithms, such as GeNorm [17], NormFinder [19], and BestKeeper [20], have recently been developed. Despite the importance of proper reference gene selection for reliable and accurate RT-qPCR assays, most reports involving sugarcane have not described, or compared, methods in order to determine the efficiency of reference genes [21–23], suggesting arbitrary criteria for this selection. To our knowledge, only one systematic study aimed to assess, standardize, and validate reference genes (GAPDH, β-tubulin, β-actin, and 25S rRNA) for tissue and genotype-specific gene expression analysis in sugarcane [24]. Additionally, this kind of study has not been carried out before under stress conditions which often alter the behavior of some genes. Thus, a rigorous selection of reference genes for expression profiling validation in sugarcane under biotic or abiotic stress was yet to be described. In the present work, screening and validation of new reliable reference genes for expression analysis in sugarcane roots were carried out. Besides, additional resources for target validation were evaluated, especially considering comprehensive transcription profiling, like those provided by HT-SuperSAGE [25], in sugarcane, revealing hundreds of candidate genes responsive against drought stress, requiring subsequent validation.
2. Materials and Methods
2.1. Plant Material and Treatments
Sugarcane drought assay: accessions were previously evaluated in a glasshouse trial conducted by the Center for Sugarcane Technology (CTC) in Piracicaba, Brazil (22° 41′ S; 47° 33′ W), aiming to identify drought-tolerant and drought-sensitive ones. Four accessions were selected as drought-tolerant (CTC6, CTC15, SP83-2847, and SP83-5073) and another four as drought-sensitive (CTC9, CTC13, SP90-3414, and SP90-1638). Some of them were previously reported as tolerant and sensitive to drought based on chlorophyll and water content measurements together with field observations [28]. Briefly, for the drought stress assay, plants of each accession were grown under glasshouse conditions (30.2 ± 5.7°C (maximum), 16.8 ± 1.9°C (average), and 9.3 ± 3.0°C (minimum) and 71.5 ± 5.1% (average) relative humidity under natural photoperiod) in 40 L pods in a randomized experimental design (comprising six repetitions) under daily irrigation (4 L·h−1) until reaching three months of age. Plants were submitted to drought conditions by continuous dehydration caused by the interruption of irrigation during 24 hours. Roots of both, stressed and unstressed plants, were collected, immediately frozen in liquid nitrogen, and stored at −80°C until total RNA extraction. On the other hand, the salinity stress assay was performed as follows: acclimated plants of the RB931011 clone in vitro cultivated (referred to as salt tolerant by the Brazilian RIDESA program of sugarcane breeding) were grown in a greenhouse (UFPE, Recife, PE, Brazil; 8° 04′S, 34° 55′W) in pots containing washed sand (washed) and they were watered daily with Hoagland solution, throughout three months. Later, NaCl (100 mM) was added to the nutritive solution as the salt stress. Roots from both stressed and nonstressed (negative control) plants were collected after stress induction (30 and 90 min) and immediately frozen in liquid nitrogen until a total RNA extraction was performed.
2.2. Total RNA Isolation, Purification, cDNA Synthesis, and HT-SuperSAGE Libraries
Total RNA was extracted with RNeasy Mini kit (Qiagen) according to the manufacturer's instructions, treated with DNAse (Qiagen), and purified with RNeasy Mini kit (Qiagen). RNA samples were quantified using Quant-iT RNA assay kit (Invitrogen) with the Qubit fluorometer (Invitrogen). RNA integrity was verified in 1.5% agarose gel electrophoresis with blue-green loading dye (LGC Biotechnology) staining. The purified RNA (1 μg) of each sugarcane accession was reverse-transcribed using QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer's instructions and resuspended in a final volume of 20 μL. The cDNA synthesis reaction was incubated at 42°C for 2 minutes (genomic DNA digestion), 42°C for 15 minutes (reverse transcription), and 95°C for 3 minutes (enzyme inactivation) and stored at −20°C. The procedures for HT-SuperSAGE library generation followed Matsumura et al. [29], including the attachment of library-specific adaptors carried out by GenXPro GmbH (Frankfurt, Germany), allowing the identification of library-specific reads after SOLEXA sequencing. Concerning the drought stress, four libraries were generated as described by Kido et al. [25]: the bulk of tolerant accessions under stress and the respective negative control and the bulk of sensitive accessions after stress and the respective negative control. The bulks were composed by equimolar amounts of poly-A+ mRNA from all accessions comprising the respective library. In relation to the salt stress, equimolar amounts of total RNA from each sample/time were assembled to compose the two bulks used to generate the SuperSAGE libraries (stressed and control).
2.3. Primer Design, Amplification Efficiency, and RT-qPCR Analysis
Sugarcane ESTs (Table 1) from dbEST database (http://www.ncbi.nlm.nih.gov/nucest), related to independent pathways as an attempt to minimize the effects of coregulation, were used for primers design with the Primer 3 software [30] with minor modifications: the amplicon length range was set to 70–200 bp, the melting temperatures were between 40°C (minimum), 50°C (optimum), and 60°C (maximum), and the CG content ranged from 45 to 55% (optimum of 50%). In relation to H1 and SAMDC, the primers sequences were obtained from the literature [23]. All primer pairs were synthesized by Bioneer Corporation (South Korea) and some details of these primers are given in Table 1. An initial standard PCR was performed with the potential reference genes, using the sugarcane cDNA samples (bulk of tolerant and sensitive accessions), in order to investigate the PCR products. Amplicons were analyzed on 1.5% agarose gel electrophoresis followed by blue-green loading dye staining (LGC Biotechnology). Additionally, a dissociation curve analysis was made, in RT-qPCR assay, to confirm the specificity of the amplification by the candidate genes. Calibration curves using a dilution series of the cDNA pool (1, 10−1, 10−2, 10−3, and 10−4) were made to calculate the PCR amplification efficiencies (E = 10−1/slope) [27] for each quantified candidate gene, their respective correlation coefficients (R), and y interceptors. The RT-qPCR amplifications were performed on LineGene 9660 model FQD-96A (Bioer), using SYBR Green detection. Each reaction mixture comprised 1 μL of template cDNA (diluted 5-fold), 5 μL of HotStart-IT SYBR Green qPCR Master Mix 2x (USB), 0.05 μL of ROX (normalization dye), 1.95 μL of ultrapure water, and 1 μL of each primer (0.05 μM), forming a final volume of 10 μL. Three biological and three technical replicates were used in each run for RT-qPCR analysis, and a no template control (NTC) was also included. The reactions were subjected to an initial denaturation step of 95°C for 2 min, followed by 40 cycles at 95°C for 15 s, 58°C for 30 s, and 72°C for 30 s in 96-well reaction plates. The dissociation curves were analyzed at 65–95°C for 20 min after 40 cycles. The baseline and quantification cycle (Cq) were determined using the LineGene 9600 software (version 1.1.10).
Table 1.
