

Significance
Chronic viral infections evolved to avoid destruction by the immune system. Lymphocytic choriomeningitis virus (LCMV) clone 13, which causes a chronic infection, induces expression of Fas death receptor protein by antigen-presenting cells, such as dendritic cells (DCs). When Fas is ligated by Fas ligand expressed by T cells, DCs die. This event blocks further T-cell activation. To break this mechanism and to sustain antiviral T-cell responses, we used mice that lost Fas from DCs. These animals cleared the infection, whereas Fas-sufficient mice did not. Moreover, a transfer of Fas-negative DCs into infected mice supported the clearance of LCMV infection. Thus, we suggest a novel strategy for stimulation of T cells to achieve the clearance of persistent viruses in infected animals and humans.
Abstract
Chronic viral infections incapacitate adaptive immune responses by “exhausting” virus-specific T cells, inducing their deletion and reducing productive T-cell memory. Viral infection rapidly induces death receptor CD95 (Fas) expression by dendritic cells (DCs), making them susceptible to elimination by the immune response. Lymphocytic choriomeningitis virus (LCMV) clone 13, which normally establishes a chronic infection, is rapidly cleared in C57Black6/J mice with conditional deletion of Fas in DCs. The immune response to LCMV is characterized by an extended survival of virus-specific effector T cells. Moreover, transfer of Fas-negative DCs from noninfected mice to preinfected animals results in either complete clearance of the virus or a significant reduction of viral titers. Thus, DC-specific Fas expression plays a role in regulation of antiviral responses and suggests a strategy for stimulation of T cells in chronically infected animals and humans to achieve the clearance of persistent viruses.
Adaptive immune responses are tightly regulated to reduce the damage inevitably invoked by activated T cells. A combination of T-cell–intrinsic and T-cell–extrinsic mechanisms drive the attrition of expanded T cells (1–4). Proapoptotic signaling receptor CD95 (Fas) regulates the fate of immunocytes, and animals and humans with systemic deficiency of Fas or its ligand (FasL) have been shown to develop lymphoproliferative diseases (5–8). Analysis of animals with tissue-specific disruption of Fas expression revealed that antigen-presenting cells (APCs), such as dendritic cells (DCs) (9) and B cells (9, 10), and T cells (9) could contribute to different features of systemic autoimmunity. An important conclusion from these studies, however, was that APCs are sensitive to Fas-mediated killing during immune activation, presumably to prevent the bystander triggering of self-reactive T cells. Indeed, an extension of DC lifespan by overexpression of antiapoptotic proteins (11, 12) or elimination of Fas (9) led to survival of autoreactive T and B cells and to systemic autoimmunity. The result also suggested that DC elimination by activated T cells could be the major mechanism of T-cell attrition during the normal immune response to a pathogen. Because systemic autoimmunity can be viewed as reactivity to persistent antigens, we reasoned that the ability of DCs to resist elimination would enable them to induce a sterilizing T-cell response to a persistent viral infection. We find that indeed mice genetically lacking Fas in DCs or transferred with Fas-negative DCs showed accelerated clearance of the infection with a persistent virus.
Results
Mice Lacking Fas in DCs Clear a Persistent Viral Infection.
To test the idea that Fas-negative DCs could promote immunity against pathogens that normally establish a persistent infection, we used the clone 13 variant of lymphocytic choriomeningitis virus (LCMV). It induces a persistent viral infection, resulting in exhaustion of the T-cell response characterized by a loss of cytokine production, loss of activation markers, expression of negative regulators, such as PD-1, and subsequent deletion of virus-reactive T cells (13, 14). The clearance of LCMV-clone 13 infection from lymphoid organs takes up to 60 d in C57Black6/J (B6) mice; after that the infection still persists in parenchymatous organs such as kidneys (13, 15). In contrast, infection with the Armstrong variant of LCMV is rapidly cleared, does not cause exhaustion of T cells, and results in the formation of a good memory T-cell response. Animals carrying loxP-flanked Fas exon IX (B6.FasKI) and CD11c-driven Cre recombinase (B6.CD11c-Cre) were infected with LCMV clone 13. Cre-negative FasKI mice as well as mice with a T-cell–specific deletion of Fas (B6.Lck-Cre.FasKI) served as controls (Fig. 1). Viral titers were determined in the secondary lymphoid organs and kidneys. Mice with Fas-negative DCs (B6.CD11c-Cre.FasKI) cleared the virus from the spleens between 2 and 4 wk after infection (Fig. 1A), whereas the control animals were still infected. Moreover, CD11c-Cre.FasKI mice but not the control mice cleared the virus from the kidneys (Fig. 1B). The loss of Fas from T cells (in B6.Lck-Cre.FasKI mice) did not make the mice resistant to infection with LCMV clone 13.
Fig. 1.
