

Significance
Many drugs used to treat pain, epilepsy, and cardiac arrhythmias target human voltage-gated sodium-selective channels. Surprisingly, we found that a bacterial voltage-gated sodium channel is also inhibited by many eukaryotic sodium channel antagonists. This bacterial channel was crystallized with several brominated blocker compounds, and the high-resolution structures reveal a common antagonist binding site in the cavity of the pore. Electrophysiology studies of channels with mutations at adjacent residues validate the site. These results suggest that despite millions of years of evolution separating human and bacterial sodium channels, these simple bacterial channels can be a valuable tool for screening and rational design of human drugs.
Keywords: crystal structure, pharmacology
Abstract
Voltage-gated sodium channels are important targets for the development of pharmaceutical drugs, because mutations in different human sodium channel isoforms have causal relationships with a range of neurological and cardiovascular diseases. In this study, functional electrophysiological studies show that the prokaryotic sodium channel from Magnetococcus marinus (NavMs) binds and is inhibited by eukaryotic sodium channel blockers in a manner similar to the human Nav1.1 channel, despite millions of years of divergent evolution between the two types of channels. Crystal complexes of the NavMs pore with several brominated blocker compounds depict a common antagonist binding site in the cavity, adjacent to lipid-facing fenestrations proposed to be the portals for drug entry. In silico docking studies indicate the full extent of the blocker binding site, and electrophysiology studies of NavMs channels with mutations at adjacent residues validate the location. These results suggest that the NavMs channel can be a valuable tool for screening and rational design of human drugs.
Nine highly homologous human voltage-gated sodium channel isoforms have been identified (1). They are composed of single polypeptide chains containing four pseudorepeated domains (designated DI to DIV), each of which is composed of six transmembrane helical segments (S1 to S6); the pore region is formed from S5 to S6, including the intervening loop and selectivity filter (SF), from all four domains. Prokaryotic sodium channels, in contrast, are homotetramers of four identical polypeptide chains, each of which is equivalent to, and homologous with, one of the eukaryotic domains. Although there are as yet no crystal structures of eukaryotic sodium channels, crystal structures of several prokaryotic sodium channels in different conformational states have been determined, including ones with closed (2), partially (3) and fully (4) open pores, and two potentially inactivated forms (5, 6). Mutations in human sodium channels (hNavs) have been linked to channelopathies such as epilepsy, cardiac arrhythmia, and chronic pain syndromes; consequently sodium channel blockers have been developed as anticonvulsant, antiarrhythmic, and local anesthetic drugs (7–10). Several eukaryotic calcium channel blocker drugs have previously been found to bind and block prokaryotic sodium channels (11–13).
Results
Drug Antagonism of Sodium Currents in NavMs and Human Nav1.1 Channels.
In this study, we have shown (Fig. 1A) that the clinically important antiepileptic sodium channel drug lamotrigine blocks both the prokaryotic NavMs (from M. marinus) and the eukaryotic hNav1.1 channels at similar (∼100 μM) potencies (Fig. 1C and Table 1) and that the block is reversible in both (Fig. 1D). This suggested that the NavMs channel could be a suitable analog for characterization and identification of other eukaryotic channel blockers and could be used to identify drug binding sites within the channel by crystallographic means.
Fig. 1.

Comparison of lamotrigine and PI1 effects on NavMs and human Nav1.1 channels. (A) HEK-293T cells transfected with either (Left) NavMs or (Right) hNav1.1 were patch clamped in the whole-cell configuration. (Insets) Voltage-gated Na+ currents activated by a 0.2-Hz train of 0.5-s depolarizations to −30 from −180 mV for NavMs and 0.1-s depolarizations to −10 from −120 mV for Nav1.1. The onset of block was assessed after 2 min extracellular application of drug at concentrations as indicated by the colored boxes. The white boxes are the control application of 0.1% DMSO at the maximum concentration used as a vehicle for the compound. Percentage of current recovery is denoted by the dotted lines (±SEM, n = 4–5 cells). (B) PI1 effects on NavMs and hNav1.1 channel functions (same conditions as in A). (C) Concentration–INa block relationships (±SEM, n = 4–6 cells) for both lamotrigine and PI1 for both NavMs and hNav1.1 channels are shown. IC50 was estimated by fitting the average percent current block to the Hill equation. The IC50 values for Nav1.1 block by lamotrigine and PI1 were 196 μM and 373 nM, respectively. The IC50 values for NavMs block by lamotrigine and PI1 were 273 μM and 178 nM, respectively. (D) Percent recovery of sodium current after <70% current block by 1 mM lamotrigine or 1 μM PI1. Examples of current recovery are denoted by the red text and dotted lines in A and B (±SEM, n = 4–6 cells).