Potential reference genes and target genes with the individual accession numbers, annotations, and primer sequences.
| Gene (accession numbers) | Source* | Predicted function | Description | Primer sequences (forward/reverse) |
|---|---|---|---|---|
| Reference genes | ||||
|
| ||||
| αTUB (CN607271) | dbEST | Structural constituent of cytoskeleton | Alpha-tubulin | (F) CCATTGGCAAGGAGATTGTT (R) TCCACCAACTGCATTGAAGA |
|
| ||||
| GAPDH (CA254672) | dbEST | Glycolysis, gluconeogenesis | Glyceraldehyde 3 phosphate dehydrogenase | (F) GGTTCACTTGAAGGGTGGTG (R) TGAGGTGTACCTGTCCTCGTT |
|
| ||||
| H1 (CA116806) | [23] | Chromatin condensation | Histone H1 | (F) CGCACACGCACACTGAAAG (R) CGGTGGCCATGATCAAAAA |
|
| ||||
| SAMDC (CA127376) | [23] | Polyamine and triamine biosynthesis | S-Adenosylmethionine decarboxylase | (F) TGCTGCTGAAGACGCTGTTG (R) TCGCCTTCAAAGCAGTGTAGAAC |
|
| ||||
| UBQ (CA077378) | dbEST | Protein degradation | Ubiquitin | (F) ACCGAAGGTTGCATTCAAGAC (R) GGGTTTGGGTCCGTTAGAAG |
|
| ||||
| 25S rRNA (BQ536525) | dbEST | Translation | 25S ribosomal RNA | (F) GCAGCCAAGCGTTCATAG (R) CGGCACGGTCATCAGTAG |
|
| ||||
| Target genes | ||||
|
| ||||
| PFPα1 (XM_004973200.1) | dbEST | Carbohydrate metabolism, glycolysis | Pyrophosphate fructose-6-phosphate 1-phosphotransferase alpha subunit | (F) TTATGAGTTGCTGCGAGAGAAG (R) TATCTCAATGTCGCCCATGTAG |
|
| ||||
| AS (FM212633.1) | dbEST | Asparagine synthesis | Glutamine-dependent asparagine synthetase | (F) CCAGAGAACACACCCACAAC (R) ATGCCACACTAGGACCTCCA |
|
| ||||
| PIP1-1 (CF572112) | dbEST | Integral to membrane, water transport | Plasma membrane intrinsic protein 1-1 | (F) GTTCCTATCCTTGCCCCACT (R) AGGCGTGATCCCTGTTGTAG |
|
| ||||
| ACC oxidase (TC127289) | SoGI | Ethylene biosynthesis | 1-Aminocyclopropane-1-carboxylate oxidase | (F) GGGACCTCTTGCAGATAATGTC (R) CTCTGGCAATGGTCCATAGAA |
*Databases: dbEST (NCBI; http://www.ncbi.nlm.nih.gov/) and Gene Index (SoGI; http://compbio.dfci.harvard.edu/tgi/); Rodrigues et al. [23]. αTUB: alpha-tubulin; GAPDH: glyceraldehyde 3 phosphate dehydrogenase; H1: histone H1; SAMDC: S-adenosylmethionine decarboxylase; UBQ: ubiquitin; 25S rRNA: 25S ribosomal RNA; PFPα1: pyrophosphate fructose-6-phosphate 1-phosphotransferase alpha subunit (EC 2.7.1.90); AS: glutamine-dependent asparagine synthetase (EC 6.3.5.4); PIP1-1: plasma membrane intrinsic protein; ACC oxidase: 1-aminocyclopropane-1-carboxylate oxidase (EC 1.14.17.4).
2.4. Data Analysis
The potential reference genes were ranked, and the number of candidate genes required for an optimal normalization was indicated according to their gene expression stability using sugarcane cDNA samples, after being analyzed by the GeNorm (version 3.5) [17], NormFinder (version 0.953) [19], and BestKeeper (version 1) [20] software. The GeNorm and NormFinder input data were based on relative quantities applying the ΔΔCq method [31]. The GeNorm software determines the reference gene stability measurement (M) as the average pairwise variation of each reference gene with all the other reference genes and enables the elimination of the least stable gene and the recalculation of the M values, resulting in the ranking of the most stable genes. The average expression stability value (M-value) was a parameter for quantification of stable reference gene candidates, in which a low M-value indicated a more stable expression [17]. The NormFinder tool was applied to identify and rank the most suitable genes for RT-qPCR normalization from the set of candidates, considering intragroup and intergroup variations, in a model-based approach of mixed linear effect modeling [19]. The BestKeeper software, using raw Cq values as input, was applied to identify the most stable expressed genes by a Pearson correlation coefficient (geometric mean of Cq values of candidate genes), calculating the standard deviation (SD) of Cq values among the entire data set. The relative gene expression levels (based on the relative quantities after the ΔΔCq method) were evaluated with the REST© tool (Relative Expression Software Tool, version 2.0.13), which bases its performance on pairwise comparisons using randomization and bootstrapping techniques (Pairwise Fixed Reallocation Randomization Test©) [32]. The normalization of the RT-qPCR was performed by taking the geometric averages of the combined reference genes, using the negative control to normalize this relative expression, and testing the hypothesis of significant differences between the control and treatment. With the input of multiple target and reference genes and based on the normalized values of the target genes, the software indicates the direction of the difference between the groups, as well as their P value. Also, the MIQE guidelines (The Minimum Information for Publication of Quantitative Real-Time PCR Experiments) [26] were followed in order to increase transparency and reliability of the results obtained.
3. Results
3.1. RNA Integrity, Specificity, and Efficiency Amplifications
All the reference candidate genes (αTUB, GAPDH, H1, SAMDC, UBQ, and 25S rRNA) amplified the cDNAs generated from the RNAs samples (Supplemental Figure 1(a) in Supplementary Material available online at http://dx.doi.org/10.1155/2014/357052) using the proposed primers. Based on standard PCR amplifications, only a single product was observed with a specific primer pair (Supplemental Figure 1(b)) and these results were supported by the dissociation curve analysis (Supplemental Figure 1(c)). Based on the standard curves using a serial dilution of the cDNA pool (Supplemental Figure 2), the real-time PCR amplification efficiency (E), considering the selected six candidate genes, ranged from 98.34% to 100.89%, with correlation coefficients (R) varying from 0.984 to 0.999, while the slopes ranged from −3.50 to −3.21 (Table 2). Considering the efficiency of 100%, the value of the expected slope should be −3.32, while slopes ranging from −3.10 to −3.58 would represent efficiency comprising 90% to 110%, thereby characterizing acceptable reactions. These parameters derived from the RT-qPCR analysis, and others in accordance with the MIQE Guidelines, are shown in Tables 2 and S2. The results showed favorable conditions for amplification, efficiency in successive dilutions, and acceptable variations in gene expression across samples, representing potential for choosing a suitable reference gene. Thus, in gene expression studies, fluctuations due to pipetting errors, variations in the quantification of samples, or the concentration of reagents could be normalized with the aid of these suitable reference genes [33].
Table 2.
