

Significance
Osteoblast precursors residing in the bone marrow experience hypoxia, which leads to stabilization and increased activity of hypoxia-inducible factor 1α (HIF1α). However, whether an increase in HIF1α activity directly impacts bone formation is not known. Two well-known functions of HIF1α are to promote angiogenesis and to stimulate glycolysis in the cell. Here, we demonstrate that HIF1α stabilization in osteoblast precursors of postnatal mice markedly increases osteoblast number and bone mass. Furthermore, the greater bone mass was not secondary to increased angiogenesis but rather dependent on the up-regulation of glycolysis in osteoblast precursors. These results provide strong evidence that alterations in cellular metabolism directly influence cell differentiation.
Abstract
The bone marrow environment is among the most hypoxic in the body, but how hypoxia affects bone formation is not known. Because low oxygen tension stabilizes hypoxia-inducible factor alpha (HIFα) proteins, we have investigated the effect of expressing a stabilized form of HIF1α in osteoblast precursors. Brief stabilization of HIF1α in SP7-positive cells in postnatal mice dramatically stimulated cancellous bone formation via marked expansion of the osteoblast population. Remarkably, concomitant deletion of vascular endothelial growth factor A (VEGFA) in the mouse did not diminish bone accrual caused by HIF1α stabilization. Thus, HIF1α-driven bone formation is independent of VEGFA up-regulation and increased angiogenesis. On the other hand, HIF1α stabilization stimulated glycolysis in bone through up-regulation of key glycolytic enzymes including pyruvate dehydrogenase kinase 1 (PDK1). Pharmacological inhibition of PDK1 completely reversed HIF1α-driven bone formation in vivo. Thus, HIF1α stimulates osteoblast formation through direct activation of glycolysis, and alterations in cellular metabolism may be a broadly applicable mechanism for regulating cell differentiation.
Molecular oxygen is required by all eukaryotic cells, and variations in oxygen levels can have profound consequences on cellular metabolism, differentiation, and survival (1, 2). The principle mechanism by which cells cope with low oxygen is through stabilization of transcription factors, the hypoxia-inducible factors (HIFs). Oxygen is a cofactor for prolyl hydroxylases that hydroxylate HIFα proteins and mark them for recognition by the E3 ubiquitin ligase von Hippel–Lindau Factor (VHL), which transports HIFα to the proteosome for degradation (3–5). Low oxygen levels allow HIFα to escape degradation and enter the nucleus, where it interacts with ubiquitously expressed HIF1β (ARNT) and cofactors to drive the expression of target genes containing hypoxia response elements (HREs) (6, 7).
Among the hundreds of validated direct HIF target genes are multiple regulators of angiogenesis and cellular metabolism (8–10). Most differentiated cell types in mammals meet their energetic requirement principally through mitochondrial oxidative phosphorylation when oxygen is replete, but shift to lactate-producing glycolysis in the cytosol when oxygen levels are low and stabilized HIF proteins up-regulate key glycolytic enzymes. This metabolic switch allows cells to maintain ATP synthesis under hypoxic conditions. In contrast to normal cells, cancer cells exhibit excessive glycolysis even when oxygen is abundant, a phenomenon known as aerobic glycolysis, or the “Warburg effect” (11). Pathologically stabilized HIF proteins are known to contribute to such an effect in certain cancer types (12). Aerobic glycolysis has been proposed to provide metabolic intermediates for generating nucleotides, amino acids, and lipids, all of which are necessary for the rapid proliferation of cancer cells (13).
HIF activity in osteoblasts has previously been linked to promotion of angiogenesis and coupling between osteogenic and angiogenic processes. Genetic deletion of VHL in mature osteoblasts with osteocalcin-Cre stabilized endogenous HIF1α and HIF2α, resulting in high trabecular bone mass and increased bone vascular density associated with increased levels of VEGF (14). Conversely, deletion of HIF1α in mature osteoblasts decreased bone volume (14, 15). Changes in bone vascular density has been proposed as a main mechanism responsible for HIF-mediated effects on bone mass (14). However, when HIF1α and HIF2α were deleted separately in osteoblasts, although both affected vascular density in long bones, only HIF1α deletion had an appreciable effect on bone formation (15). More recently, a VEGF-independent effect of HIF1α was demonstrated in mouse fetal cartilage (16). Thus, HIF1α may play important direct roles in osteoblasts, independent of effects on angiogenesis caused by up-regulation of VEGF.