Table 1.
Sodium channel antagonist chemical and pharmacological properties
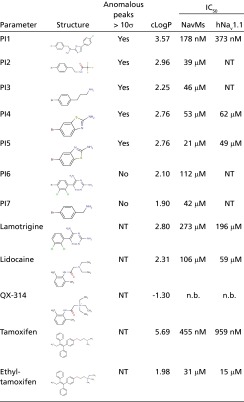 |
Octanol-water partition coefficient (clog P) was calculated using Molinspiration software. The IC50 for each molecule is listed for the NavMs and human Nav1.1 sodium channels. PI1, 2-(4-bromophenyl)-1-(5-(4-chlorophenyl)-1H-imidazol-2-yl)ethanamine; PI2, N-[2-(4-bromophenyl)-ethyl]-2,2,2-trifluoro-acetamide; PI3, 3-(4-bromophenyl)propanamine; PI4, amino-6-bromobenzothiazole; PI5, amino-5-bromobenzothiazole; PI6, 4-bromo-lamotrigine; PI7, 4-bromobenzylamine; QX-314, charged lidocaine; NT, not tested; n.b., no block observed using a range of 10–1,000 μM.
Channel blocking compounds bind to eukaryotic channels with 1:1 stoichiometries (14); because these channels are composed of four similar but nonidentical domains, this corresponds to only a single drug binding for every four domains present in the pseudotetrameric channel. For prokaryotic channels, the corresponding drug-to-channel stoichiometry would be one drug per homotetrameric channel. Because there are four equivalent sites in each tetramer, only one of those sites will be occupied at random, whereas the others remain empty, presumably because binding at one site would prevent binding at the other three by either steric clash or electrostatic repulsive forces. Consequently, in a prokaryotic channel/drug complex crystal structure, the maximal drug occupancy at any monomer in the tetramer is likely to be 0.25, which is too low for clear crystallographic identification. Hence, to be able to locate the sites of channel blockers in crystal structures, it was necessary to examine compounds that contain bromine atoms as part of their structures because bromines produce anomalous diffraction signals clearly visible in anomalous electron density maps, even at low occupancy.
The bromine-containing compound chosen for initial diffraction studies, 2-(4-bromophenyl)-1-(5-(4-chlorophenyl)-1H-imidazol-2-yl)ethanamine (designated compound PI1) (Table 1), was tested for its functional effects on both the NavMs and Nav1.1 channels; it was found to block both types of channels (Fig. 1B) with potencies >1,000 times those for lamotrigine (Fig. 1C and Table 1). Development of current block for lamotrigine follows single exponential time courses (τ = 450 ms), whereas that for PI1 is a double exponential (τ1 = 490 ms, τ2 = 3.7 s) (Fig. S1). The faster rate constants are similar for the two channel blockers, but the slower rate constant for PI1 block likely reflects an irreversible interaction with the channels. This suggests that PI1 binding is tight and thus a good candidate for producing cocrystals.
Blocker Binding Site in the NavMs-Pore/Drug Cocrystal Structure.
Crystalline complexes of PI1 and the NavMs-pore (in the fully open conformation) (4) (Table 2) produced anomalous electron density maps, which showed four peaks inside the cavity (Fig. 2A), near the fenestrations (Fig. 2B). Refinement of the anomalous signal for the bromines indicated that the structure contains the PI1 compound, with each site having an occupancy of ∼0.3 (±0.02), closely consistent with the expected occupancies of 0.25. There were no significant difference densities seen in the protein regions of the map calculated using the apo pore structure and the PI1-bound data, suggesting that drug binding does not cause conformational changes in the channel. However, the cocrystals do exhibit considerably less density in the middle of the SF (Fig. 2C) than seen in the apo structure (Fig. 2D). The density that is missing has previously been proposed to be due to sodium ions (3), suggesting that block in the cavity may inhibit sodium ion occupancy of the entire channel.
Table 2.