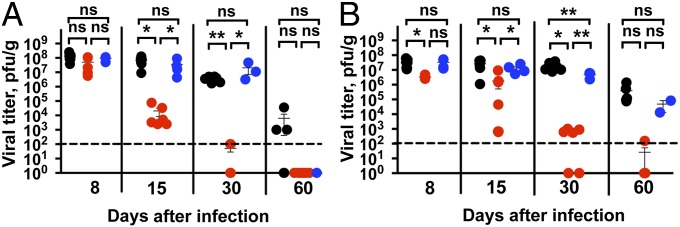
Rapid clearance of chronic LCMV infection in the presence of Fas-deficient DCs. Mice lacking Fas in DCs (B6.CD11c-Cre.FasKI, red dots) or in T cells (B6.Lck-Cre.FasKI, blue dots) and control (B6.FasKI, black dots) mice were infected with LCMV-clone 13 and viral titers were estimated by plaque assay in the spleens (A) and kidneys (B) of infected mice at the indicated timepoints after infection. Data are compiled from two independent experiments with 2–7 mice per time point. Dotted line indicates the limit of detection. Error bars represent SEM. Statistical significance was determined by ordinary one-way ANOVA (*P < 0.05; **P < 0.005; ns, not significant).
Survival of Fas-Negative DCs Is the Primary Cause of Efficient Anti-LCMV Response.
For the Fas molecule to matter in the survival or demise of DCs during an immune response, it should be inducible by viral infection. Previous studies have shown rapid up-regulation of type I IFN and cell surface costimulatory molecules, such as major histocompatibility complex (MHC) II, CD80, and CD86 by 24 h after LCMV infection (16–18). We have also shown that Fas expression can be rapidly induced in DCs by multiple ligands of innate signaling receptors and by type I IFN (9). Moreover, Fas up-regulation in human DCs by a paramyxovirus (measles) has been reported (19). To address the issue, splenic DCs from infected B6 mice were tested for induction of Fas by LCMV. Fas was rapidly induced, and equally well, by both LCMV clone 13 and LCMV-Armstrong (Fig. 2A). Generation of LCMV-specific T cells has been shown to occur within 24 h of an LCMV infection and precedes a marked depletion of DCs in the spleen of infected mice (18). The loss of DCs during LCMV infection has been noted to occur as early as day 3 after infection with LCMV (20). To show that the lack of Fas expression correlated with better survival of DCs during LCMV-clone 13 infection, the numbers of activated DCs (MHC class II high, CD11c+ cells) were compared between B6.FasKI and B6.CD11c-Cre.FasKI mice at different time points after LCMV-clone 13 infection (Fig. 2B). Whereas the DC numbers were similar before infection, they differed between the two strains of mice as early as day 3 after infection, with B6.CD11c-Cre.FasKI mice preserving more DCs. The rates of DC infection with LCMV were not drastically different between the two strains (Fig. S1).
Fig. 2.

Fas is rapidly up-regulated upon LCMV infection and reduces dendritic cell survival. (A) Splenic DCs from B6 mice infected with LCMV-clone 13 or LCMV-Armstrong were analyzed for CD95(Fas) expression at 2 and 5 d after infection. Data represent two independent experiments. (B) LCMV-clone 13–infected B6.FasKI and B6.CD11-Cre.FasKi mice were analyzed at 0, 3, 8, 10, and 21 d after infection, and the total number of splenic DCs were estimated. Data are compiled from two independent experiments with 3–8 mice per time point. Error bars represent SEM. Statistical significance was determined by unpaired Student t test. (*P < 0.05; **P < 0.005; ns, not significant.) (C) DCs from mice in B were analyzed for surface expression of MHC class I, CD80, and CD86 on day 5 after infection. (D) Purified T cells from OT-1 and OT-2 mice were stimulated with indicated doses of OVA protein presented by Mitomycin-treated DCs from B6.FasKI (black symbols) or B6.CD11c-Cre.FasKI (red symbols) mice. Stimulation index is the ratio between 3H-Thymidine incorporation in the presence of OVA vs. incorporation in the absence of antigen. One of two similar experiments is shown. Error bars are SEM. (E) Volcano plot of significance versus change of gene expression between day 5 LCMV-clone 13–infected splenic DCs from B6.FasKI and B6.CD11c-Cre.FasKI mice. The two vertical (red) lines indicate twofold change, and the horizontal (red) line indicates the 0.05 P value level.