Here, we have investigated the effects mediated by stabilized HIF1α in osteoblast precursors. We report that expression of stabilized HIF1α in SP7 (also known as osterix)-positive cells dramatically increased bone formation in postnatal bones. We further show that the increase in bone mass is independent of VEGF-associated angiogenesis, but rather requires HIF-directed enhanced glycolysis in bone cells.
Results
Bone Marrow Osteoblast Precursors Experience Low Oxygen Tension in Vivo.
The marrow cavity of long bones, although well-vascularized, has been suggested to contain regions of low oxygen concentrations (17). To determine whether osteoblast-lineage cells within the marrow cavity experience hypoxia, we used hypoxyprobe (pimonidazole hydrochloride) to detect cells experiencing oxygen partial pressures of <10 mmHg in the long bones of 6-wk-old mice (18). Immunofluorescence staining revealed hypoxyprobe labeling throughout the bone marrow cavity in both primary and secondary centers (Fig. 1A). Importantly, a strong signal was detected at the chondro–osseous junction of the primary ossification center, an area known to contain mostly osteoblast precursors (Fig. 1 B and B’) (19). As predicted, most of these cells were negative for osteocalcin, the mature osteoblast marker, although occasional cells doubly positive for osteocalcin and hypoxyprobe were detected on the nascent bone trabeculae (Fig. 1B, arrow). On the other hand, the mature osteoblasts associated with the diaphyseal cortical bone were largely negative for hypoxyprobe (Fig. 1 C and C’). The specificity of the hypoxyprobe staining was confirmed by the largely negative background when the reagent was not injected into the mouse (Fig. 1 D and D’). Therefore, osteoblast precursors at the primary ossification center, but not the mature osteoblasts associated with the cortical bone, experience low oxygen tension in vivo.
Fig. 1.

Osteoblast precursors experience hypoxia in vivo. (A) Immunofluorescence staining on femur cryosection from a 6-wk-old wild-type mouse labeled with 60 mg/kg hypoxyprobe for 75 min. Green, hypoxyprobe; red, osteocalcin; blue, DAPI. White boxes indicate regions of interest shown at higher magnification in B, B’, C, and C’. Note that the bright green signal scattered in the marrow in A is not reproducible. (B and B’) Proximal cancellous region. White arrowhead denotes a double-positive cell for hypoxyprobe and osteocalcin. (C and C’) Cortical bone region. Note the bone surface osteoblasts stained positive for osteocalcin but negative for hypoxyprobe. (D and D’) Negative control for hypoxyprobe antibody staining (no hypoxyprobe injection). (B’, C’, and D’) Hypoxyprobe channel only.
Stable HIF1α Increases Cancellous Bone Formation in Postnatal Mice.
The low oxygen tension in osteoblast precursors observed above would be expected to stabilize HIFα proteins in those cells. To gain insights about the cellular effect of such stabilization, we decided to express an oxygen-stable form of HIF1α (alanine substitutions at P402, P562, and N803, hereafter HIF1-PPN) in those cells in postnatal mice. For this purpose, we used a doxycycline (dox) regulatable system. The SP7-tTA transgenic line expresses the tet-transactivator tTA (inactive in the presence of dox) that in the absence of dox drives the expression of tetO-Cre::GFP from the same transgene in SP7-positive cells, and has been shown to target effectively early-stage osteoblast-lineage cells in the mouse embryo (20). To assess the efficacy of SP7-tTA in postnatal mice, we raised SP7-tTA mice on dox from conception until 3 wk of age before withdrawing dox for15 d, and then assessed GFP expression. GFP was detected prominently in osteoblast precursors at the chondro–osseous junction, as well as bone-surface osteoblasts and osteocytes in either cancellous or cortical bone (Fig. S1). Thus, SP7-tTA is an effective tool for targeting osteoblast precursors in postnatal mice.