Parameters derived from RT-qPCR data analysis*, including PCR amplification efficiency (E) established by calibration curves for each quantified target.
| Gene | Tm (°C) | Product size (bp) | Average Cq | E (%) | NTC (Cq) | Correlation coefficient (R) | Slope | Y intercept |
|---|---|---|---|---|---|---|---|---|
| αTUB | 75.9 | 104 | 27.48 | 99.53 | 35.82 | 0.998 | −3.33 | 37.34 |
| GAPDH | 81.8 | 100 | 23.69 | 100.89 | N.D. | 0.984 | −3.30 | 41.40 |
| H1 | 78.0 | 57 | 24.76 | 97.41 | 33.59 | 0.999 | −3.39 | 37.92 |
| SAMDC | 79.0 | 60 | 22.74 | 99.66 | 36.62 | 0.992 | −3.33 | 34.36 |
| UBQ | 81.2 | 153 | 24.64 | 98.34 | N.D. | 0.999 | −3.36 | 37.86 |
| 25S rRNA | 82.9 | 108 | 14.00 | 99.82 | N.D. | 0.999 | −3.33 | 35.54 |
| PFPα1 | 83.6 | 151 | 21.92 | 92.99 | 32.90 | 0.988 | −3.50 | 40.44 |
| AS | 79.8 | 112 | 24.94 | 105.05 | N.D. | 0.990 | −3.21 | 40.26 |
| PIP1-1 | 84.6 | 134 | 24.47 | 95.55 | N.D. | 0.995 | −3.43 | 37.50 |
| ACC oxidase | 82.2 | 152 | 30.21 | 93.52 | N.D. | 0.990 | −3.49 | 44.65 |
*Based on MIQE Guidelines [26]. Tm: melting temperature (°C); N.D.: not detected; NTC: no template control; αTUB: alpha-tubulin; GAPDH: glyceraldehyde 3 phosphate dehydrogenase; H1: histone H1; UBQ: ubiquitin; SAMDC: S-adenosylmethionine decarboxylase; 25S rRNA: 25S ribosomal RNA; PFPα1: pyrophosphate fructose-6-phosphate 1-phosphotransferase alpha subunit (EC 2.7.1.90); AS: glutamine-dependent asparagine synthetase (EC 6.3.5.4); PIP1-1: plasma membrane intrinsic protein; ACC oxidase: 1-aminocyclopropane-1-carboxylate oxidase (EC 1.14.17.4).
3.2. Gene Expression Stability of the Reference Gene Candidates
The six candidate genes selected for normalization (αTUB, GAPDH, H1, SAMDC, UBQ, and 25S rRNA) in RT-qPCR tests showed Cq values ranging from 13.06 to 28.00 (Supplemental Table S1). Most of the candidate genes presented Cq values with slight variations (below one cycle), except UBQ and SAMDC. Based on these values, 25S rRNA was the most abundantly transcribed gene (average Cq = 14.00), while αTUB was the least abundant (average Cq = 27.48). These data, in order to assess the gene expression stability of the reference gene candidates, were used in GeNorm [17], NormFinder [19], and BestKeeper [20] analysis.
3.2.1. GeNorm Analysis
Considering the average expression stability values (M-value), αTUB (M = 0.61), GAPDH (M = 0.62), and histone H1 (M = 0.63) were the most stable genes while SAMDC represented the most variable (M = 0.87) gene. However, all of them showed an expressive high stability with M-values below 1 (Figure 1), suggesting that all the six candidates may be adequate for normalizing gene expression data under the conditions used in the present work. Besides, based on the pairwise variation (V) data (Figure 2), it was possible to determine the optimal number of reference genes required for the relative quantification analysis and to investigate the effect of gene addition in this normalization. The data suggested that the addition to the two most stable genes (αTUB and GAPDH) considering a third gene (V 2/3 = 0.15; Figure 2), a fourth (V 3/4 = 0.14), or even more genes (V 4/5 and V 5/6; Figure 2) still exhibited desired values (below 0.15 as proposed by Vandesompele et al. [17]). To normalize the gene expression in the above mentioned sugarcane samples, αTUB, GAPDH, and H1 seem to be sufficient (Figure 1).
Figure 1.
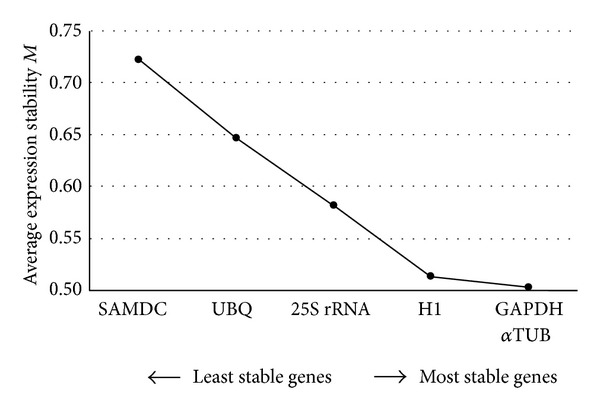
Average gene expression stability values (M) of six sugarcane potential reference genes (αTUB: alpha-tubulin; GAPDH: glyceraldehyde 3 phosphate dehydrogenase; H1: histone H1; SAMDC: S-adenosylmethionine decarboxylase; UBQ: ubiquitin; 25S rRNA: 25S ribosomal RNA) based on the GeNorm analysis [17].
Figure 2.
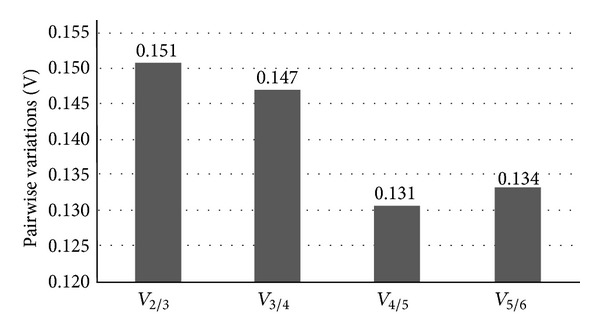
Pairwise variation (V) analysis for six potential reference genes of sugarcane (α-tubulin, glyceraldehyde 3 phosphate dehydrogenase, histone H1, S-adenosylmethionine decarboxylase, ubiquitin, and 25S rRNA) based on the GeNorm analysis [17]. The addition to the two most stable genes (αTUB and GAPDH) of a random third gene (V 2/3 = 0.15), a fourth gene (V 3/4 = 0.14), or even more (V 4/5 and V 5/64) still exhibited desirable values (basically below than 0.15).
3.2.2. NormFinder and BestKeeper Analysis
Basically, the gene expression stability ranking provided by the NormFinder and BestKeeper software exhibited the same order, with only the first two candidates switching places comparing the ranking (Table 3). The two software programs identified histone H1 (M = 0.28; CV ± SD = 1.06 ± 0.26) and αTUB (M = 0.32; CV ± SD = 1.06 ± 0.29) as the most stable genes, followed by GAPDH, 25S rRNA, UBQ, and SAMDC (see respective values in Table 3). Again, SAMDC showed the highest instability, in agreement with the GeNorm results. All the three software programs presented fairly consistent results showing the first three (H1, αTUB, and GAPDH) as the most stable and reliable genes for RT-qPCR data normalization. Two of them (H1 and αTUB) are reported as RT-qPCR normalizing genes suitable for sugarcane roots under abiotic stress for the first time.
Table 3.
