

Significance
It is widely accepted that prosurvival B-cell lymphoma 2 (Bcl-2) family members not only inhibit apoptosis but also negatively regulate autophagy by binding to Beclin 1. Herein, we challenge this view and provide genetic and biochemical evidence that the effects of prosurvival Bcl-2 family members on autophagy are instead an indirect consequence of their inhibition of apoptosis mediators Bcl-2–associated X (Bax) and Bcl-2 homologous antagonist/killer (Bak). We show that in the absence of Bax and Bak, antagonizing or altering the levels of prosurvival Bcl-2 family members has no detectable impact on autophagy. Because several inhibitors of both autophagy and Bcl-2 are in clinical trials for the treatment of cancer, it is important to understand the cross-talk between these pathways.
Keywords: ABT-737, BH3 mimetic, Bim, LC3B, LC3
Abstract
Antiapoptotic B-cell lymphoma 2 (Bcl-2) family members such as Bcl-2, myeloid cell leukemia 1 (Mcl-1), and B-cell lymphoma-X large (Bcl-xL) are proposed to inhibit autophagy by directly binding to the BH3 domain of Beclin 1/Atg6. However, these Bcl-2 family proteins also block the proapoptotic activity of Bcl-2–associated X (Bax) and Bcl-2 homologous antagonist/killer (Bak), and many inducers of autophagy also cause cell death. Therefore, when the mitochondrial-mediated apoptosis pathway is functional, interpretation of such experiments is complicated. To directly test the impact of the endogenous antiapoptotic Bcl-2 family members on autophagy in the absence of apoptosis, we inhibited their activity in cells lacking the essential cell death mediators Bax and Bak. We also used inducible lentiviral vectors to overexpress Bcl-2, Bcl-xL, or Mcl-1 in cells and subjected them to treatments that promote autophagy. In the absence of Bax and Bak, Bcl-2, Bcl-xL, and Mcl-1 had no detectable effect on autophagy or cell death in myeloid or fibroblast cell lines. On the other hand, when Bax and Bak were present, inhibiting the prosurvival Bcl-2 family members stimulated autophagy, but this correlated with increased cell death. In addition, inhibition of autophagy induced by amino acid starvation, etoposide, or interleukin-3 withdrawal did not affect cell death in the absence of Bax and Bak. These results demonstrate that the antiapoptotic Bcl-2 family members do not directly inhibit components of the autophagic pathway but instead affect autophagy indirectly, owing to their inhibition of Bax and Bak.
Autophagy is a process in which cellular material is degraded so that homeostasis can be maintained when nutrients are scarce. During macroautophagy (henceforth referred to as autophagy), cytoplasm is enveloped by the formation of the autophagosome, which when fused to the lysosome forms the autophagolysosome. This organelle degrades the enclosed cellular material and returns “building blocks” such as amino acids back to the cytoplasm. Autophagy was initially studied in yeast and subsequently in mammalian cells, where it has been proposed to be not only a mechanism to promote cell survival in conditions of starvation but also a mechanism by which cells can commit suicide (1).
Much of our understanding of the molecular mechanisms of autophagy has come from studying the highly conserved Atg (autophagy-related) proteins. As autophagy progresses, microtubule-associated protein 1 light chain 3 beta (LC3B)-I (Atg8 in yeast) in the cytoplasm is conjugated with phosphatidylethanolamine to form LC3B-II, which becomes associated with the autophagosomal membrane and is involved in its elongation. An increase in LC3B-II (and concomitant decrease in LC3B-I) is commonly used as a marker of autophagy. Because LC3B is also one of the only autophagy-associated proteins that remain attached to the autophagosome throughout the entire process, it is commonly used to visualize autophagosomes and autophagolysosomes.
In yeast, Vacuolar protein sorting 30 (Vps30)/autophagy-related protein 6 (Atg6) is a core component of the class III phosphatidylinositol 3-kinase (Vsp34) complex required for nucleation and assembly of the autophagosomal membrane. In a similar way, the mammalian Atg6 homolog, Beclin 1, is important for the formation of a complex with the mammalian PI3K Vps34 and nucleation of the autophagosome membrane. Beclin 1 was identified in yeast two-hybrid experiments using the antiapoptotic protein B-cell lymphoma 2 (Bcl-2) as bait (2). It has been proposed that when nutrients are abundant, Bcl-2 and the related proteins Bcl-xL and myeloid cell leukemia 1 (Mcl-1) bind to Beclin 1 via Beclin 1’s BH3 domain and thereby inhibit induction of autophagy (Fig. 1A) (3–5). According to this model, when nutrients are scarce, Bcl-2 is phosphorylated by JNK1, which prevents its binding to Beclin 1 and allows it to initiate formation of autophagosomes (6).