The surviving Fas-negative DCs may sustain the antiviral response of T cells by two mechanisms that are not mutually exclusive. Either Fas-negative DCs resist Fas-mediated destruction by T cells, persist longer, and continue to provide T cells with survival (MHC-peptide and costimulatory) signals, or antigen-presenting function is altered in Fas-negative DCs protecting T cells from exhaustion. Several approaches were applied to compare the properties of DCs from infected control and B6.CD11c-Cre.FasKI mice. First, staining of DCs from LCMV-clone 13–infected mice (day 5 after infection) with antibodies to MHC class I, CD80, and CD86 did not reveal differences between the two DC populations (Fig. 2C). Second, we tested the ability of bone marrow-derived DCs (BMDCs) from CD11c-Cre–positive and –negative mice to present antigens to T cells in vitro. Chicken ovalbumin (OVA)-specific OT-1 (CD8+) and OT-2 (CD4+) T cells were activated in vitro with different concentrations of the whole protein (Fig. 2D). No difference was found in ability of two DC types to process and present antigens. Third, gene expression microarray analysis was performed by using DCs isolated on day 5 after LCMV infection from the spleens of B6.FasKI and B6.CD11c-Cre.FasKI mice. Very few (less than 10) genes with the known expression in DCs were differentially expressed in these two subsets (Fig. 2E and Table S1). This result argued that beyond their better survival because of the lack of Fas-mediated killing, DCs did not undergo major physiological changes in B6.CD11c-Cre.FasKI mice compared with control Fas-sufficient DCs.
Antiviral Immune Responses Do Not Exhaust in LCMV-Clone 13–Infected Mice with Fas-Negative DCs.
In persistent viral infections, viral epitope-specific T cells become “exhausted,” undergo deletion, and do not leave T-cell memory (13, 21). To show that the viral clearance was linked to a more efficient immune response in mice with Fas-negative DCs, the fate of virus-specific CD8+ T cells was followed for 60 d by staining with tetramers of MHC class I molecules carrying LCMV-derived peptides and with antibodies to inflammatory cytokines. Several tetramers were used to exclude a selective maintenance of responses to privileged epitopes by Fas-negative DCs. Fig. 3 demonstrates the dynamics of the numbers of CD8+ tetramer+ T cells during the 2-mo period after LCMV-clone 13 infection. The number of tetramer-positive CD8+ T cells in B6.CD11cCre.FasKI mice (with Fas-negative DCs) was maintained at higher levels throughout the observation period compared with B6.FasKI and B6.Lck-Cre.FasKI animals (Fig. 3A and Fig. S2A). Because in LCMV-clone 13 infection the presence of antigen-specific T cells does not necessarily correlate with their functionality, the ability of these cells to produce multiple cytokines (IFN-γ and TNF-α), a commonly accepted sign of their immune competence (13, 15), was also measured. The total numbers (Fig. 3B and Fig. S2B) and proportions (Fig. S2C) of IFN-γ and TNF-α secreting tetramer-positive CD8+ T cells were significantly increased and stably maintained in B6.CD11cCre.FasKI mice, whereas they were rapidly lost in control animals. Interestingly, the rate of deletion of Fas-negative, LCMV-specific T cells was slower compared with Fas-sufficient animals (Fig. 3A and Fig. S2A); however, the ability of Fas-negative T cells to simultaneously secrete multiple cytokines was reduced quickly compared with T cells from B6.CD11c-Cre.FasKI mice that maintained this feature (Fig. 3B and Fig. S2B). Thus, Fas-mediated T-cell deletion contributes to the reduction of T-cell numbers during the immune response (22, 23), but the functional inactivation of T cells by a persistent viral infection happens independently of Fas-expressed by T cells.
Fig. 3.

Enhanced anti-LCMV T-cell response to LCMV-clone 13 in the presence of Fas-negative DCs, but not Fas-negative T cells. Splenic T cells responses to LCMV Db GP33 epitope were estimated in LCMV-clone 13–infected mice by MHCI tetramer staining (A) and by intracellular cytokine staining for IFN-γ, TNF-α double-producing T cells after 5 h in vitro stimulation with GP33 peptide (B). (C) LCMV Db GP276 tetramer-positive CD8+ splenic T cells from LCMV-clone 13–infected mice were analyzed for expression of KLRG1 and PD1 at 8, 15, 30, and 60 d after infection. Error bars represent SEM. Statistical significance was determined by ordinary one-way ANOVA (*P < 0.05; **P < 0.005; ns, not significant).
T Cells Persisting in LCMV-Infected B6.CD11cCre.FasKI Mice Are Activated Effectors.
To determine the type of T cells persisting in mice with Fas-negative DCs, virus-specific T-cell populations were characterized on the basis of expression of activation and differentiation markers (14, 24). The expansion of T cells with an effector phenotype (CD44hi, KLRG1hi, PD1lo) became obvious (Fig. 3C and Fig. S2D). Importantly, B6.CD11c-Cre.FasKI mice acquired expression of KLRG1 simultaneously with T cells in control animals but maintained its expression throughout the experiment, whereas KLRG1 expression was gradually lost in the control mice, possibly due to deletion of these cells. Moreover, the effector T cells in B6.CD11c-Cre.FasKI mice lost PD1, a negative regulator of the T-cell response associated with the exhaustion of the anti-LCMV response (14), during the second week of infection (Fig. 3C and Fig. S2D). Thus, the loss of Fas by DCs led to the clearance of a persistent viral infection accompanied by reduced deletion of T cells and markedly reduced signs of T-cell exhaustion (stable production of multiple cytokines, maintenance of an activation marker, and loss of a marker of negative regulation).