We next used SP7-tTA to activate expression of tetO-HIF1-PPN with the same dox regimen described above (21) (Fig. 2A). Mice carrying both transgenes (designated SH) were maintained on dox from conception until 3 wk of age, and then either weaned off (SH−dox) or continued on dox (SH+dox) for 15 d before harvest (Fig. 2 B–E). As an additional control, mice from the same litters carrying only SP7-tTA were treated in the same way as the SH−dox mice (SP7−dox). Imaging of the long bones by microcomputed tomography (μCT) revealed a marked increase in cancellous bone within the primary ossification center of the SH−dox over the SH+dox or the SP7−dox mice (Fig. 2 C–E). Quantification of the μCT data indicated that the cancellous bone volume normalized to tissue volume (BV/TV) was increased in SH−dox by ∼500% or 300% over SP7−dox or SH+dox, respectively (Fig. 2F). The increase in bone mass was largely due to changes in trabecular thickness, whereas trabecular number or separation was not significantly altered in the SH−dox mice (Fig. 2 G–I). These changes seen in the primary ossification center were not present in the secondary ossification center, the bone shaft (cortical bone) of the long bones, or the calvarium (Fig. S2). Thus, transient stabilization of HIF1α in osteoblast precursors in postnatal mice increases cancellous bone mass within the primary ossification center.
Fig. 2.

Expression of HIF1-PPN in SP7-positive cells increases cancellous bone volume. (A) Schematic of transgenes used to yield SP7;HIF1-PPN (SH) mice. The tetO-driven HIF1-PPN is only expressed in cells actively transcribing the SP7 promoter-driven tTA. (B) Treatment schematic. The experimental window began at 3 wk of age with dox removal. Mice were analyzed 15 d later. (C–E) µCT analyses of 1.6 mm of cancellous bone immediately under the growth plate of the tibia. Mouse genotypes are as labeled. In D, SH mice were maintained on dox throughout the experimental window to evaluate leakiness in the suppression system. (F) Bone volume per total volume of the regions shown in C–E. There was no difference between SP7-dox and SH+dox mice but a significant increase (P = 0.0015) in SH-dox animals. (G) Trabecular number per millimeter, as quantified from µCT scans. (H) Trabecular thickness by µCT (P = 0.0010). (I) Trabecular separation. n = 7 (SP7-dox); n = 6 (SH+dox); n = 6 (SP7-dox).
We next determined the cellular basis for the HIF1α-induced increase in bone mass. Histological sections of the tibia confirmed a marked increase in cancellous bone within the primary ossification center of the SH−dox mice (Fig. 3 A–C, Upper). Histomorphometric analyses indicated an increase in osteoblast number when normalized to the cancellous bone surface in SH−dox mice compared with the control mice (Fig. 3D). Calcein double-labeling experiments did not detect a significant change in the mineral apposition rate (MAR). Consistent with more osteoblasts, serum osteocalcin levels were higher in SH−dox mice (Fig. 3E). Interestingly, although the number of osteoclasts per bone surface was reduced in SH−dox mice, collagen type I C-telopeptide (CTX-I) level, which reflects overall bone resorption activity in the mouse, was actually higher in these mice (Fig. 3 F and G and Fig. S3). The increase in total bone resorption activity was likely due to more active osteoclasts, as the total osteoclast number within the trabecular bone region was similar between the SH and SH−dox mice (Fig. 3H). The altered osteoclast activity was expected to be indirect, as SP7-tTA did not target the osteoclast lineage, but the exact mechanism was not pursued here. Overall, these results suggest that the increase in osteoblast number is likely the main cause for the higher bone mass induced by HIF1α stabilization.
Fig. 3.

Osteoblast numbers increase upon HIF1-PPN expression. (A–C) Histological sections of tibiae. Boxed regions are shown in higher magnification in the lower panels. (D) Quantification of osteoblast numbers per bone surface, as evaluated by histomorphometry. P = 0.0425, n = 3 per group. (E) Serum osteocalcin measurements (ng/mL). P = 0.0125, n = 6 (SH+dox); n = 8 (SH-dox). (F) Number of osteoclasts per bone surface, as evaluated by histomorphometry of TRAP-stained sections. P = 0.0015, n = 3 per group. (G) Serum CTX-I concentrations. P = 0.0488, n = 6 (SH+dox); n = 8 (SH-dox). (H) Total osteoclast numbers within the entire cancellous bone region on TRAP-stained sections. Three medial sections from each of three mice per group were counted.