Fig. 1.

Inhibiting the prosurvival Bcl-2 family members does not promote nonapoptotic cell death or LC3B lipidation in the absence of Bax and Bak. (A) A simplified model illustrating the proposed role of the Bcl-2 family members. (B) MEFs were cotreated with 34 μM etoposide (VP-16) and indicated concentrations of ABT-737 for 96 h. Viability relative to cells not treated with ABT-737 was measured by the absence of PI uptake. The mean ± SD of two independent cell lines are shown relative (n = 4). (C) ABT-737 only promotes LC3B lipidation when Bax or Bak are present. Western blot of MEFs after a 4-h treatment with 1 μM ABT-737 or HBSS. (D) Induction of Bims does not alter LC3B levels. Bax−/−Bak−/− MEFs were treated with 1 μg/mL dox for 48 h or were cultured in HBSS for 4 h.
In apoptosis, the roles of Bcl-2, Bcl-2–like protein W (Bcl-w), Mcl-1, and B-cell lymphoma-X large (Bcl-xL) are well established. They inhibit apoptosis in two ways: first by directly binding the proapoptotic effector proteins Bcl-2–associated X (Bax) and Bcl-2 homologous antagonist/killer (Bak), and second by binding to BH3-only proteins such as Bim, thereby preventing them from activating Bax and Bak (7, 8). The prosurvival Bcl-2 family members bind to Bim, Bak, and Bax and the BH3 mimetic compound ABT-737 via the BH3 binding groove, the same region as the proposed binding site for Beclin 1, and they bind competitively (Fig. 1A). In contrast, the roles for prosurvival Bcl-2 family members in the regulation of autophagy have been less well characterized, not least because their inhibition or knockdown can also trigger Bax/Bak-dependent apoptosis. Furthermore, many of the pivotal studies on the role of Bcl-2 in autophagy were performed using overexpression of one or both binding partners, putting into question the physiological relevance of the interactions. Because of these caveats, we decided to further investigate whether the prosurvival Bcl-2 family members can inhibit autophagy in cells unable to undergo mitochondrial-mediated apoptosis owing to deletion of genes for the essential apoptosis effector proteins Bax and Bak.
To definitively determine whether the prosurvival Bcl-2 family members can regulate autophagy, we investigated whether inhibiting endogenous Bcl-2, Bcl-xL, and Mcl-1 could block autophagy in cells lacking Bax and Bak. We found that in both fibroblast and interleukin-3 (IL-3) dependent myeloid cell lines, treatment with the BH3 mimetic ABT-737, or altering the levels of Bcl-2, Bcl-xL, or Mcl-1, had no discernable effects on autophagy. Indeed, the prosurvival Bcl-2 proteins only affected LC3B lipidation when Bax and Bak were present and cells were undergoing apoptosis. These results do not support the model that direct interactions between Beclin 1 and the antiapoptotic Bcl-2 family members inhibit autophagy. Instead, they suggest that previously reported cellular effects are likely to be a consequence of inducing or modifying apoptotic events triggered by Bax or Bak (4, 9–11).
Results
Inhibiting the Antiapoptotic Bcl-2 Family Members Does Not Promote Nonapoptotic Cell Death or LC3B Lipidation in the Absence of Bax and Bak.
Structural studies have shown that a BH3 region of Beclin 1 can bind to the same pocket in the prosurvival Bcl-2 family proteins that interacts with the BH3-only proteins such as Bim (3). To investigate this potential mechanism, we initially inhibited the interaction of the endogenous proteins in fibroblasts using the BH3 mimetic ABT-737 (12). On the basis of previous reports (10), we hypothesized that ABT-737 would be able to compete with Beclin 1 for the prosurvival Bcl-2 family members, thereby releasing Beclin 1 to promote autophagy (Fig. 1A). Because ABT-737 has a much higher affinity for Bcl-2, Bcl-w, and Bcl-xL than for Mcl-1 (13), we took advantage of cells lacking the latter in case it could compensate for the others. The prosurvival protein A1 is inherently absent in mouse embryonic fibroblasts (MEFs) (14).