Persistence of Antigen-Specific T Cells in Mice with Fas-Negative DCs Is Independent of the Type of Infection or Antigen Persistence.
It has been suggested that the nature of infection (acute, caused by LCMV-Armstrong, vs. persistent, caused by LCMV-clone 13) and the rate of viral clearance (i.e., length of antigenic stimulation) (25–27) affect T-cell responses. To test whether the nature of the virus was important for the persistence of T-cell response supported by Fas-negative DCs, B6.CD11c-Cre.FasKI animals were infected with LCMV-Armstrong and followed for 60 d. The total number of tetramer-positive (GP276 and NP396 epitopes) cells was consistently higher in the spleens (Fig. 4A and Fig. S3A) and lymph nodes (Fig. 4B and Fig. S3B) of B6.CD11c-Cre.FasKI mice infected with LCMV-Armstrong than of infected control animals. H-2Db--GP33 epitope-specific P14 CD8+ T cells (28) were transferred (105 purified naïve T cells per mouse) into B6.FasKI controls and B6.CD11c-Cre.FasKI mice 1 d before infection with LCMV-Armstrong and followed for 60 d. The total number of P14 T cells recovered from the spleens of B6.CD11c-Cre.FasKI was consistently higher compared with controls (Fig. S3C). This experiment shows that recognition of another viral epitope follows the same rule, and also that the enhanced anti-LCMV T-cell response in B6.CD11c-Cre.FasKI mice is T-cell extrinsic and depends on the absence of Fas on DCs.
Fig. 4.

Extended effector T-cell response in CD11c-FasKI mice is independent of antigen persistence. B6.FasKI and B6.CD11c-Cre.FasKI mice infected with LCMV-Armstrong were compared for the total Db NP396 tetramer-positive T numbers in spleen (A) and lymph nodes (B) at 12, 19, and 60 d after infection. Data are compiled from two independent experiments, 3–5 mice per time point. (C) B6.FasKI and B6.CD11c-Cre.FasKi mice (both CD45.2+, Thy1.2+) were infected with LCMV-Armstrong and injected on day 19 after infection with a 1:1 mixture of CFSE-labeled naïve purified P14 T cells (CD45.1+) and OT1 T cells (Thy1.1+). Injection of the same mix into B6 mice at day 3 after infection served as a positive control. Proliferation of T cells was tested in the spleens of recipient mice 3 d after injection. (D and E) Db NP396 tetramer-positive splenic CD8+ T cells from B6.FasKI and B6.CD11c-Cre.FasKI mice infected with LCMV-Armstrong were analyzed at indicated time points for the composition of memory T cells: SLECs (CD44hi, KLRG1hi, CD127lo, CD62Llo) in blue, MPECs (CD44hi, KLRG1lo, CD127hi, CD62Llo) in gray, and TCM (CD44hi, CD127hi, KLRG1lo, CD62Lhi) in green. Data are compiled from two independent experiments, 3–5 mice per time point. (F) Naïve CD45.1+ P14 T cells (105) were transferred into B6.FasKI and B6.CD11c-Cre.FasKI mice, 1 d before infection with LCMV-Armstrong. Eight days later, 4 × 106 activated P14 T cells were isolated from the spleens of infected mice by magnetic cell separation and transferred into infection-matched B6 recipient mice. Data represent the number of P14 T cells with SLEC (blue) and TCM (green) phenotype recovered from recipient B6 mice 11 d later (day 19 of initial infection). Data are compiled from two independent experiments, 5–8 mice per time point. The experiment is also diagramed in Fig. S4. (G) The percent of Ki-67–positive KLRG1+ and KLRG1− T cells were identified among tetramer-positive (GP33 +GP34 +GP276 + NP396) CD8+ T cells from the spleens of B6.FasKI and B6.CD11c-Cre.FasKi mice infected with LCMV-Armstrong for 21 d. Error bars represent SEM. Statistical significance was determined by unpaired Student t test (*P < 0.05; **P < 0.005; ns, not significant).