Expression stability values for sugarcane candidate calculated by NormFinder and BestKeeper software.
| Ranking | NormFinder analysis | BestKeeper analysis | ||
|---|---|---|---|---|
| Gene name | M | Gene name | CV ± SD | |
| 1 | αTUB | 0.11 | H1 | (1.06 ± 0.26) |
| 2 | H1 | 0.16 | αTUB | (1.06 ± 0.29) |
| 3 | GAPDH | 0.19 | GAPDH | (1.33 ± 0.31) |
| 4 | 25S rRNA | 0.28 | 25S rRNA | (3.89 ± 0.55) |
| 5 | UBQ | 0.31 | UBQ | (2.04 ± 0.50) |
| 6 | SAMDC | 0.33 | SAMDC | (2.53 ± 0.57) |
M: average expression stability value; CV: coefficient of variance; SD: standard deviation; αTUB: alpha-tubulin; H1: histone H1; GAPDH: glyceraldehyde 3 phosphate dehydrogenase; 25S rRNA: 25S ribosomal RNA; UBQ: ubiquitin; SAMDC: S-adenosylmethionine decarboxylase.
3.3. Normalization of Target Genes
In order to assess the applicability of the recommended histone H1, αTUB, and GAPDH as reference genes in relative expression studies using RT-qPCR, four targets based on sugarcane SuperSAGE unitags annotated as glutamine-dependent asparagine synthetase (AS, EC 6.3.5.4), pyrophosphate fructose-6-phosphate 1-phosphotransferase alpha subunit (PFPα1, EC 2.7.1.90), plasma membrane intrinsic protein (PIP1-1), and 1-aminocyclopropane-1-carboxylate oxidase (ACC oxidase, EC 1.14.17.4) were evaluated (Table 4). HT-SuperSAGE survey pointed SD282748 unitag as a potential AS being upregulated 1.92 times in the drought-tolerant bulk after the stress (24 h of continuous dehydration) as compared to the unstressed control while no relevant unitag frequency change was observed (P < 0.05) with the sensitive contrast (Table 4). The RT-qPCR relative quantification results confirmed the overexpressed status in relation to both cDNA bulks with 1.473-fold change for the tolerant bulk compared to its negative control (Figure 3(a)) and no significant change (1.038 times) considering the sensitive bulk in the comparative contrast (Figure 3(b)). In turn, SD179780 unitag (annotated as PFPα1) did not respond (P < 0.05) to water deficit stimulus in any contrast analyzed involving sugarcane drought-tolerant or -sensitive accessions, in agreement with the RT-qPCR results, showing constitutive expression of this gene (Figures 3(a) and 3(b), resp.). In an attempt to explore the use of the proposed reference genes, a target relative to the ASS122537 unitag (ACC oxidase) from the salt HT-SuperSAGE libraries, which was induced (UR) by the salt-tolerant accession (1.95 times after the salt stress exposition, NaCl 100 mM), showed overexpression after 30 min according to RT-qPCR results (Figure 3(c) and Table 4). Another target relative to the ASS140030 unitag (PIP1-1) from sugarcane after salt stress presented RT-qPCR results confirming the constitutive expression observed in silico (P < 0.05), even after the bulk has been opened in two times of salt exposition (30 and 90 min, Figures 3(c) and 3(d) and Table 4). The same PIP1-1 had been validated by RT-qPCR with cDNAs from the drought-tolerant and -sensitive sugarcane accessions (24 h of continuous dehydration) using GAPDH and 25S rRNA as the reference genes, in a previous work, showing differential expressions as expected by the SuperSAGE analysis [34].
Table 4.
Relative expression rates of target genes (PFPα1, AS, PIP1-1, and ACC oxidase) based on RT-qPCR with roots, cDNAs of sugarcane accessions under abiotic stress, and respective unitag regulation by SuperSAGE analysis covering droughta stress (24 h of continuous dehydration) or saltb stress (100 mM NaCl).
| Unitag | Annotation | SuperSAGE [FC/Regulation*] | RT-qPCR∗& | ||
|---|---|---|---|---|---|
| Tolerant | Sensitive | Tolerant | Sensitive | ||
| SD282748a | AS | 1.92#/UR | −1.10#/ns | 1.473#/UR | 1.038#/ns |
| D179780a | PFPα1 | 1.99#/ns | −1.07#/ns | 0.756#/ns | 1.403#/ns |
| ASS122537b | ACC oxidase | 1.95/UR | — | 2.174/UR (30′) 1.830/ns (90′) |
— |
| ASS140030b | PIP1-1 | −1.31/ns | — | 0.805/ns (30′) 1.057/ns (90′) |
— |
AS: glutamine-dependent asparagine synthetase (EC 6.3.5.4); PFPα1: pyrophosphate fructose-6-phosphate 1-phosphotransferase alpha subunit (EC 2.7.1.90); ACC oxidase: 1-aminocyclopropane-1-carboxylate oxidase (EC 1.14.17.4); PIP1-1: plasma membrane intrinsic protein. #Bulk with four accessions by each library; FC: fold change [ratio of the frequencies (normalized to 1,000,000) observed in the stressed library in relation to the control library]; &relative expression level by REST software (v.2.0.13) after the ΔΔCq method, *P < 0.05 [27]; UR: upregulated; ns: not significant at P < 0.05. The time in the parentesis represents the salt stress exposition (min).
Figure 3.
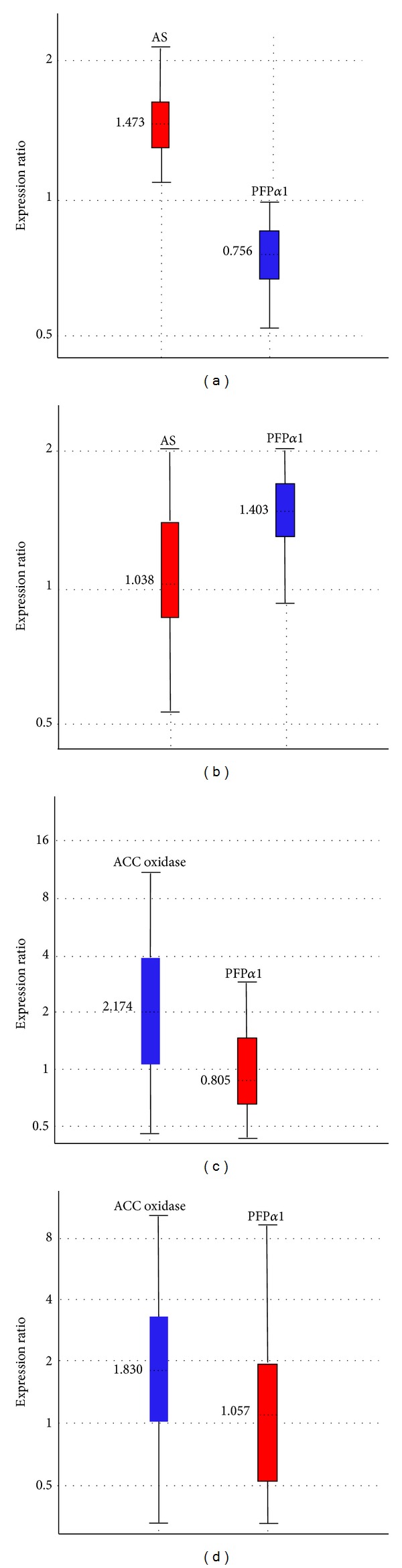
Relative expression of glutamine-dependent asparagine synthetase (AS), pyrophosphate-fructose 6-phosphate 1-phosphotransferase (PFPα1), plasma membrane intrinsic protein 1-1 (PIP1-1), and 1-aminocyclopropane-1-carboxylate oxidase (ACC oxidase) by the REST software v. 2.0.13 (after the ΔΔCq method) in cDNAs of sugarcane roots under abiotic stress (24 h of continuous dehydration or salt stress; 100 mM NaCl), normalized by the reference genes H1, αTUB, and GAPDH. (a) Tolerant bulk (CTC6, CTC15, SP83-2847, and SP83-5073 accessions) compared to its negative control. (b) Susceptible bulk (CTC9, CTC13, SP90-1638, and SP90-3414) compared to its negative control. (c) Salt-tolerant accession (RB931011) after 30 min of salt stress compared to its negative control. (d) RB931011 after 90 min of salt stress compared to its negative control. Relative expression with the median value (horizontal dotted line at the colored box) and range comprising 100% of the observations (horizontal bars), being 50% of them in the confidence interval at 95% (colored box).