Data collection and refinement statistics (molecular replacement) for the wild-type and T207A/F214A mutant proteins in the apo form and in complex with PI1
| Parameter | Wild-type apo | Wild-type PI1 complex | Wild-type PI1 complex* | T207A/F214A apo | T207A/F214A PI1 complex |
| Protein Data Bank ID | 4CBC | 4P9O | 4PA9 | 4P2Z | 4P30 |
| Data collection | |||||
| Space group | C2221 | C2221 | C2221 | C2221 | C2221 |
| Cell dimensions | |||||
| a, b, c (Å) | 79.90, 331.7, 79.93 | 80.45, 328.46, 80.40 | 80.32, 330.55, 80.25 | 80.01 333.04 80.39 | 80.26, 334.26, 80.04 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 50.0–2.67 (2.8–2.67) | 50.0–2.89 (3.1–2.89) | 50.0–3.43 (3.7–3.43) | 45.68–3.08 (3.30–3.08) | 45.73–3.31 (3.57–3.31) |
| Rpim | 0.064(0.296) | 0.191 (0.714) | 0.100 (0.244) | 0.133 (0.615) | 0.090 (0.363) |
| I/σI | 11.7 (2.7) | 12.7 (3.6) | 13.2 (4.3) | 8.4 (1.6) | 7.3 (2.4) |
| Completeness (%) | 99.5 (96.2) | 99.8 (99.0) | 99.7 (97.7) | 93.6 (71.1) | 98.8 (98.1) |
| Redundancy | 19.7 (7.8) | 13.5 (13.9) | 31.0 (13.2) | 5.1 (2.1) | 3.3 (3.4) |
| Refinement | |||||
| Resolution (Å) | 45.5–2.67 (2.75–2.67) | 43–2.89 (2.95–2.89) | 45.4–3.43 (3.55–3.43) | 45.68–3.08 (3.195–3.085) | 31.03–3.31 (3.428–3.309) |
| No. reflections | 30,860 (2,906) | 24,368 (2,372) | 18,472 (2,690) | 18,903 (1,275) | 16,314 (1,556) |
| Rwork/Rfree | 27.6/29.9 (31.5/35.2) | 21.4/25.1 (20.8/23.7) | 28.7/29.4 (23.4/22.6) | 26.8/29.35 (35.62/42.22) | 21.22/23.96 (31.62/38.10) |
| No. atoms | |||||
| Protein | 2,912 | 2,856 | 2,856 | 2,832 | 2,839 |
| Ligand/ion | 125 | 124 | 156 | 117 | 126 |
| Water | 88 | 253 | 18 | 18 | 25 |
| B-factors | |||||
| Protein | 70.7 | 61.5 | 61.2 | 63.0 | 74.6 |
| Ligand/ion | 78.3 | 75.3 | 76.2 | 86.0 | 95.5 |
| Water | 55.2 | 49.2 | 15.0 | 20.9 | 57.8 |
| rmsd | |||||
| Bond lengths (Å) | 0.005 | 0.010 | 0.010 | 0.016 | 0.018 |
| Bond angles (Å) | 0.93 | 1.08 | 1.09 | 1.85 | 1.86 |
Crystals were obtained after cocrystallizing the wild-type protein with PI1. All of the other compound/complex structures derive from soaking experiments.
Fig. 2.

Binding site of PI1 in the NavMs pore. (A) Crystal structure of the NavMs-pore in complex with PI1. The four monomers are depicted in different colors in surface representation. The view is a slice through the middle of the structure, in the cavity region, viewed from the bottom of the pore. The anomalous difference map (which indicates the locations of the bromine atoms at the top of the cavity) is overlaid as a red mesh contoured at 3 σ and corresponds with ∼0.3 occupancy/site. (B) Side view of the pore showing the anomalous difference density location adjacent to the entrance of one of the transmembrane fenestrations, between two monomers. For reference, the black bars indicate the approximate locations of the top and bottom of the bilayer. (C) Side view slice through the middle of the pore, showing the lack of density in the SF (indicated by the black box in D) for the PI1 cocrystals. The protein structure (in cartoon, ribbon, and stick representation) is overlaid with (2Fo-Fc) and (Fo-Fc) difference electron density maps contoured at 1.5 σ (blue) and 3 σ (green), respectively. The anomalous difference map contoured at 5 σ is shown in red. The best docked position of PI1 is shown in stick representation, for reference. (D) View as in C but for the apo crystals. The density in the center of the SF corresponds to sodium ions (28). (E and F) In silico docking results using the apo NavMs-pore structure and PI1. The position of the best predicted site (in stick representation) is overlaid on a surface representation of the protein crystal structure, with the position of the bromine in the cocrystals indicated as a solid red ball) and the anomalous density map (red mesh). The distal end of PI1 protrudes into the bottom of the SF. E corresponds to a side view of a slice through the center of the channel (corresponding to the direction in A), whereas F corresponds to a slice through the center from the perpendicular direction (which corresponds to the direction of the view seen in B). (G) Detailed view of the PI1 binding pocket. The locations of the residues that were mutated for the functional studies (T207 and F214, which effect block, and T206 and I215, which do not), and their distances to the crystallographically-located bromine atom are indicated by orange dashed lines. The hydrogen bond between the imidazole nitrogen of PI1 and the Thr176 main chain carbonyl group predicted from docking is shown as a black dashed line.