We performed these experiments in cells that also lack Bax and Bak so the effects on autophagy would not be confounded by the concurrent induction of apoptosis. This was important because many inducers of autophagy, including the chemotherapeutic agent etoposide (VP-16) or nutrient starvation by culturing in HBSS, are also potent inducers of apoptosis (compare Fig. S1A vs. Fig. 1B) (5, 15, 16). Because there are conflicting reports claiming autophagy can promote both cell survival and death, we measured the ability of the BH3 mimetic compound ABT-737 to affect cell death either positively or negatively when autophagy was induced by etoposide (5, 9, 17). After 96 h, etoposide induced moderate cell death in Bax−/−Bak−/− fibroblasts, as has been reported elsewhere (Fig. 1B) (5). Treatment with ABT-737 alone caused an expected dose-dependent decrease in viability in cells lacking Mcl-1 when Bax and Bak genes were intact, but in Bax−/−Bak−/− cells, ABT-737 neither increased nor decreased the amount of cell death induced by etoposide (Fig. 1B).
Because nonapoptotic cell death was unaffected by ABT-737, we investigated whether this compound could directly regulate autophagy, by monitoring conjugation of LC3B-I with phosphatidylethanolamine to form LC3B-II, a standard measure of autophagy (16). Both etoposide and amino acid starvation increased the ratio of LC3B-II to -I, as expected (Fig. S1B). If Bcl-2, Bcl-w, or Bcl-xL bind to and block Beclin 1, inhibiting this interaction with ABT-737 should promote autophagy. In contrast, we found that addition of ABT-737 to Bax−/−Bak−/− or Bax−/−Bak−/−Mcl-1−/− fibroblasts had no effect on LC3B conversion, whether the cells were undergoing basal levels of autophagy or had the autophagy pathway stimulated with etoposide or HBSS (Fig. S1B). By performing experiments in Bax/Bak doubly deficient cells, we avoided confounding factors caused by induction of apoptosis and by the ability of the prosurvival Bcl-2 family members to inhibit this type of cell death. Because the BH3-mimetic was able to kill Mcl-1−/− MEFs under these conditions (Fig. S1C), it is clear that under this experimental condition Bcl-2, Bcl-w, and Bcl-xL were efficiently inhibited. Indeed, ABT-737 was able to induce LC3B lipidation in Mcl-1−/− cells that were actively undergoing apoptosis but not in the equivalent lines lacking Bax and Bak (Fig. 1C).
To confirm that inhibiting the endogenous prosurvival Bcl-2 family via their BH3 binding groove does not induce autophagy in the absence of apoptosis, we overexpressed the BH3-only protein Bims, which strongly binds all of the prosurvival Bcl-2 family members. We found that although culture in HBSS promoted the lipidation of LC3B in MEFs lacking Bax and Bak, high levels of Bims did not (Fig. 1D).
Inhibiting the Antiapoptotic Bcl-2 Family Does Not Increase Autophagic Flux in the Absence of Bax and Bak.
LC3B-II levels were unchanged when the antiapoptotic Bcl-2 family members were inhibited in Bax/Bak-null cells. However, under some circumstances LC3B lipidation status can be independent of overall autophagic flux, which is the dynamic process from autophagosome formation to the ultimate degradation of its contents (16, 18). To determine whether inhibiting the antiapoptotic Bcl-2 family members could directly influence the later stages of autophagy, we engineered Bax−/−Bak−/−Mcl-1−/− MEFs to express an mCherry-EGFP-LC3B fusion protein as a marker for autophagolysosome formation and function. In resting cells, when LC3B is in the cytoplasm or bound to autophagosomes, this fusion protein emits both green and red fluorescence, but when autophagy is induced and lysosomes fuse with autophagosomes, the pH drops inside the organelle, and there is a reduction in the EGFP signal due to its pH sensitivity (16).
A linear relationship between the EGFP and mCherry fluorescence was observed in the majority of untreated cells (Fig. S2), as expected for the fusion protein in the cytosol or autophagosomes. Culture in amino acid free conditions decreased the green fluorescence but not the red, indicating an increase of LC3B present in autophagolysosomes, and hence an increase in autophagic flux (Fig. 2A and Fig. S2). As expected, both chloroquine and bafilomycin A1, which inhibit autophagolysosomal function, were able to prevent the decrease in EGFP signal after amino acid starvation. Although culturing cells in HBSS was able to reduce EGFP fluorescence, addition of ABT-737 did not, either in cells cultured in normal media, or cultured in HBSS (Fig. 2A and Fig. S2), indicating that in the absence of Mcl-1 and apoptosis, inhibition of Bcl-2, Bcl-w, and Bcl-xL via their BH3 binding groove does not affect autophagolysosome formation or function.
Fig. 2.