4. Discussion
The understanding of sugarcane physiology under environmental stress remains under intensive research, due to the socioeconomic importance of this crop and the increasing unpredictability of environmental conditions worldwide. In this regard, gene expression analysis is an attractive approach to dissect plant physiological response to stress conditions. Nevertheless, reference gene selection has received limited attention in sugarcane. RT-qPCR is currently one of the most used techniques for gene expression analysis, due to its rapid, specific, and highly sensitive parameters. However, problems with RNA samples variations, standardization, and protocols efficiency (RNA extraction, RT, and qPCR) have routinely been observed [35]. Furthermore, the choice of normalizing genes remains one of the most time consuming and difficult steps in RT-qPCR. It requires reference genes to be constitutively expressed under external stimuli. Additionally, it needs to exhibit little or no behavior change in different cell types or tissues, as well as in specific developmental stages and experimental conditions [36–38]. Stal Papini-Terzi et al. [21] described transcriptional profile of signal transduction events in different sugarcane tissues, using reference genes selected based upon the literature (tubulin and actin), microarray data, and ESTs (polyubiquitin and 14-3-3 proteins). To individually normalize gene expression in sugarcane under certain conditions, Rocha et al. [22] relied upon four reference genes (14-3-3, polyubiquitin, GAPDH, and 25S rRNA). Moreover, Rodrigues et al. [23] used β-tubulin as the reference gene based on previous data [24]. To our knowledge, Iskandar et al. [24] represented an attempt to prospect stable sugarcane reference genes by checking the reliability of four genes (β-actin, β-tubulin, GAPDH, and 25S rRNA) in leaf, root, and internode tissues of some sugarcane cultivars and representatives of Saccharum genus, but none of them under abiotic stress. According to the authors, GAPDH was the most stable gene (CV = 51%) comparing different tissues, followed by β-actin and β-tubulin (CV = 81% and 68%, resp.); regarding species, β-actin showed the lowest coefficient of variation (31%) followed by GAPDH (33%). Although these methods are useful for prospecting candidate reference genes [21] or addressing gene expression using validated reference genes for target tissues [22, 23], selection using more appropriate statistical approaches should be the method of choice for identification of new reliable reference genes. In this way, software programs like GeNorm, NormFinder, and BestKeeper have assisted researchers by indicating reference genes suitable for expression profiling normalization studies [39]. In the present study, a group of potential reference genes (αTUB, GAPDH, H1, SAMDC, UBQ, and 25S rRNA) were evaluated by the three software programs, in order to evaluate their reliability for expression profiles normalization in sugarcane roots under abiotic stress (24 h of continuous dehydration). Basically, all three software programs pointed histone H1, αTUB, and GAPDH as the most reliable reference genes, with some of them switching places in the ranking. This set of genes was employed here as reference genes to validate sugarcane cDNAs relative to unitags from SuperSAGE libraries composed of roots of plants after stress exposition. Thus, the gene expression stability ranking provided by NormFinder and BestKeeper software showed the same order after the third place. Besides, based on the GeNorm analysis and M-values, all the six candidates may be suitable for normalizing gene expression data as presented here. But, combining the two best candidates (αTUB and H1) with the most variable one (SAMDC, M = 0.87), as reference genes normalizing the target AS (induced in SuperSAGE analysis), the REST software did not detect the expected overexpression (comparison 1, Table 5) due to the SAMDC largest standard deviation (SD) influencing the P value calculated, consequently leading to a false negative and possible misinterpretation of data. Alias, any other combination including SAMDC as reference gene did not reveal the alleged overexpression (comparisons 2–5, Table 5), indicating that this gene is not suitable for gene expression normalization in roots of sugarcane accessions under the evaluated stress. However, Hong et al. [40] reported SAMDC as the most reliable reference gene in grass Brachypodium distachyon when evaluated under four abiotic stress conditions (high salt (300 mM), cold (4°C for 5 h), heat (42°C for 2 h), and drought (400 mM mannitol)). In addition, Li and Chen [41], when describing SAMDC as a target gene, verified that this gene was induced in roots of rice seedlings at three leaf stages (after application of 171 mM salt for 24 h and 20 mM exogenous abscisic acid (ABA) and dehydration using 15% PEG6000). These results highlight the need to choose appropriate reference genes for each experiment, especially under stress conditions.
Table 5.
Potential reference gene combinations (and number of genes involved in each comparison) used in gene expression normalization of glutamine-dependent asparagine synthetase (AS), with cDNAs of sugarcane accessions (root under drought stress, 24 h of continuous dehydration).
| Comparison | Gene combinations | Number of genes | Expression* value | P value | Regulation |
|---|---|---|---|---|---|
| 1 | αTUB, H1, and SAMDC | 3 | 1.431 | 0.112 | ns |
| 2 | GAPDH, 25S rRNA, and SAMDC | 3 | 1.482 | 0.009 | ns |
| 3 | αTUB, H1, GAPDH, 25S rRNA, and SAMDC | 5 | 1.496 | 0.113 | ns |
| 4 | αTUB, H1, UBQ, and SAMDC | 4 | 1.557 | 0.105 | ns |
| 5 | UBQ and SAMDC | 2 | 1.595 | 0.305 | ns |
| 6 | αTUB and H1 | 2 | 1.519 | 0.000 | UR |
| 7 | GAPDH and 25S rRNA | 2 | 1.600 | 0.017 | UR |
| 8 | αTUB, H1, GAPDH, and 25S rRNA | 4 | 1.559 | 0.011 | UR |
| 9 | αTUB, H1, and UBQ | 3 | 1.667 | 0.033 | UR |
| 10 | GAPDH, 25S rRNA, and UBQ | 3 | 1.725 | 0.017 | UR |
| 11 | αTUB, H1, GAPDH, 25S rRNA, and UBQ | 5 | 1.640 | 0.017 | UR |
*REST software analysis after the ΔΔCq method. UR: upregulated; ns: not significant at P < 0.05; αTUB: alpha-tubulin; H1: histone H1; SAMDC: S-adenosylmethionine decarboxylase; GAPDH: glyceraldehyde 3 phosphate dehydrogenase; 25S rRNA: 25S ribosomal RNA; UBQ: ubiquitin.