The nonbromine atoms of PI1 could not be confidently placed into the difference electron density map, owing to the difficulty of interpreting electron density for light atoms at low occupancies. This problem is further exacerbated because the compound replaces partially occupied water sites at the top of the cavity, resulting in compensating loss of water density and gain of compound density. Consequently, the complete location of the compound was examined using in silico docking into the crystal structure of the apo pore. Because a racemic mixture (the asymmetric carbon is indicated by * in Table 1) was used in the crystallizations, both R- and S- enantiomers were used in the docking calculations. The highest affinity for the R- enantiomer (Fig. 2 C, E, and F) was −8.7 kcal/mol (corresponding to an apparent KD of ∼300–400 nM, for temperatures ∼21–23 °C), which was significantly better than expected for a random ligand and similar to the IC50 value determined experimentally for the R- enantiomer (Table 1). It is notable that the S- enantiomer is far less efficacious (IC50 ∼200-fold greater) than the R- enantiomer (Fig. S2), consistent with the docking result. No positional constraints were applied to the bromine during docking calculations, but the top hits placed the bromine very close to the crystallographically observed location (Fig. 2 E and F). The best hit places the remainder of the bromophenyl end of the PI1 structure adjacent to residues T207 and F214 (Fig. 2G) (numbering according to the full-length NavMs sequence; Fig. 3A). The distal (chlorine) end of the compound extends into the SF (Fig. 2 E and F), forming a hydrogen bond between its imidazole nitrogen and the main chain carbonyl oxygen of Thr176 at the bottom of the SF (Fig. 2G), which would potentially block transmembrane translocation of sodium ions. Placing the compound in the equivalent sites in all four monomers shows the distal ends would clash in the region of the SF (Fig. S3A), thus precluding there being more than one compound present per tetramer. Nevertheless, despite the low occupancy of the compound at any one site when the electron density map is contoured at low levels (Fig. S3B), there is some poorly defined density in the SF at a completely different location than that for the ions. This could correspond to partial occupancy by the distal ends of the PI1 compound in four different monomers that overlap, but not in a way that reinforces the signal of a single structure. Consequently it is evidence for the location of this end of PI1, but is not interpretable on a detailed molecular level.
Fig. 3.

Mutational effects on NavMs blocker efficacy. (A) Multiple sequence alignments of the S6 helices of NavMs (UnitProt A0L5S6) and the four domains of human Navs (UnitProt P35498 for Nav1.1, Q99250 for Nav1.2, Q9NY46 for Nav1.3, P35499 for Nav1.4, Q14524 for Nav1.5, Q9UQD0 for Nav1.6, Q15858 for Nav1.7, Q9Y5Y9 for Nav1.8, and Q9UI33 for Nav1.9). The residues in the human channels shown by site-directed mutagenesis to be important for drug binding are highlighted by the color of their domains (blue bar for domain I, dark gray for domain II, dark green for domain III, and purple for domain IV). Residues where the NavMs and human Navs are identical are denoted by “*” in the bottom rows; conservative substitutions are denoted by “:” and “.” Residue names are colored by residue type. Residues mutated in NavMs in this study are indicated by black arrows (those that produce effects) and gray arrows (those that do not produce effects). (B) Effects of PI1 on mutated channels T207A (Top), F214A (Middle), and the T207A:F214A (Bottom) double mutant. F214A and T207A:F214A channels were depolarized for 1 s to compensate for the slower inactivation intrinsic to the mutated channels. (Insets) Sodium currents activated by a 0.2-Hz train of 0.5-s depolarizations to −30 from −180 mV in control and in conditions where extracellular PI1 was applied (colored boxes). Graphs depict the time course of INa block by 2–3 min applications of PI1. (C) (Left) Reduction in potency for PI1 due to the T207A and the F214A and the T207:F214 double mutations (±SEM, n = 4–6 cells). (Right) Magnitudes of recovery from block by PI1 for each of these mutants after bath exchange for 3–5 min.
The bromine sites are close to the transmembrane fenestrations in the sides of the channel (Fig. 2B). When the 10 top R- enantiomer hits from the docking studies are superposed onto the NavMs structure, their positions form a continuous series starting from the transmembrane region near the fenestration, through the fenestration, into the center of the cavity (Fig. S3C). Such an entry path for hydrophobic drugs was originally proposed by Hille (15) in the absence of structural information; these crystallographic/computational data show that such a pathway would not require any rearrangement in the pore structure to accommodate blocker entry and binding, nor would the entry or binding be in anyway impeded by the presence of the voltage sensor or S4-S5 linker.
Correspondence of the NavMs Binding Site with Mutationally Defined Eukaryotic Drug Binding Sites.