LCMV-Armstrong is normally cleared within 8 d after infection, and it is agreed that no virus persists past that time point (13, 29). That was true for both B6.FasKI and B6.CD11c-Cre.FasKI mice (Fig. S3D). However, in mice with manipulated properties of DCs, the extended lifespan of viral antigens, although unlikely, could not be excluded. To test this possibility, B6.FasKI controls and B6.CD11cCre.FasKI mice were infected with LCMV-Armstrong and injected on day 19 after infection with a mixture of CFSE-labeled P14 T cells and cells from transgenic mouse carrying ovalbumin-specific OT-1 TCR (30) to account for nonspecific proliferation. Neither of these cells proliferated when injected on day 19 after infection, whereas P14 expanded strongly (and OT-1 showed some by-stander activation) in control B6 mouse on day 3 after LCMV infection (Fig. 4C). Thus, virus-specific T cells persisted in B6.CD11cCre.FasKI mice despite the lack of cognate antigens capable of driving T-cell proliferation, although the presence of very low levels of antigen capable of supporting T-cell survival could not be experimentally excluded.
Fas-Negative DCs Enhance Effector CD8 T-Cell Commitment Without Altering the Generation of Long-Lived Memory T Cells.
The tetramer+ CD8 T cells were next analyzed for the presence of various effector/memory T cells subsets induced by LCMV-Armstrong infection in mice with Fas-sufficient or Fas-negative DCs. Similar to the observation with LCMV-clone 13 infection, Fas-negative DCs induced increased numbers of short-lived effector T cells (SLEC) (CD44hi, KLRG1hi, CD127lo, CD62Llo) and memory precursor effector T cells (MPEC) (CD44hi, KLRG1lo, CD127hi, CD62Llo) (31, 32) (Fig. 4 D and E and Fig. S4 A–C). This increase altered the ratio of effector to central memory T cells (TCM) (CD44hi, CD127hi, KLRG1lo, CD62Lhi); however, the absolute numbers of TCM cells were not different between mice with Fas-negative and Fas-sufficient DCs (Fig. 4E and Fig. S4B). A similar enhancement to effector T-cell commitment was observed in P14 T cells primed in the presence or absence of Fas on DCs (Fig. S4C). In agreement with the surface marker phenotype, the total number of IL-2+ long-lived memory CD8+ T cells were similar between mice with Fas-sufficient or Fas-negative DCs (Fig. S4 D and E). Thus, extended engagement with stimulation-competent DCs during the early stages of infection promotes the expansion of effector-type CD8+ T cells, whereas the generation of TCM cells is independent of these events, supporting the idea of the early dichotomy of effector/memory generation, rather than the continuum of effector→memory differentiation (33–35).
CD8+ cells with an effector phenotype persisted in B6.CD11c-Cre.FasKI mice long after clearance of the virus had been achieved. However, whether the presence of Fas-negative DCs was required for the prolonged persistence of LCMV-activated effector T cells or “initial hit” with Fas-negative DCs was sufficient to induce the persistence of effector T cells was not clear. To address this issue, naïve P14 T cells were activated by LCMV-Armstrong infection either in B6.CD11c-Cre.FasKI or in control B6.FasKI mice and 8 d after infection transferred into infection matched B6 animals, and analyzed 11 d later (Fig. S5). P14 T cells failed to persist in B6 recipients independently of whether they were activated by viral infection in B6.CD11c-Cre.FasKI or control B6.FasKI mice (Fig. 4F).
Finally, to test whether the maintenance of effector virus-specific CD8+ T cells in B6.CD11c-Cre.FasKI required their proliferation, tetramer-positive (stained with a mixture of four LCMV-specific MHC tetramers) T cells from B6.FasKI and B6.CD11c-Cre.FasKI mice infected 3 wk before were additionally stained with antibodies to activation markers and to Ki-67 (Fig. 4G). The proportion of proliferating cells was not different between control and experimental groups, and KLRG1+ T cells did not proliferate stronger than KLRG1− counterparts.
Thus, the presence of Fas-negative DCs past the acute phase of the immune response was required to support the persistence of antiviral effectors, but the process did not depend on (detectable) persistence of viral antigens and was not due to an excessive proliferation of the effector T cells.
DC Transfer for Antiviral Therapy.
Mice with Fas-negative DCs efficiently cleared LCMV-clone 13 infection from secondary lymphoid organs and visceral organs (Fig. 1). To test the possibility of a therapeutic application of Fas-negative DCs, experiments were performed in which splenic DCs from uninfected animals were transferred to LCMV-clone 13–infected B6 mice (Fig. 5A). Animals receiving Fas-negative DCs were able to completely clear the infection from spleens and, importantly, from kidneys in 3 wk when injected on day 8 of LCMV-clone 13 infection. As expected, they also demonstrated the presence of a higher number of virus-reactive T cells (Fig. 5B) and higher numbers of IFN-γ + TNF-α double-producing CD8+ T cells (Fig. 5C). We next assessed the ability of in vitro GM-CSF–BMDCs to affect viral clearance upon transfer into LCMV-clone 13–infected mice, at day 8 after infection. Mice injected with Fas-negative BMDCs also demonstrated significantly lower viral titers (Fig. 5D), significant protection from deletion (Fig. 5E), and enhanced cytokine response (Fig. 5F). In addition, the DC-treated mice did not develop antinuclear antibodies when examined 3 wk after DC transfer (Fig. S6); suggesting that short-term therapy with Fas-negative DCs does not induce systemic autoimmunity.