Autophagic flux remains constant after inhibition of the prosurvival Bcl-2 family members in the absence of Bax and Bak. (A) Bax−/−Bak−/−Mcl-1−/− MEFs expressing the fusion protein mCherry-EGFP-LC3B were treated with 1 μM ABT-737, 50 μM CQ, or 0.1 μM bafilomycin A1 in complete media or HBSS. After 24 h, cells were harvested and fluorescence measured by flow cytometry. The mean ± SD is shown for two independent cell lines. Shapes represent independently performed experiments. (B–D) LC3B turnover assay in fibroblasts after a 4-h treatment with 10 μM CQ with or without (B) HBSS, (C) 1 μM ABT-737, or (D) 48 h pretreatment with 1 μg/mL dox to overexpress Bims.
To confirm that autophagic flux was unchanged, we measured rate of LC3B-II formation by inhibiting the autophagolysosome function with chloroquine. When chloroquine was added, LC3B-II levels increased because the protein was no longer degraded by the autophagolysosome (Fig. 2B). When autophagy was induced upstream, as with culturing in HBSS, the ratio of LC3B-II to LC3B-I increased even further (Fig. 2B). In contrast, addition of BH3 mimetic (Fig. 2C) or over-expression of Bims (Fig. 2D), did not affect LC3B-II levels either in the presence or absence of chloroquine, indicating that inhibition of Bcl-2, Bcl-w, Bcl-xL, and Mcl-1 does not increase autophagic flux. Together with the previous results, these experiments show that the combined inhibition of endogenous prosurvival Bcl-2 family is insufficient to promote autophagy in fibroblasts in the absence of mitochondrial-mediated apoptosis.
Overexpression of Bcl-2, Mcl-1, or Bcl-xL Does Not Directly Inhibit Autophagy in Fibroblasts.
Because inhibition of endogenous Bcl-2 and Bcl-xL did not directly affect autophagy in MEFs, we hypothesized that the interaction between Beclin 1 and the prosurvival Bcl-2 family was too weak to have a detectable effect. We therefore tested whether increasing the levels of Bcl-2, Bcl-xL, or Mcl-1 (the family members that have been reported to regulate autophagy) by overexpressing them using inducible lentiviral vectors could inhibit autophagy. As a positive control, we inhibited autophagy by expressing an shRNA targeting autophagy related 5 (ATG5). Whereas knocking down ATG5 reduced conversion of LC3B-I to LC3B-II in Bax/Bak-deficient MEFs, increasing the expression of the prosurvival Bcl-2 family members in a sorted cell population did not (Fig. S3 A–C). These experiments demonstrate that substantial changes in the levels of Bcl-2, Bcl-xL, or Mcl-1 have no detectable effect on LC3B lipidation when the prosurvival proteins cannot regulate apoptosis (i.e., in the absence of Bax and Bak).
It has been proposed that by phosphorylating Bcl-2, JNK 1 can prevent the prosurvival protein from binding to Beclin 1 (6). To determine whether this event could explain why Bcl-2 did not inhibit autophagy, we mutated the JNK 1 phosphorylation sites (T69A S70A S84A) of Bcl-2 and measured LC3B conversion in cells with basal or stimulated autophagy rates. Similar to its wild-type counterpart, high levels of the Bcl-2 mutant had a negligible effect on LC3B (Fig. S3D).
To determine whether overexpression of the antiapoptotic Bcl-2 family members could regulate autophagic flux in the absence of apoptosis, we induced their expression and measured LC3B-II levels in the presence of chloroquine. Once again, no difference was observed in the ratio of LC3B-II to LC3B-I when any of the Bcl-2 family proteins were overexpressed (Fig. 3 A–C and Fig. S3E). These experiments demonstrate that substantial changes in the levels of Bcl-2, Bcl-xL, or Mcl-1 did not affect autophagy when the prosurvival proteins are not regulating apoptosis.
Fig. 3.
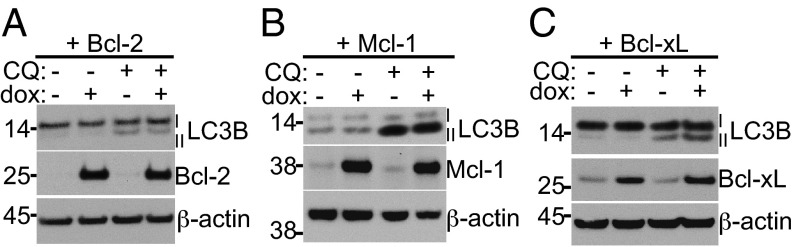
The LC3B-II turnover assay is unchanged by prosurvival Bcl-2 family overexpression in Bax−/−Bak−/− fibroblasts. Western blot after 48 h treatment of 1 μg/mL dox to overexpress (A) Bcl-2, (B) Mcl-1, or (C) Bcl-xL, followed by 10 μM CQ for 4 h to inhibit LC3B-II degradation.