Residues F1774 and Y1781 of DIV (and residues in DI and DIII) of eukaryotic sodium channels have been identified as important for binding channel-blocker compounds (7–10, 16–18) and use-dependence drug-binding (8, 17). The residues in DIV correspond to residues T207 and F214 in NavMs (Figs. 2G and 3A). To directly test whether blocker binding involves these residues in NavMs, we mutated T207 and F214 and examined their function in the presence (Fig. 3B) and absence (Fig. S4) of the PI1 compound. As a control, we also mutated T206 and I215 that do not appear to face the blocker binding site (Fig. 2G). Both T207A and F214A significantly reduce the potency of PI1 (Fig. 3 B and C), with the double mutant T207A:F214A resulting in an increase in IC50 of more than 100-fold. F214A alone and the double mutant also produce significant effects on the kinetics of inactivation (Fig. S4), although T207A does not. The T206A and I215A control mutants have essentially no effect on PI1 kinetics (Fig. S5 A and B) or potency (Fig. S5C).
The crystal structures of the double mutant in the presence and absence of PI1 (Table 2) are very similar to the PI1-containing crystals of wild-type pores, except for small changes associated with the side chain replacements. However, for the PI1 double mutant cocrystals there was no anomalous signal visible, suggesting greatly diminished (or no) binding, consistent with the electrophysiological studies.
The T207A:F214A and T214A mutants also show significant reduction in lamotrigine binding with respect to wild-type channels (albeit to a lesser extent than they do for the PI1 compound) (Fig. S6), suggesting that PI1 binds in a similar region as this clinically used drug. However, the T207A mutant has essentially no effect on the potency of lamotrigine, indicating that the receptor sites for PI1 and lamotrigine may overlap but are not identical. This correlates with mutational studies of rat Nav1.1 channels (16), where local anesthetics and antiepileptic drugs were proposed to have different but overlapping channel blocker sites.
Structural and Functional Characterization of Other Channel Blockers.
To identify characteristic features present in other effective channel blockers, parallel electrophysiology studies (Fig. 4A and Table 1) and cocrystallizations (Table S1) were undertaken on six related hydrophobic compounds (PI2 to PI7; Table 1) that contain covalently bound bromines. The compounds all have a log P (octanol–water partition coefficient) of ≥2. Binding in the crystal was defined by an anomalous signal of ≥10 σ. Two of the structurally related compounds (PI6 and PI7) do not bind to the crystals, despite blocking the NavMs channel at midmicromolar potency (Fig. 4A and Table 1). Interestingly, only these two compounds exhibit a high percentage of sodium current recovery after block (Fig. 4B, Lower), indicative of reversible binding. The bromine atoms of all PI compounds for which an anomalous signal is observed are in very similar positions to those of the bromines of PI1 (Fig. 4C). Even the short analog (PI3) did not exhibit a stronger anomalous signal, which would have indicated a higher occupancy, which suggests that although it was not sterically prevented from entering the SF, electrostatic repulsion (due to the distal amino group) was sufficient to prevent occupancy of more than one binding site in the tetramer. Although none of the other brominated compounds were as effective as PI1 in blocking NavMs (178 nM), they had IC50 values ranging from 21 to 112 μM, and all were more potent than lamotrigine (273 μM) (Fig. 4A and Table 1). The least potent of the brominated compounds was a derivative of lamotrigine (PI6), one of the compounds that did not produce a significant anomalous signal. Common features of the brominated compounds that produced functional block were that they contained a bromine attached to a phenyl ring with an amino or amido moiety separated from the bromine by approximately six to seven atoms. Their similar potencies toward the NavMs and Nav1.1 channels (Fig. 4B and Table 1) further support the parallel nature of blocker binding by the NavMs and eukaryotic channels.
Fig. 4.

Binding of other channel blockers. (A) Comparisons of the channel-blocking effects of compounds listed in Table 1. Plots of drug concentrations versus block of the NavMs current were fit by the Hill equation. The IC50s are listed in Table 1 (±SEM, n = 4–8 replicates). (B) Comparisons of channel blocker potencies, hydrophobicities, and recoveries from block for compounds listed in Table 1. (Upper) The potencies (IC50) for NavMs (filled circles) and hNav1.1 (open circles), and octanol:water partition coefficient log P (blue squares) are plotted for each drug listed in the lower panel. (Lower) Percentage of current recovered after ≥60% sodium current block. (C) The structure of the NavMs-pore (ribbon representation with the same monomer colors as in Fig. 2) overlaid with the anomalous difference maps contoured at 5 σ for compounds PI1 to PI5 (red for PI1, cyan for PI2, magenta for PI3, yellow for PI4, green for PI5), showing the similarity of the positions of the bromine atoms.