On the other hand, the other five genes (αTUB, GAPDH, H1, UBQ, and 25S rRNA) could be successfully employed in the normalization analyses, composing different combinations of reference genes (comparisons 6–11, Table 5), with similar results to that observed for the proposed set (H1, αTUB, and GAPDH). In relation to the 25S rRNA gene, it was the most abundant transcript (Cq 13.06), in agreement with results previously obtained from rice (Cq values of 15 [42]) and sugarcane (Cq values of 16.6 [24]). This can be explained by the fact that rRNA comprises the majority of total RNA present in a cell and, thus, further dilution for its use in RT-qPCR approaches [43] would be required. In the present work, only a 1 : 5 dilution was applied. Furthermore, the abundance of transcripts can affect the stability and, therefore, the normalizing results for the reference gene candidates [44]. In turn, GAPDH was also one of the most stable genes, confirming it to be an appropriate reference gene for experiments involving sugarcane roots under water deficit conditions. Concerning αTUB, it was indicated by GeNorm as the most stable gene using the bulks of accessions, both under regular irrigation and after 24 h of continuous dehydration. By the NormFinder and BestKeeper analysis, this gene was the second most appropriate reference gene. However, Fan et al. [38] assessing the reliability of reference genes in 14 different tissues and developmental stages of Phyllostachys edulis observed that αTUB showed a larger variation (M = 1.94) among all candidates. Similar results were reported by Zhong et al. [45] with litchi (Litchi chinensis Sonn.) under several experimental conditions (tissues, organs, developmental stages, and varieties), showing αTUB as the most variable gene among 10 candidates. Under biotic and abiotic stresses αTUB also displayed instability, as demonstrated by Die et al. [39], Hong et al. [40], and Zhu et al. [46]. Thus, the selection of suitable reference genes to normalize gene expression in sugarcane and other plant species seems to be essential because reference genes may be differently regulated in different species, displaying particular gene expression patterns [43]. The proposed reference genes (H1, αTUB, and GAPDH) validated the gene expression of sugarcane cDNAs related to SuperSAGE unitags, showing upregulation or even constitutive basis, in the mentioned drought assay. Among those targets, AS is a crucial component of the asparagine synthesis, acting as a key member in nitrogen assimilation, recycling, and storage in higher plants [47]. The overexpression results observed in the present study supported those found in a previous microarray [48] showing AS induction in wheat accession considered tolerant to drought after 36 h of irrigation suppression. AS were also detected by RT-PCR, using mRNA samples from roots and shoots (two-week-old plantlets), significantly induced after salinity (250 mM), osmotic stress (using mannitol 5.0% (w/v)), and exogenous abscisic acid (ABA) application (20 mM) [49]. Altogether, these results indicate the involvement of the AS gene in response to several stresses. Considering PFPα1, the constitutive expression observed in the present assay has been reported previously and was in agreement with Lim et al. [50] who demonstrated by semiquantitative RT-PCR that the expression of PFPα1 in wild-type Arabidopsis was also constitutive in different tissues (roots, leaves, and flowers) and also in distinct developmental stages (15, 25, and 45 days after planting). PFPα1 is responsible for the addition of phosphate to the second D-fructose 6-phosphate in the glycolysis pathway [51] and is essential for maintenance of carbohydrate metabolism and other processes in plant cells [50]. In sugarcane, PFPs are known to play a prominent role in sucrose accumulation, especially in immature and metabolically active tissues, taking part in glycolysis and in carbon compartmentalization [52].
The effort to explore the use of the proposed reference genes (H1, αTUB, and GAPDH) normalizing the expressions of sugarcane cDNAs (associated with SuperSAGE unitags) also from roots of plants under salt stress (100 mM) was effective in a preliminary study. This way, the induction of ASS122537 unitag (annotated as ACC oxidase, enzyme responsible to convert the ethylene precursor ACC to ethylene, [53]), as revealed by in silico SuperSAGE analysis, was confirmed by RT-qPCR after 30 minutes of salt exposition (tolerant accession), suggesting that rapid ethylene production is an adaptive response to the new conditions imposed by the salt stress to the evaluated genotype. Unfortunately this overexpression was not detected during the 90 min of stress exposition. Nevertheless, there is evidence that a variety of stressful conditions trigger the synthesis of ethylene [54]. Regarding salt stress, it has been observed that this hormone signaling may be required for triggering the tolerance process. Yang et al. [55], when evaluating Arabidopsis mutants (ein2-5, ein3-1, and ctr1-1) and wild plants ecotype Col-0, found that mutants insensitive to ethylene (ein2-5 or ein3-1) were more sensitive to saline stress when compared to their wild counterpart. The opposite was found in the mutant sensitive to ethylene (ctr1-1), which showed significant tolerance to salt stress. Concerning the PIP1-1, the RT-qPCR results showed no significant differences in any of the two sampled times, confirming the SuperSAGE results with the bulk comprising both sampling times. The same target in RT-qPCR assay confirmed differential expressions expected by the SuperSAGE analysis, with root cDNAs from the drought-tolerant and -sensitive sugarcane accessions (24 h of continuous dehydration) and GAPDH and 25S rRNA as the reference genes [34]. An explanation could be that the time intervals used for stress exposition were not enough for PIP1-1 expression. In rice, Guo et al. [56] reported PIP1-1 expression in response to salt stress (250 mM NaCl) after 2 h of stress exposition.
5. Conclusions
The potential of the six proposed reference genes (αTUB, GAPDH, H1, SAMDC, UBQ, and 25S rRNA) was confirmed after they were tested with cDNAs from sugarcane roots under drought stress (24 h of continuous dehydration) and analyzed by three different software programs (GeNorm, NormFinder, and BestKeeper). With the exception of SAMDC, all the other candidate genes seem to be suitable for sugarcane expression profiling normalization, but three of them (αTUB, H1, and GAPDH) were considered as the best reference genes. In this study, two new reference genes were reported for the first time for sugarcane (αTUB and H1), to undergo a RT-qPCR validation study involving expression in roots under abiotic stresses. Also, the present work pointed GAPDH and 25S rRNA genes, both indicated by Iskandar et al. [24], as reference genes in a previous study, also suitable for use with sugarcane root under abiotic stress. Using the proposed set of reference genes (αTUB, H1, and GAPDH), it was confirmed that the relative expression profile, with the aid of the REST software, of cDNAs was associated with unitags (26 bp) and annotated as AS and PFPα1, using a bulk of cDNAs relative to the drought-tolerant sugarcane accessions (four accessions, 24 h of continuous dehydration), in agreement with the HT-SuperSAGE data. Another two unitags (associated with ACC oxidase and PIP1-1) had their expression profiles validated by RT-qPCR, using cDNAs from sugarcane roots after salt stress exposition (100 mM NaCl), in an attempt to explore other possibilities using these reference genes. In conclusion, this set of reference genes will be useful for dissecting gene expression in sugarcane roots, especially in advanced transcriptomic studies using SuperSAGE or RNAseq approaches covering abiotic stresses.
Supplementary Material
Supplementary Material showing the quality of the total RNAs from sugarcane roots under drought stress (FIGURE S1 - A), the amplicons of expected size for each gene used in the study, displaying primer specificity as required for RT-qPCR amplification (FIGURE S1 - B), and the melting curves showing a single peak for six potential reference genes (αTUB; GAPDH; H1; SAMDC; UBQ and 25S rRNA) and four targets (PFPα1; AS; PIP1-1 and ACC oxidase).
Supplementary Material (FIGURE S2) showing standard curves using a dilution series (100, 10−1, 10−2, 10−3 e 10−4) of six potential reference genes (αTUB; GAPDH; H1; SAMDC; UBQ and 25S rRNA) for sugarcane roots under drought stress and the respective curves for four targets (PFPα1; AS; PIP1-1 and ACC oxidase).