Inhibiting Autophagy Does Not Alter Bax/Bak-Independent Cell Death.
There are conflicting reports in the literature: some articles conclude that autophagy is a prosurvival mechanism, whereas others state it is a mechanism for programmed cell death (19, 20). Because we could induce autophagy in cell lines incapable of undergoing mitochondrial apoptosis, we returned to our original experiment (Fig. 1B) to determine whether inhibiting autophagy itself could affect cell death when Bax and Bak were absent.
When ATG5 was knocked down with RNAi in MEFs lacking Bax and Bak, autophagy was strongly inhibited as shown by the reduction of LC3B-II to nearly undetectable levels (Fig. 4A); however, the knockdown had no appreciable effect on the viability of these cells, whether they were cultured in complete media, HBSS, or treated with etoposide (Fig. 4B). Similarly, chemical inhibition of late-stage autophagy with chloroquine also failed to significantly affect the amount of cell death (Fig. 4C and Fig. S4A). We found no evidence that autophagy can affect viability in fibroblasts by a Bax/Bak-independent mechanism, in contrast to earlier reports (5).
Fig. 4.

Autophagy does not alter cell death in cells lacking Bax and Bak. (A) Knockdown of ATG5 inhibits conversion of LC3B-I to LC3B-II even when autophagy is stimulated. Western blot of Bax−/−Bak−/−Mcl-1−/− MEFs expressing an shRNA against ATG5 or Renilla (negative control) were treated with 34 μM VP-16 or cultured in HBSS for 4 h. (B) Stable knockdown of ATG5 does not modulate viability when autophagy is induced. Viability of MEFs after treatment with 34 μM VP-16 or culture in HBSS for 96 h. Shapes represent individual experiments (closed, Bax−/−Bak−/−; open, Bax−/−Bak−/−Mcl-1−/−; two independent lines per genotype). (C) MEF viability after a 96-h treatment with 34 μM VP-16 (Left) or HBSS (Right) with or without chloroquine (10 μM) to inhibit autophagy (three to four independent lines per genotype; closed circles, Bax−/−Bak−/−; open circles, Bax−/−Bak−/−Mcl-1−/−). (D) Wild-type IL-3–dependent FDM cells expressing shRNA against Renilla or ATG5 were infected with dox-inducible Bcl-2, and the viability was determined after 48 h dox (1 μg/mL) treatment and/or IL-3 withdrawal. (E) IL-3–dependent Bax−/−Bak−/− FDM cells were infected with a plasmid expressing inducible Bcl-2 and an shRNA against ATG5 where indicated. IL-3 was removed, and 1 μg/mL dox was added 4 d before analysis. (F) The concentration of viable IL-3–dependent FDM cells lacking Bax and Bak was determined by trypan-blue exclusion (n = 9; two independent cell lines). The mean ± SD is shown in all graphs.
To determine whether our findings were relevant to another cell type, we tested whether the prosurvival Bcl-2 family members could regulate autophagy in IL-3–dependent (factor-dependent) myeloid (FDM) cell lines. We chose to work with this cell type in particular because it has been reported that autophagy maintains cell viability after IL-3 withdrawal (9). In FDM cells with intact Bax and Bak genes, removal of IL-3 reduced viability by approximately half within 24 h, and to approximately 25% within 48 h (Fig. S4 B and C). Elevating levels of Bcl-2, Mcl-1, or Bcl-xL was able to prevent death after IL-3 withdrawal (Fig. 4D and Fig. S4 D and E; compare the third and fourth column of each figure). In contrast, blocking autophagy by knockdown of ATG5 had no effect on cell survival whether IL-3 was present or not (Fig. 4D and Fig. S4 D and E; compare columns 1–4 with 5–8). Similar results were obtained when autophagy was chemically inhibited with chloroquine (Fig. S4 F–H).
To exclude the possibility that the effects on autophagy were hidden by apoptosis, we measured LC3B levels in Bax−/−Bak−/− FDM cells. Similar to MEFs, LC3B-II levels were not decreased when Bcl-2, Bcl-xL, or Mcl-1 were induced (Fig. 4E and Fig. S4I). Similarly, autophagic flux was unchanged by high levels of expression of the prosurvival Bcl-2 family members (Fig. S5). Taken together, neither Bcl-2, Mcl-1, nor Bcl-xL had detectable effects on autophagy in FDM cells in any of the circumstances we examined. Because FDM cells had such high levels of LC3B-II at basal conditions (Fig. 4E), we did not attempt to induce autophagy by inhibiting the prosurvival proteins.