A number of other known blockers of human sodium channels were also tested for their potency and channel blocking effects on the two types of channels (Fig. 4B and Table 1). A clear correspondence in binding to NavMs and hNav1.1 is found for all of the channel blockers, including those with different mechanisms that are believed to have overlapping but not identical sites to lamotrigine, such as the analgesic lidocaine. The cancer drug tamoxifen, which has documented effects on human sodium channels (19), also shows similar behavior on both types of channels. The charged homolog of lidocaine, QX-314 (20), does not inhibit sodium currents from either channel (Table 1), whereas the charged version of tamoxifen (ethyl tamoxifen) is several orders of magnitude less potent in both types of channels than tamoxifen itself (Fig. 4B and Table 1). Strikingly, the IC50s determined for all of the 12 compounds tested are within a factor of 3 (half-log unit) for the NavMs and hNav1.1 channels (Fig. 4B and Table 1), demonstrating that the drug potencies for the two channels are very similar, and strongly suggesting that the prokaryotic NavMs and eukaryotic hNav1.1 channels have similar binding sites for small molecule channel blockers. Higher log P values correlate with increased potency for both channels, suggesting that hydrophobic compounds better access the transmembrane blocker sites within the channels, consistent with access via the transmembrane fenestrations rather than through the SF or via extramembranous access to the channel pore. Nevertheless, although these findings are encouraging for using the NavMs as a model for mammalian sodium channel pharmacology, it should be kept in mind that there are also likely to be differences in the effects of drugs that block mammalian Nav channels in the fast inactivated state, which is not found in prokaryotic channels.
Discussion
Despite millions of years of evolution and substantial alterations to the molecular structure of sodium channels (including quaternary structure and SF changes), the blocking mechanisms and affinities of the prokaryotic sodium channel NavMs and the human sodium channel Nav1.1 for small hydrophobic compounds are highly correlated. Clinically important eukaryotic channel drugs, such as lamotrigine and lidocaine, and other related compounds produce remarkably similar channel blocking effects in NavMs and in human Nav1.1 channels. Crystallographic and docking studies suggest that the PI1 blocker compound binds at the top of the pore cavity, near one of the fenestrations, which would act as a portal to enable hydrophobic drug entry into the pore through the transmembrane region of the bilayer by a mechanism that does not require traversing the SF or entry through the channel gate. The proximity of the binding site to equivalent residues that have been identified as being important for drug binding in eukaryotic channels, and the validation of these sites by combined mutational/electrophysiological/crystallographic studies of NavMs, suggest that this prokaryotic channel can provide a valuable 3D template for the design of new candidate channel blocking drugs for human voltage-gated sodium channels, augmenting traditional pharmacological methods.
Materials and Methods
Materials.
2-(4-bromophenyl)-1-(5-(4-chlorophenyl)-1H-imidazol-2-yl)ethanamine (designated PI1 in the Protein Data Bank file), N-[2-(4-bromophenyl)-ethyl]-2,2,2-trifluoro-acetamide (PI2), 3-(4-bromophenyl) propanamine (PI3), 2-amino-6-bromobenzothiazole (PI4), 4-bromo lamotrigine (PI6), and 4-bromobenzylamine (PI7) were provided by Pfizer Neusentis. 2-amino-5-bromobenzothiazole (PI5), thallium(I) nitrate, lidocaine, QX-314 (N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide), and lamotrigine were bought from Sigma–Aldrich. Ethylbromide tamoxifen was synthesized by Dr. A. Christy Hunter (University of Brighton School of Pharmacy, Brighton, United Kingdom). Molinspiration software (www.molinspiration.com) was used to calculate the log P values in Table 1 and Fig. 4B. MarvinSketch version 6.1.5 (www.chemaxon.com) was used for drawing the chemical structures.
Protein Expression, Purification, and Crystallization.
The NavMs-pore, including its full-length C-terminal domain (NavMs-pore-FL), was purified according to Bagnéris et al. (4). The crystals were grown as previously described (4) with the following modifications: crystals used for soaking experiments were obtained by preincubating the protein (15 mg/mL) overnight at 4 °C with thallium(I) nitrate (100 mM stock in 100% DMSO) in a 10 molar excess before crystallization. Drops (0.5 μL) containing crystals with large dimensions (∼50–200 μm) were soaked with 0.5 μL of 5 mM solutions of the compounds made using 100% DMSO stocks diluted with stabilizing solution to a final concentration of 5% (vol/vol) DMSO. The stabilizing solution contained 1 volume of gel filtration buffer (10 mM Tris, 100 mM NaCl, 0.52% Hega10, pH 7.5) and 1 volume of crystallization solution [0.1 M trisodium citrate, 0.1 M Tris⋅HCl (pH 8), 34% (vol/vol) PEG400]. The crystals obtained by cocrystallization with drug compounds (100 mM in 100% DMSO) were produced by incubating the protein (15 mg/mL) overnight at 4 °C with the drug in a 10 molar excess before crystallization.