Supplementary Material (TABLE S2) based on MIQE checklist for authors, reviewers, and editors.
Supplementary Material (TABLE S1) showing Cq values of potential reference genes for gene expression normalization (RT-qPCR) with cDNAs from sugarcane roots under drought stress (24 h of continuous dehydration).
Acknowledgments
The authors acknowledge the Brazilian institutions FINEP (Financiadora de Estudos e Projetos), FACEPE (Fundação de Amparo à Ciência e Tecnologia do Estado de Pernambuco), and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) for financial support and fellowships. They also thank Dr. Björn Rotter, Ruth Jungmann, Dr. Peter Winter (GenXPro GmbH, Germany), and Eng. Ernestino Ciambarella (Labtrade do Brasil) for scientific and technical advice.
Conflict of Interests
The authors have declared that they have no conflict of interests.
References
- 1.Welbaum GE, Meinzer FC. Compartmentation of solutes and water in developing sugarcane stalk tissue. Plant Physiology. 1990;93(3):1147–1153. doi: 10.1104/pp.93.3.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rae AL, Grof CPL, Casu RE, Bonnett GD. Sucrose accumulation in the sugarcane stem: pathways and control points for transport and compartmentation. Field Crops Research. 2005;92(2-3):159–168. [Google Scholar]
- 3.Dillon SL, Shapter FM, Henry RJ, Cordeiro G, Izquierdo L, Lee LS. Domestication to crop improvement: genetic resources for Sorghum and Saccharum (Andropogoneae) Annals of Botany. 2007;100(5):975–989. doi: 10.1093/aob/mcm192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.FAOSTAT. FAO Statistical Databases. 2011. Food and Agriculture Organization of the United Nations. http://faostat.fao.org/ [Google Scholar]
- 5.D'Hont A, Grivet L, Feldmann P, Rao S, Berding N, Glaszmann JC. Characterization of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Molecular and General Genetics. 1996;250(4):405–413. doi: 10.1007/BF02174028. [DOI] [PubMed] [Google Scholar]
- 6.Grivet L, Arruda P. Sugarcane genomics: depicting the complex genome of an important tropical crop. Current Opinion in Plant Biology. 2002;5(2):122–127. doi: 10.1016/s1369-5266(02)00234-0. [DOI] [PubMed] [Google Scholar]
- 7.Jannoo N, Grivet L, Chantret N, et al. Orthologous comparison in a gene-rich region among grasses reveals stability in the sugarcane polyploid genome. The Plant Journal. 2007;50(4):574–585. doi: 10.1111/j.1365-313X.2007.03082.x. [DOI] [PubMed] [Google Scholar]
- 8.Manners JM, Casu RE. Transcriptome Analysis and Functional Genomics of Sugarcane. Tropical Plant Biology. 2011;4(1):9–21. [Google Scholar]
- 9.Gachon C, Mingam A, Charrier B. Real-time PCR: what relevance to plant studies? Journal of Experimental Botany. 2004;55(402):1445–1454. doi: 10.1093/jxb/erh181. [DOI] [PubMed] [Google Scholar]
- 10.Guénin S, Mauriat M, Pelloux J, van Wuytswinkel O, Bellini C, Gutierrez L. Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. Journal of Experimental Botany. 2009;60(2):487–493. doi: 10.1093/jxb/ern305. [DOI] [PubMed] [Google Scholar]
- 11.Rizzi F, Belloni L, Crafa P, et al. A novel gene signature for molecular diagnosis of human prostate cancer by RT-qPCR. PLoS ONE. 2008;3(10) doi: 10.1371/journal.pone.0003617.e3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao G, Chernock RD, Gay HA, et al. A novel RT-PCR method for quantification of human papillomavirus transcripts in archived tissues and its application in oropharyngeal cancer prognosis. International Journal of Cancer. 2013;132(4):882–900. doi: 10.1002/ijc.27739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yong SYC, Choong CY, Cheong PL, et al. Analysis of ESTs generated from inner bark tissue of an Acacia auriculiformis x Acacia mangium hybrid. Tree Genetics and Genomes. 2011;7(1):143–152. [Google Scholar]
- 14.Golisz A, Sugano M, Hiradate S, Fujii Y. Microarray analysis of Arabidopsis plants in response to allelochemical L-DOPA. Planta. 2011;233(2):231–240. doi: 10.1007/s00425-010-1294-7. [DOI] [PubMed] [Google Scholar]
- 15.Molina C, Zaman-Allah M, Khan F, et al. The salt-responsive transcriptome of chickpea roots and nodules via deepSuperSAGE. BMC Plant Biology. 2011;11, article 31 doi: 10.1186/1471-2229-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bleeker PM, Spyropoulou EA, Diergaarde PJ, et al. RNA-seq discovery, functional characterization, and comparison of sesquiterpene synthases from Solanum lycopersicum and Solanum habrochaites trichomes. Plant Molecular Biology. 2011;77(4-5):323–336. doi: 10.1007/s11103-011-9813-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vandesompele J, de Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology. 2002;3(7) doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez L, Mauriat M, Guénin S, et al. The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnology Journal. 2008;6(6):609–618. doi: 10.1111/j.1467-7652.2008.00346.x. [DOI] [PubMed] [Google Scholar]
- 19.Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Research. 2004;64(15):5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 20.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: bestKeeper—excel-based tool using pair-wise correlations. Biotechnology Letters. 2004;26(6):509–515. doi: 10.1023/b:bile.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 21.Stal Papini-Terzi F, Riso Rocha F, Zorzetto Nicoliello Vêncio R, et al. Transcription profiling of signal transduction-related genes in sugarcane tissues. DNA Research. 2005;12(1):27–38. doi: 10.1093/dnares/12.1.27. [DOI] [PubMed] [Google Scholar]
- 22.Rocha FR, Papini-Terzi FS, Nishiyama MY, Jr., et al. Signal transduction-related responses to phytohormones and environmental challenges in sugarcane. BMC Genomics. 2007;8, article 71 doi: 10.1186/1471-2164-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodrigues FA, de Laia ML, Zingaretti SM. Analysis of gene expression profiles under water stress in tolerant and sensitive sugarcane plants. Plant Science. 2009;176(2):286–302. [Google Scholar]
- 24.Iskandar HM, Simpson RS, Casu RE, Bonnett GD, Maclean DJ, Manners JM. Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Molecular Biology Reporter. 2004;22(4):325–337. [Google Scholar]
- 25.Kido EA, Ferreira Neto JRC, Silva RLO, et al. New insights in the sugarcane transcriptome responding to drought stress as revealed by Super SAGE. The Scientific World Journal. 2012;2012:14 pages. doi: 10.1100/2012/821062.821062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 27.Rasmussen R. Quantification on the light cycler. In: Meuer S, Wittwer C, Nakagawara K, editors. Rapid Cycle Real-Time PCR, Methods and Applications. Heidelberg, Germany: Springer; 2001. pp. 21–34. [Google Scholar]
- 28.Thiebaut F, Grativol C, Carnavale-Bottino M, et al. Computational identification and analysis of novel sugarcane microRNAs. BMC Genomics. 2012;13(2):p. 290. doi: 10.1186/1471-2164-13-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsumura H, Krüger DH, Kahl G, Terauchi R. SuperSAGE: a modern platform for genome-wide quantitative transcript profiling. Current Pharmaceutical Biotechnology. 2008;9(5):368–374. doi: 10.2174/138920108785915157. [DOI] [PubMed] [Google Scholar]
- 30.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods in Molecular Biology. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic acids research. 2002;30(9):p. e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apllied Biosystems. Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR. 2004.