Because the antiapoptotic Bcl-2 family members protected cells with intact Bax and Bak genes from death induced by IL-3 withdrawal but did not regulate autophagy, we inferred that their death was solely due to mitochondrial-mediated apoptosis. To confirm this, we removed IL-3 from the media of FDM cells lacking Bax and Bak. Although Lum et al. had reported that inhibition of autophagy led to the death of Bax−/−Bak−/− FDM cells (9), we found that inhibition of autophagy with the hairpin against ATG5 did not reduce viability even after 9 d of culture in the absence of IL-3 (Fig. S6A). Similarly, chloroquine did not affect viability of Bax−/−Bak−/− FDM cells in the absence of IL-3 (Fig. S6B). To rule out the possibility that the cells were dying without becoming stained with propidium iodide (PI), we monitored the concentration of viable cells via trypan blue staining (Fig. 4F) and CellTitre-Glo (Fig. S6C). The fluorescence signal corresponding to ATP concentration started to slightly decline (by 7 d) when measured with CellTitre-Glo, but this was most likely due to depletion of ATP stores rather than cell death. Nevertheless, we detected no change in cell viability when autophagy was inhibited, even after a week. Therefore, in these IL-3–dependent mouse myeloid cells the antiapoptotic Bcl-2 family members were able to efficiently prevent apoptosis, but in the absence of Bax and Bak they did not affect autophagy, and the rate of autophagy did not seem to affect cell viability.
Discussion
It is widely believed that the prosurvival Bcl-2 family members Bcl-2, Bcl-xL, and Mcl-1 can inhibit autophagy by binding to Beclin-1 and blocking its function (21, 22). However, many of the experiments characterizing this interaction were overexpression studies or undertaken in conditions whereby some of the cells might have been dying by Bax/Bak-dependent apoptosis (4, 11). We tested this model in cells lacking Bax and Bak to eliminate confounding effects due to apoptosis.
If Beclin 1 is inhibited when Bcl-2, Bcl-xL, or Mcl-1 bind to its BH3 domain under physiological conditions, then occluding the BH3 binding groove should release Beclin 1 and stimulate autophagy. We therefore treated cells mutant for Bax and Bak with the BH3 mimetic ABT-737 and measured autophagy by several different means. We observed no stimulation of autophagy by measuring LC3B lipidation (Fig. 1C), LC3B turnover (Fig. 2C), or autophagolysosome formation and function (Fig. 2A), at either basal autophagy or when the pathway was activated by multiple stimuli (5). Indeed, ABT-737 was incapable inducing autophagy even in Bax−/−Bak−/−Mcl-1−/− MEFs, excluding the possibility that Mcl-1 (and A1), which only weakly bind ABT-737, could compensate for the function of the other Bcl-2 family members (Fig. 2A and Fig. S1B). Similar results were observed when the BH3-binding pockets of the endogenous prosurvival Bcl-2 family were occupied by the BH3-only protein Bims (Figs. 1D and 2D). These results suggest that endogenous levels of the Bcl-2 family cannot directly regulate autophagy and/or that Beclin 1 does not bind to the BH3 binding groove with sufficient strength to have a physiological effect.
It has been reported that addition of ABT-737, or overexpression of BH3 proteins such as Bad, can induce autophagy (10, 11, 23), but these studies used cell lines that have intact apoptotic machinery. To determine whether induction of autophagy by ABT-737 was indirect and as a consequence of apoptosis, we compared Mcl-1−/− and Bax−/−Bak−/−Mcl-1−/− cells. Induction of autophagy by ABT-737 correlated with apoptosis, as indicated by PI staining and conversion of LC3B-I to LC3B-II, respectively (Fig. 1C and Fig. S1C). Therefore, the previously reported effects of the Bcl-2 family members and BH3-mimetics on autophagy might not be a consequence of reduced inhibition of Beclin 1 but due to Bax/Bak-mediated apoptosis (Figs. 1C and 5 and Fig. S1C). This alternative model is consistent with previous data showing that conversion to LC3B-II was most pronounced only after extended treatment (12 h) (23), whereas other Beclin 1-dependent inducers of autophagy are able to increase LC3B lipidation within a few hours independently of cell death (Fig. 4A) (24). It would be interesting to determine at which stage after Bax and Bak activation autophagy is induced, whether it is dependent on caspase activity, and whether cell death mechanisms that are independent of the mitochondria, such as those requiring caspases or MLKL (e.g., death receptor-induced apoptosis, pyroptosis, or necroptosis), also induce autophagy. Indeed, some proteins involved in death receptor pathways have already been implicated in regulating autophagy, albeit not via cell death (25, 26).