Crystal Data Collection, Processing, Refinement, and Display.
Multiple data sets were collected for each crystal type at beamlines IO3, IO4, or IO4-1 (Diamond Light Source), beamline PROXIMA1 (Soleil), or beamline ID23-1 (European Synchrotron Radiation Facility). All data were indexed and integrated with XDS (21) and scaled with Aimless (22). Multiple datasets of comparable resolution for one crystal type were combined with the help of Blend (J. Foadi and P. Aller, Diamond Light Source). All subsequent data analyses, including the calculation of anomalous difference maps, were carried out with the CCP4 suite (23). The structures were solved by rigid-body refinement in Buster (24), using Protein Data Bank ID 3ZJZ (4); subsequently all atom refinement was undertaken using Phenix (25) and/or Buster. Bromine positions were identified with Phaser (26) using the known partial structure in the phase calculation. Refinement of anomalous occupancies depends heavily on the B value used for the bromines. This was fixed to that of the Wilson B factor for the protein and hence may be inexact but indicative. The same reflections were omitted for all datasets for the calculation of Rfree to avoid model bias in this statistic. The resolution limits of the crystals (Table 2 and Table S1) range from 2.67 Å to 3.43 Å.
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 4CBC (apo structure), 4P9O and 4PA9 (PI1 complexes), 4P2Z (double mutant apo), 4P30 (double mutant PI1 “complex”), and 4OXS, 4PA3, 4PA4, 4P9P, 4PA6, and 4PA7 (PI2–PI7 complexes, respectively).
Bioinformatics.
Multiple sequence alignments used Clustal-Omega (27). Docking experiments were carried out with Glide (version 5.9, Schrödinger, LLC) via the Maestro interface using the Protein Data Bank ID 4P9O structure, after removal of the bromine atoms and water molecules. An extended grid (14 Å per side for the inner box, and 39 Å per side for the outer box) was used, centered on the bromine atom position in one of the monomers. Glide Emodel scores were used for ranking. PI1 was prepared using LigPrep (version 2.6, Schrödinger, LLC).
Electrophysiology.
HEK293T cells were transiently transfected with C-terminally His-tagged NavMs or hNav1.1, seeded onto glass coverslips, and placed in a perfusion chamber for experiments in which extracellular conditions could be altered. The exchange rate within the bath was 3–5 mL/min. All cells were voltage clamped in the whole-cell configuration at 21–23 °C. For the experiments in Fig. S1, rapid exchange was used in which patched cells were moved into the path of a stream of perfused drug to achieve rapid transition from control to drug conditions. Unless otherwise noted, extracellular solutions contained (in mM) NaCl (150); CaCl2 (2); MgCl2 (1); HEPES (10); pH 7.4; and the intracellular (pipette) solution contained (in mM): CsMES (90); NaCl (20); HEPES (20); 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA)-tetracesium (20); MgCl2 (2); pH 7.3. CaCl2 was added to achieve 100 nM free Ca2+. Data were analyzed by Igor Pro-6.00 (Wavemetrics). Residual leak (> −100 pA) and capacitance were subtracted using a standard P/-4 protocol. Current–voltage relationships were fit with (V − Vrev)/{1 + exp[(V − V1/2)/k]}, where Vrev is the extrapolated reversal potential. The equation for the exponential fits used in Fig. S1 was: f(x) = B + A•exp[(1/τ)x], where τ is the time constant of current block. All drug stocks were initially formulated in DMSO and diluted 100- to 1,000-fold into extracellular saline solutions. Percent INa block was calculated by (Idrug − Icontrol/Icontrol) × 100, where Icontrol is the average current measured during the minute before drug application, and Idrug is the amount of current 2–4 min after drug application. Drug concentration–INa block relationships were fit to the Hill equation to estimate drug potency (IC50). Percent current recovery was calculated by (Irecovery − Idrug) × 100, where Irecovery is the recovered current measured 3–5 min after removal.