- 34.Silva MD, Silva RLO, Ferreira Neto JRC, et al. Expression analysis of sugarcane aquaporin genes under water deficit. Journal of Nucleic Acids. 2013;2013:14 pages. doi: 10.1155/2013/763945.763945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nature Protocols. 2006;1(3):1559–1582. doi: 10.1038/nprot.2006.236. [DOI] [PubMed] [Google Scholar]
- 36.Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. BioTechniques. 2004;37(1):112–119. doi: 10.2144/04371RR03. [DOI] [PubMed] [Google Scholar]
- 37.Banda M, Bommineni A, Thomas RA, Luckinbill LS, Tucker JD. Evaluation and validation of housekeeping genes in response to ionizing radiation and chemical exposure for normalizing RNA expression in real-time PCR. Mutation Research—Genetic Toxicology and Environmental Mutagenesis. 2008;649(1-2):126–134. doi: 10.1016/j.mrgentox.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Fan C, Ma J, Guo Q, Li X, Wang H, Lu M. Selection of reference genes for quantitative real-time PCR in bamboo (Phyllostachys edulis) PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0056573.e56573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Die JV, Román B, Nadal S, González-Verdejo CI. Evaluation of candidate reference genes for expression studies in Pisum sativum under different experimental conditions. Planta. 2010;232(1):145–153. doi: 10.1007/s00425-010-1158-1. [DOI] [PubMed] [Google Scholar]
- 40.Hong S-Y, Seo PJ, Yang M-S, Xiang F, Park C-M. Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biology. 2008;8, article 112 doi: 10.1186/1471-2229-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z-Y, Chen S-Y. Differential accumulation of the S-adenosylmethionine decarboxylase transcript in rice seedlings in response to salt and drought stresses. Theoretical and Applied Genetics. 2000;100(5):782–788. [Google Scholar]
- 42.Li Q-F, Sun SSM, Yuan D-Y, Yu H-X, Gu M-H, Liu Q-Q. Validation of candidate reference genes for the accurate normalization of real-time quantitative RT-PCR data in rice during seed development. Plant Molecular Biology Reporter. 2009;28(1):49–57. [Google Scholar]
- 43.Jain M, Nijhawan A, Tyagi AK, Khurana JP. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochemical and Biophysical Research Communications. 2006;345(2):646–651. doi: 10.1016/j.bbrc.2006.04.140. [DOI] [PubMed] [Google Scholar]
- 44.Hu R, Fan C, Li H, Zhang Q, Fu Y-F. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Molecular Biology. 2009;10, article 1471:p. 93. doi: 10.1186/1471-2199-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong H-Y, Chen J-W, Li C-Q, et al. Selection of reliable reference genes for expression studies by reverse transcription quantitative real-time PCR in litchi under different experimental conditions. Plant Cell Reports. 2011;30(4):641–653. doi: 10.1007/s00299-010-0992-8. [DOI] [PubMed] [Google Scholar]
- 46.Zhu X, Li X, Chen W, et al. Evaluation of new reference genes in papaya for accurate transcript normalization under different experimental conditions. PLoS ONE. 2012;7(8) doi: 10.1371/journal.pone.0044405.e44405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaufichon L, Reisdorf-Cren M, Rothstein SJ, Chardon F, Suzuki A. Biological functions of asparagine synthetase in plants. Plant Science. 2010;179(3):141–153. [Google Scholar]
- 48.Mohammadi M, Kav NNV, Deyholos MK. Transcriptional profiling of hexaploid wheat (Triticum aestivum L.) roots identifies novel, dehydration-responsive genes. Plant, Cell and Environment. 2007;30(5):630–645. doi: 10.1111/j.1365-3040.2007.01645.x. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Liu D, Sun J, Zhang A. Asparagine synthetase gene TaASN1 from wheat is up-regulated by salt stress, osmotic stress and ABA. Journal of Plant Physiology. 2005;162(1):81–89. doi: 10.1016/j.jplph.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 50.Lim H, Cho M-H, Jeon J-S, Bhoo SH, Kwon Y-K, Hahn T-R. Altered expression of pyrophosphate: fructose-6-phosphate 1-phosphotransferase affects the growth of transgenic Arabidopsis plants. Molecules and Cells. 2009;27(6):641–649. doi: 10.1007/s10059-009-0085-0. [DOI] [PubMed] [Google Scholar]
- 51.Reeves RE, South DJ, Blytt HJ, Warren LG. Pyrophosphate: D-fructose 6 phosphate 1 phosphotransferase. A new enzyme with the glycolytic function of 6 phosphofructokinase. Journal of Biological Chemistry. 1974;249(24):7737–7741. [PubMed] [Google Scholar]
- 52.Groenewald J-H, Botha FC. Down-regulation of pyrophosphate: fructose 6-phosphate 1-phosphotransferase (PFP) activity in sugarcane enhances sucrose accumulation in immature internodes. Transgenic Research. 2008;17(1):85–92. doi: 10.1007/s11248-007-9079-x. [DOI] [PubMed] [Google Scholar]
- 53.Kende H. Ethylene biosynthesis. Annual Review of Plant Physiology and Plant Molecular Biology. 1993;44(1):283–307. [Google Scholar]
- 54.Druege U. Ethylene and plant responses to abiotic stress. In: Kahn NA, editor. Ethylene Action in Plants. New York, NY, USA: Springer; 2006. pp. 81–118. [Google Scholar]
- 55.Yang L, Zu Y-G, Tang Z-H. Ethylene improves Arabidopsis salt tolerance mainly via retaining K+ in shoots and roots rather than decreasing tissue Na+ content. Environmental and Experimental Botany. 2013;86:60–69. [Google Scholar]
- 56.Guo L, Zi YW, Lin H, et al. Expression and functional analysis of the rice plasma-membrane intrinsic protein gene family. Cell Research. 2006;16(3):277–286. doi: 10.1038/sj.cr.7310035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material showing the quality of the total RNAs from sugarcane roots under drought stress (FIGURE S1 - A), the amplicons of expected size for each gene used in the study, displaying primer specificity as required for RT-qPCR amplification (FIGURE S1 - B), and the melting curves showing a single peak for six potential reference genes (αTUB; GAPDH; H1; SAMDC; UBQ and 25S rRNA) and four targets (PFPα1; AS; PIP1-1 and ACC oxidase).
Supplementary Material (FIGURE S2) showing standard curves using a dilution series (100, 10−1, 10−2, 10−3 e 10−4) of six potential reference genes (αTUB; GAPDH; H1; SAMDC; UBQ and 25S rRNA) for sugarcane roots under drought stress and the respective curves for four targets (PFPα1; AS; PIP1-1 and ACC oxidase).
Supplementary Material (TABLE S2) based on MIQE checklist for authors, reviewers, and editors.
Supplementary Material (TABLE S1) showing Cq values of potential reference genes for gene expression normalization (RT-qPCR) with cDNAs from sugarcane roots under drought stress (24 h of continuous dehydration).