Fig. 5.

Transfer of Fas-negative DCs rapidly clears an established LCMV-clone 13 infection. B6 mice infected with LCMV-clone 13 for 8 d were injected with 2 × 106 purified splenic DCs (A–C) or GM-CSF–derived BMDCs (D–F) from uninfected B6.FasKI or B6.CD11c-Cre.FasKi animals or received no transfer. (A and D) Recipient groups of B6 mice were analyzed 3 wk after DC transfer and viral titers were estimated in the indicated organs. Dotted line indicates the limit of detection. (B and E) LCMV-specific T cells were estimated in the spleens of recipient mice by staining with indicated tetramers. (C and F) IFN-γ and TNF-α double-producing T cells numbers were identified by intracellular flow cytometric analysis after 5 h in vitro stimulation with indicated LCMV peptides. Data are compiled from two independent experiments with 5–8 mice per time point. Error bars represent SEM. Statistical significance was determined by ordinary one-way ANOVA (*P < 0.05; **P < 0.005; ns, not significant).
Discussion
The proapoptotic signaling receptor CD95 (Fas) was originally implicated in the regulation of immune responses due to unrestricted proliferation of lymphocytes in animals and humans deficient in Fas or its ligand. However, the attrition of T cells that normally follows the immune response was later attributed mostly to T-cell–intrinsic apoptotic pathways (22, 23, 36, 37). In fact, animals with double deficiency of Fas and the proapoptotic molecule Bim (Bcl2l11) suffered a more severe autoimmune disease than mice deficient in Fas alone (38, 39). In an attempt to dissect the role of different cell types in autoimmunity associated with Fas deficiency, we have shown that Fas deficiency restricted to APCs could induce the symptoms of systemic immunity (lymphoproliferation, enlargement of secondary lymphoid organs, foci of lymphopoesis in internal organs, and production of high titers of antinuclear antibodies) (9). The most likely scenario that explained the finding was that DCs are normally eliminated by the very T-cell response that they are activating. DC elimination leads to a rapid cessation of signaling necessary for T-cell activation-antigen presentation and costimulation. Why DCs are eliminated primarily (or exclusively) through a Fas-mediated mechanism is not known, but several studies have found evidence for the prevalence of Fas over other cytotoxic mechanisms (9, 11). At the same time, deficiency of another cytotoxic factor, perforin, leads in humans to a systemic autoimmune disorder, familial histocytosis (40). In mice, perforin deficiency does not induce systemic autoimmunity, but a combination of DC-specific deficiency of Fas and systemic deficiency of perforin increases the severity of the autoimmune response (41). One plausible explanation for the prevalent role of Fas is that the kinetics of the induction of cytotoxic mechanisms in T cells is such that FasL-mediated killing is activated first. Importantly, most of the DCs are Fas negative unless activated by microbial stimuli or type I IFN (9). This finding is also a strong argument in favor of the idea that Fas-mediated destruction of DCs is involved in the regulation of responses to pathogens. In the present study, we also find that Fas is rapidly up-regulated by infection with LCMV and that this up-regulation is independent of the type of LCMV virus (Fig. 2A). Thus, up-regulation of Fas expression and of FasL sensitivity of activated DCs are crucial physiological mechanisms preventing excessive bystander activation of T cells and autoimmunity during an inflammatory response to infections.
This study was initiated based on a presumption that persistent viral infection has similarities with autoimmunity, given that self-antigens can be viewed to persist chronically. When this idea was put to the test, we discovered that mice with DC-specific loss of Fas were able to sustain a strong response to the persistent LCMV-clone 13 (Fig. 1). Further analysis has revealed that CD11c-Cre.FasKI mice were capable of sustaining a bona fide immune response to the virus, and judging by their ability to produce multiple cytokines and express activation markers (CD44, KLRG1) and by the loss of the negative regulator PD1 (14, 42), their T cells were not exhausted.
Several interesting observations shed light on the regulation of the immune response during a persistent viral infection. First, Fas-negative T cells were somewhat more resistant to deletion than T cells in control mice (Fig. 3 and Fig. S2), but nevertheless underwent exhaustion with the same kinetics. Thus, Fas-mediated T-cell deletion makes a contribution to the reduction of T-cell numbers during the immune response (22, 23, 36), but the functional inactivation of T cells by a persistent viral infection happens independently of Fas expression by T cells.