Supplementary Material
Acknowledgments
We thank Florence Thomas for initial involvement in the purification and crystallization of pores with nonbrominated compounds; Dr. Sharan Bagal (Pfizer Neusentis) for help in selecting compounds for study; Cesar de Oliveira (Pfizer) for identifying the enantiomeric forms of PI1; Dr. Andrew Turnbull from Cancer Research Technology for helpful advice on soaking of compounds; Dr. Andrew Sharff from Global Phasing Limited for advice on refinement; Gregory Dick (West Virginia University School of Medicine) and Dr. A. Christy Hunter (University of Brighton School of Pharmacy) for generously providing ethylbromide tamoxifen; Dr. Ambrose Cole (Birkbeck College) for help with crystallographic data collection; and the beamline scientists at the Diamond Light Source (beamlines IO4-1, Pierpaolo Romano; IO4, Dave Hall; IO3, Katherine McAuley), Soleil (beamline PROXIMA 1, Andrew Thompson), and European Synchrotron Radiation Facility (beamline ID23-1, Alexander Popov). This work was supported by Grants BB/H01070X, BB/L006790, and BB/J020702 from the United Kingdom Biotechnology and Biological Science Research Council (to B.A.W.). P.G.D. was supported by National Institutes of Health Grant T32-HL007572.
Footnotes
The authors declare no conflict of interest.
Database deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID code 4CBC (apo structure), 4P9O and 4PA9 (PI1 complexes), 4P2Z (double mutant apo), 4P30 (double mutant PI1 “complex”), and 4OXS, 4PA3, 4PA4, 4P9P, 4PA6, and 4PA7 (P2–P7 complexes, respectively)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1406855111/-/DCSupplemental.
References
- 1.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57(4):397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 2.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCusker EC, et al. The open conformation of a voltage-gated sodium channel reveals the transmembrane pathway and mechanism of channel opening and closing. Nat Commun. 2012;3:1102. doi: 10.1038/ncomms2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagnéris C, et al. Role of the C-terminal domain in the structure and function of tetrameric sodium channels. Nat Commun. 2013;4:2465. doi: 10.1038/ncomms3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486(7401):135–139. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X, et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature. 2012;486(7401):130–134. doi: 10.1038/nature11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu G, et al. Differential interactions of lamotrigine and related drugs with transmembrane segment IVS6 of voltage-gated sodium channels. Neuropharmacology. 2003;44(3):413–422. doi: 10.1016/s0028-3908(02)00400-8. [DOI] [PubMed] [Google Scholar]
- 8.Hanck DA, et al. Using lidocaine and benzocaine to link sodium channel molecular conformations to state-dependent antiarrhythmic drug affinity. Circ Res. 2009;105(5):492–499. doi: 10.1161/CIRCRESAHA.109.198572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci USA. 1996;93(17):9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang GK, Quan C, Wang SY. Local anesthetic block of batrachotoxin-resistant muscle Na+ channels. Mol Pharmacol. 1998;54(2):389–396. doi: 10.1124/mol.54.2.389. [DOI] [PubMed] [Google Scholar]
- 11.Ren D, et al. A prokaryotic voltage-gated sodium channel. Science. 2001;294(5550):2372–2375. doi: 10.1126/science.1065635. [DOI] [PubMed] [Google Scholar]
- 12.Nurani G, et al. Tetrameric bacterial sodium channels: Characterization of structure, stability, and drug binding. Biochemistry. 2008;47(31):8114–8121. doi: 10.1021/bi800645w. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Goodchild SJ, Ahern CA. Local anesthetic inhibition of a bacterial sodium channel. J Gen Physiol. 2012;139(6):507–516. doi: 10.1085/jgp.201210779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. J Gen Physiol. 1983;81(5):613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hille B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69(4):497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarov-Yarovoy V, et al. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J Biol Chem. 2001;276(1):20–27. doi: 10.1074/jbc.M006992200. [DOI] [PubMed] [Google Scholar]
- 17.Desaphy JF, et al. Molecular determinants of state-dependent block of voltage-gated sodium channels by pilsicainide. Br J Pharmacol. 2010;160(6):1521–1533. doi: 10.1111/j.1476-5381.2010.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265(5179):1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 19.Smitherman KA, Sontheimer H. Inhibition of glial Na+ and K+ currents by tamoxifen. J Membr Biol. 2001;181(2):125–135. doi: 10.1007/s00232-001-0016-2. [DOI] [PubMed] [Google Scholar]
- 20.Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007;449(7162):607–610. doi: 10.1038/nature06191. [DOI] [PubMed] [Google Scholar]
- 21.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans PR. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 23.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bricogne G, et al. BUSTER Version 2.10.0. Cambridge, UK: Global Phasing Ltd.; 2011. [Google Scholar]
- 25.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 4):352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ulmschneider MB, et al. Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc Natl Acad Sci USA. 2013;110(16):6364–6369. doi: 10.1073/pnas.1214667110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.