Fig. 5.

Our model for the role of the Bcl-2 family in autophagy. The prosurvival Bcl-2 family does not bind directly to Beclin 1 but instead regulate autophagy by inhibiting Bax/Bak mediated apoptosis, a process that activates autophagy by a yet unknown mechanism.
Our results demonstrate that when Bax or Bak are not present, occupying the BH3 binding pocket of prosurvival Bcl-2 family members is insufficient to increase the rate of autophagy. Furthermore, when the abundance of prosurvival Bcl-2 family members is increased, which should theoretically sequester Beclin-1, we saw no reduction in the rate of autophagy (Figs. 3 and 4E). The latter data challenge the possibility that the Bcl-2 family can bind and regulate Beclin 1 independently of its BH3 binding pocket. We therefore conclude that none of the prosurvival Bcl-2 family members bind to Beclin-1 under physiological circumstances or, if they do, they do not significantly inhibit its function.
In the absence of Bax and Bak, we found that inhibiting autophagy had little impact on viability of MEFs or FDM cells when autophagy was induced by multiple stimuli (Fig. 4 B, C, and F and Fig. S6). It is doubtful that the shRNA against ATG5 was insufficient to block autophagy in fibroblasts, because LC3B-II was undetectable under these conditions (Fig. 4A). An alternative model is that Beclin 1 acts to promote apoptosis (as a BH3-only protein) when autophagy fails, but this is inconsistent with previous data that overexpression of Beclin 1 does not influence apoptosis (4). Although our data contest the idea that autophagy is an independent form of cell death (5, 19), at least in the circumstances we tested, they do not negate the possibility that autophagy cannot be a mechanism for cell death in other circumstances. Indeed, there is strong genetic evidence showing that autophagy is necessary for cell death during metamorphosis in flies (27).
In conclusion, our data demonstrate that the prosurvival Bcl-2 family of proteins does not directly regulate autophagy, but any impact they have on autophagy is indirect, via Bax and Bak activation (Fig. 5). This work supports a model that apoptosis induces autophagy and that autophagy does not significantly induce cell death in the absence of Bax and Bak.
Materials and Methods
Cell Lines, Constructs, and Compounds.
MEFs were immortalized with SV40 large T antigen and grown in DMEM supplemented with 10% (vol/vol) FCS (Bovogen), 50 mM 2-mercaptoethanol (Sigma), and 100 mM asparagine (Sigma). Wild-type cells as well as Mcl-1−/− and Bax−/−Bak−/− genotypes have been previously described (14, 28). Bax−/−Bak−/−Mcl−/− fibroblasts were derived from immortalized Bax−/− Bak−/− Mcl-1fl/fl MEFs, transiently transfected to express Cre recombinase, which was followed by screening for Mcl-1−/− clones. Factor-dependent (IL3-dependent) myeloid cells were previously described and grown in Dulbecco’s modified Eagle’s with 10% (vol/vol) FCS and 0.26 ng/mL IL-3 (29).
ATG5 (1390-AGTTTGTATTTCTGATTA) shRNA was cloned into pLMP, and the Renilla shRNA has been previously described (30). pBABE-puro mCherry-EGFP-LC3B is from Addgene. Murine Bcl-2, Mcl-1 (provided by Toru Okamoto, Osaka University, Osaka), and Bcl-xL coding sequences were cloned into pFTRE 3G rtTA GFP and human Bims into pFTRE 3G rtTA Puro (provided by Toru Okamoto) (31). The JNK 1 mutant of murine Bcl-2 (T69A/S70A/S84A) was adapted from the human sites (6) and created by subcloning a synthetically made DNA oligo into the wild-type vector (gBlocks; IDT). ABT-737 was a kind gift from AbbVie, whereas bafilomycin A and chloroquine were both purchased from Sigma.
Viability Assays.
MEFs and FDM cells were treated with 34 μM VP-16 (etoposide) or starved of amino acids by culture in HBSS to induce autophagy for indicated times. Media, PBS washes, and cells were collected, and the cell pellets were resuspended in KDS BSS (150 mM NaCl, 3.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 7.4 mM HEPES.NaOH, 1.2 mM KH2PO4, 0.8 mM K2HPO4), 2% (vol/vol) FCS, and 1% sodium azide with 10 μg/mL PI before they were analyzed using a FACSCalibur flow cytometer (Becton Dickinson). Alternatively, viable cell concentrations were determined using a Countess automatic cell counter (Invitrogen) or the CellTiter-Glo Luminescent Cell Viability Assay (Promega) following the manufacturers’ instructions.
Monitoring LC3B Lipidation and Turnover.