Second, the phenotype of activated T cells that were maintained in CD11c-Cre.FasKI mice was that of effector T cells. They were maintained independently of the presence of activating antigens (at least transferred virus-specific T cells were not able to respond to these antigens in vivo), and their lifespan exceeded the expected lifespan of SLECs (2, 31). Their long-term survival was not based on excessive proliferation but required the presence of Fas-negative DCs. The expansion of effector T cells, both SLECs and MPECs, did not lead to an expansion of central memory T cells (Fig. 4). This observation is interesting because it argues against linear models of TCM generation: if TCM were the progeny of effector cells, one would anticipate them to expand proportionally with the expansion of effector T cells. Our results are more supportive of the early dichotomy models (35, 43, 44) of memory T-cell generation.
Third, effector cells were expanded and persisted in mice with Fas-negative DCs regardless of the type of the infecting virus, and their persistence did not require chronic infection (Fig. 4) but depended solely on the presence of Fas-negative DCs.
The ability of Fas-negative DCs to stimulate a response that clears a persistent viral infection has a strong translational value. In fact, injection of Fas-negative DCs led to either complete elimination of the virus or significant reduction of its titers. The efficiency of specific DC subsets has yet to be evaluated. Gene expression profiling did not reveal significant differences between Fas-negative and Fas-sufficient DCs in the spleens of infected animals. Thus, it is likely that DCs with extended lifespan due to protection from Fas-mediated killing provide effector T cells with “tonic” survival signals, protecting them from exhaustion and subsequent deletion.
Taken together, this study demonstrates that during persistent infections, T-cell expansion, maintenance, and contraction depend on the survival of the antigen-presenting DCs. It is not clear yet why Fas-mediated death dominates over other possible mechanisms of DC destruction, but there is no doubt that it is a nonredundant antiinflammatory feedback mechanism. Very importantly, Fas-negative DCs sustain efficient antiviral immune responses and can potentially be used as a therapeutic tool to clear persistent infections.
Methods
Mice.
Mice with Exon IX of Fas flanked with LoxP sites (9) were bred to mice with Cre recombinase under the control of the proximal Lck promoter B6.Cg-Tg (Lck-cre) 548Jxm/J (Lck-Cre) or the CD11c promoter C57BL/6J-Tg (Itgax-cre,-EGFP) 4097Ach/J (CD11c-Cre) obtained from The Jackson Laboratory. C57BL/6 mice and OT1 and OT2 TCR transgenic mice were obtained from The Jackson Laboratory. B6.SJL-Ptprca/BoyAiTac (CD45.1+) and B6.Cg-Tcratm1Mom Tg (TcrLCMV) 327Sdz (P14) mice were purchased from Taconic Farms. Mice were housed in a specific pathogen-free research facility and used in accordance with institutional guidelines for animal welfare. The Biological Sciences Division Institutional Review Board at the University of Chicago approved all animal studies.
Infections.
LCMV viruses (a generous gift from R. M. Welsh, University of Massachusetts, Worcester, MA) were propagated in baby hamster kidney BHK21 cells (ATCC) and titrated by plaque assays on Vero cells (ATCC) as described (45). Six- to eight-week-old mice were infected i.v. with 2 × 106 pfu of LCMV-clone 13 or i.p. with 2 × 105 pfu of LCMV-Armstrong.
Statistical Analysis.
Statistical significance between two experimental groups was determined by using a two-tailed unpaired Student t test, where a P value of <0.05 was considered significant. For analysis of more than two groups of observations, significance was found by using ordinary one-way ANOVA with Tukey’s post hoc test, and a P value of <0.05 was considered significant. Graphs were generated and statistical analyses were performed by using GraphPad Prism (GraphPad Software). Results are displayed as mean ± SEM.
Supplementary Material
Acknowledgments
We thank Dr. R. M. Welsh for the LCMV stocks and for teaching us the relevant techniques. The work was supported by National Institutes of Health Grant R01AI072627 (to A.V.C.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: Gene expression data has been deposited with the Gene Expression Omnibus repository (accession no. GSE57443).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1401750111/-/DCSupplemental.
References
- 1.Häcker G, Bauer A, Villunger A. Apoptosis in activated T cells: What are the triggers, and what the signal transducers? Cell Cycle. 2006;5(21):2421–2424. doi: 10.4161/cc.5.21.3397. [DOI] [PubMed] [Google Scholar]
- 2.Joshi NS, Kaech SM. Effector CD8 T cell development: A balancing act between memory cell potential and terminal differentiation. J Immunol. 2008;180(3):1309–1315. doi: 10.4049/jimmunol.180.3.1309. [DOI] [PubMed] [Google Scholar]
- 3.Strasser A, Pellegrini M. T-lymphocyte death during shutdown of an immune response. Trends Immunol. 2004;25(11):610–615. doi: 10.1016/j.it.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Vella AT, Dow S, Potter TA, Kappler J, Marrack P. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95(7):3810–3815. doi: 10.1073/pnas.95.7.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rieux-Laucat F, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268(5215):1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, et al. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. 1996;98(5):1107–1113. doi: 10.1172/JCI118892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi T, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76(6):969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 8.Adachi M, et al. Enhanced and accelerated lymphoproliferation in Fas-null mice. Proc Natl Acad Sci USA. 1996;93(5):2131–2136. doi: 10.1073/pnas.93.5.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stranges PB, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26(5):629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hao Z, et al. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity. 2008;29(4):615–627. doi: 10.1016/j.immuni.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, et al. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006;311(5764):1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 12.Nopora A, Brocker T. Bcl-2 controls dendritic cell longevity in vivo. J Immunol. 2002;169(6):3006–3014. doi: 10.4049/jimmunol.169.6.3006. [DOI] [PubMed] [Google Scholar]
- 13.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77(8):4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 15.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160(2):521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teijaro JR, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340(6129):207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson EB, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340(6129):202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montoya M, Edwards MJ, Reid DM, Borrow P. Rapid activation of spleen dendritic cell subsets following lymphocytic choriomeningitis virus infection of mice: Analysis of the involvement of type 1 IFN. J Immunol. 2005;174(4):1851–1861. doi: 10.4049/jimmunol.174.4.1851. [DOI] [PubMed] [Google Scholar]
- 19.Servet-Delprat C, et al. Consequences of Fas-mediated human dendritic cell apoptosis induced by measles virus. J Virol. 2000;74(9):4387–4393. doi: 10.1128/jvi.74.9.4387-4393.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borrow P, Evans CF, Oldstone MB. Virus-induced immunosuppression: Immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J Virol. 1995;69(2):1059–1070. doi: 10.1128/jvi.69.2.1059-1070.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol. 2012;86(15):8161–8170. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mogil RJ, et al. Fas (CD95) participates in peripheral T cell deletion and associated apoptosis in vivo. Int Immunol. 1995;7(9):1451–1458. doi: 10.1093/intimm/7.9.1451. [DOI] [PubMed] [Google Scholar]
- 23.Van Parijs L, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity. 1996;4(3):321–328. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 24.Sarkar S, et al. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205(3):625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA. 2009;106(21):8623–8628. doi: 10.1073/pnas.0809818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fousteri G, et al. Increased memory conversion of naïve CD8 T cells activated during late phases of acute virus infection due to decreased cumulative antigen exposure. PLoS ONE. 2011;6(1):e14502. doi: 10.1371/journal.pone.0014502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirth TC, et al. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 2010;33(1):128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pircher H, Bürki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342(6249):559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 29.Moskophidis D, et al. Role of virus and host variables in virus persistence or immunopathological disease caused by a non-cytolytic virus. J Gen Virol. 1995;76(Pt 2):381–391. doi: 10.1099/0022-1317-76-2-381. [DOI] [PubMed] [Google Scholar]
- 30.Hogquist KA, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 31.Kaech SM, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4(12):1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 32.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27(2):281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Löhning M, et al. Long-lived virus-reactive memory T cells generated from purified cytokine-secreting T helper type 1 and type 2 effectors. J Exp Med. 2008;205(1):53–61. doi: 10.1084/jem.20071855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Opferman JT, Ober BT, Ashton-Rickardt PG. Linear differentiation of cytotoxic effectors into memory T lymphocytes. Science. 1999;283(5408):1745–1748. doi: 10.1126/science.283.5408.1745. [DOI] [PubMed] [Google Scholar]
- 35.Wakim LM, Bevan MJ. From the thymus to longevity in the periphery. Curr Opin Immunol. 2010;22(3):274–278. doi: 10.1016/j.coi.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hughes PD, et al. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 2008;28(2):197–205. doi: 10.1016/j.immuni.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hildeman DA, et al. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16(6):759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- 38.Hutcheson J, et al. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28(2):206–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 39.Weant AE, et al. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. 2008;28(2):218–230. doi: 10.1016/j.immuni.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 40.Stepp SE, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 41.Chen M, Felix K, Wang J. Critical role for perforin and Fas-dependent killing of dendritic cells in the control of inflammation. Blood. 2012;119(1):127–136. doi: 10.1182/blood-2011-06-363994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang JT, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315(5819):1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 44.Teixeiro E, et al. Different T cell receptor signals determine CD8+ memory versus effector development. Science. 2009;323(5913):502–505. doi: 10.1126/science.1163612. [DOI] [PubMed] [Google Scholar]
- 45.Welsh RM, Seedhom MO. 2008. Lymphocytic choriomeningitis virus (LCMV): Propagation, quantitation, and storage. Curr Protoc Microbiol, Chapter 15:Unit 15A 11.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.