MEFs or FDM cells were seeded in a six-well dish before treatment with 34 μM VP-16 or HBSS for indicated times. Doxycycline (dox, 1 μg/mL) was added to induce protein overexpression for 24–48 h before autophagy induction where indicated. Chloroquine (10 μM) was added for 4 h to monitor autophagic flux. Cells and media were harvested, washed in PBS, and lysed in 20 mM Tris (pH 7.5), 135 mM NaCl, 1.5 mM EDTA, 10% (vol/vol) glycerol, and 1% Triton X-100 including protease inhibitors (Roche). Supernatant was retained after centrifugation, and equal amounts of protein were separated on a 10% (wt/vol) NUPAGE Bis Tris gel (Invitrogen). After transfer to PVDF or nitrocellulose, the membrane was blotted with the antibodies raised against LC3B (D11; Cell Signaling), ATG5 (D1G9; Cell Signaling), Bcl-2 (3F11; WEHI), Mcl-1 (19C4;WEHI), Bcl-xL (44; BD Biosciences), Bim (3C5; WEHI), Bax (49F9-13–13;WEHI), Bak (Sigma), and HSP70 (N6; gift from W. Welch, University of California, San Francisco), or β-actin (AC-15; Sigma) as loading controls.
Monitoring Autophagolysosome Formation and Function.
Bax−/−Bak−/−Mcl-1−/− MEFs infected with pBABE puro mCherry-EGFP-LC3B were treated with 1 μM ABT-737, 50 μM chloroquine, or 0.1 μM bafilomycin A1 in complete media or starved in HBSS for 24 h. Cells were harvested by typsonization, washed, and subsequently analyzed by flow cytometry to determine EGFP and mCherry fluorescence. Cells that were positive for mCherry were selected, and the percentages of cells with EGFP in a linear relationship with mCherry were calculated (details of gating in Fig. S2).
Supplementary Material
Acknowledgments
We thank Andreas Strasser for helpful discussions and H. Ierino for technical assistance. Funding for this project was provided by National Health and Medical Research Council (NHMRC) Grants 1016701 and 1020136. L.M.L. is a Bisby Fellow and holds an NHMRC Peter Doherty Early Career Fellowship (1035502) and a Canadian Institutes of Health Research (CIHR) Post-PhD Fellowship. NHMRC Research Fellowships are held by both D.C.S.H. (1043149) and D.L.V. (1020136). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC Independent Research Institute Infrastructure Support Scheme (IRIISS) Grant 361646.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1406425111/-/DCSupplemental.
References
- 1.Baehrecke EH. Autophagy: Dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6(6):505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 2.Liang XH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72(11):8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282(17):13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 4.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6(12):1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 8.Youle RJ, Strasser A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 9.Lum JJ, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 10.Maiuri MC, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3(4):374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 11.Maiuri MC, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26(10):2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 13.van Delft MF, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willis SN, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindqvist LM, et al. Translation inhibitors induce cell death by multiple mechanisms and Mcl-1 reduction is only a minor contributor. Cell Death Dis. 2012;3:e409. doi: 10.1038/cddis.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zalckvar E, et al. A systems level strategy for analyzing the cell death network: Implication in exploring the apoptosis/autophagy connection. Cell Death Differ. 2010;17(8):1244–1253. doi: 10.1038/cdd.2010.7. [DOI] [PubMed] [Google Scholar]
- 18.Matsui Y, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 19.Denton D, Nicolson S, Kumar S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012;19(1):87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gump JM, Thorburn A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011;21(7):387–392. doi: 10.1016/j.tcb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine B, Sinha S, Kroemer G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malik SA, et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene. 2011;30(37):3918–3929. doi: 10.1038/onc.2011.104. [DOI] [PubMed] [Google Scholar]
- 24.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15(3):1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27(1):135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell BD, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA. 2008;105(43):16677–16682. doi: 10.1073/pnas.0808597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denton D, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19(20):1741–1746. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee EF, et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008;180(2):341–355. doi: 10.1083/jcb.200708096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekert PG, et al. Cell death provoked by loss of interleukin-3 signaling is independent of Bad, Bim, and PI3 kinase, but depends in part on Puma. Blood. 2006;108(5):1461–1468. doi: 10.1182/blood-2006-03-014209. [DOI] [PubMed] [Google Scholar]
- 30.Zuber J, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol. 2011;29(1):79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brumatti G, et al. HoxA9 regulated Bcl-2 expression mediates survival of myeloid progenitors and the severity of HoxA9-dependent leukemia. Oncotarget. 2013;4(11):1933–1947. doi: 10.18632/oncotarget.1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.