Histological examination of the primary ossification center at a higher magnification revealed a second feature specific to the SH−dox mice. Instead of hematopoietic cells normally present in this region, a large number of fibroblast-like cells occupied the space between the bone trabeculae in the SH−dox mice (Fig. 3 A–C, Lower). These cells were negative for CD45 or CD11b, but stained positive for PDGFRα (also known as CD140A), and were therefore likely of a mesenchymal origin (Fig. S4 A and B). Lineage-tracing experiments with SP7-tTA (also expressing Cre upon dox removal) and the Rosa-Ai9 reporter revealed that a subset but not all of the fibrotic cells were derived from cells expressing SP7 during the 15 d of dox withdrawal (Fig. S4B) (22). The SP7-derived fibrotic cells were negative for osteocalcin, and therefore not mature osteoblasts (Fig. S4C). Moreover, these cells produced a reticulin-positive and trichrome-negative matrix indicative of type III collagen (Fig. S4 D and E). Thus, in addition to bone formation, HIF1α stabilization also increases fibrosis within the primary ossification center.
HIF1α Does Not Require VEGFA to Induce Bone Formation.
HIF1α is known to induce VEGFA that activates angiogenesis, and the latter has been postulated to drive ossification in other mouse models (14, 23). Moreover, overexpression of an isoform of VEGFA (VEGF164) was reported to cause bone marrow fibrosis similar to what we observed here in the SH−dox mice (24). Therefore, we tested whether VEGFA up-regulation is responsible for the HIF1α-driven bone and fibrosis phenotypes. To this end, we introduced two floxed alleles of VEGFA into the SH mice (genotype Sp7-tTA;tetO-HIF1-PPN and VEGFAf/f, designated SHΔV) and subjected them to the same dox regimen as the SH−dox mice (25). Dox withdrawal was expected to delete VEGFA in the same cells expressing HIF1-PPN in the SHΔV−dox mice. Remarkably, histology of the long bones indicated that loss of VEGFA did not reduce the overall increase in cancellous bone mass caused by HIF1-PPN over the control (Fig. 4 A–C, Upper). However, the intertrabecular fibrosis observed in SH−dox was greatly ameliorated with deletion of VEGFA in the SHΔV−dox mice (Fig. 4 A–C, Lower, and H). Quantification by μCT confirmed that the cancellous BV/TV in the proximal tibia was similarly elevated in SH−dox and SHΔV−dox compared with control (Fig. 4G). Thus, VEGFA contributes to HIF1α-induced bone marrow fibrosis but is dispensable for HIF1α-driven cancellous bone formation.
Fig. 4.

Concomitant deletion of VEGF does not impact HIF1-PPN–induced bone accrual. (A–C) Histological sections of tibiae. Boxed regions are shown in higher magnification in the lower panels. (D–F) Evaluation of bone vasculature by Microfil casting and µCT. The avascular growth plate is at the top of each cast. Note the dense accumulation of small-caliber vessels in SH-dox mice compared with control or SHΔV-dox. (G) Quantification of bone volume per total volume of tibia cancellous bone via µCT. There was no difference between SH-dox and SHΔV-dox mice. (H) Quantification of marrow fibrosis. Fibrotic marrow area normalized to total marrow area on three H&E sections per mouse. *P < 0.05, n = 4 (control); n = 4 (SH-dox); n = 3 (SHΔV-dox). Control mice were sex-matched littermates without SP7-tTA, subjected to same dox regime as experimental groups.
In contrast to bone formation, deletion of VEGFA notably altered the vascular architecture in the cancellous bone region in the SHΔV−dox mice. We analyzed the bone vasculature with μCT following microfil perfusion. In SH−dox mice, the vessels in the proximal metaphysis of the tibia were numerous, dense, and thin, compared with the control mouse (Fig. 4 D and E). In contrast, the SHΔV−dox vessels more closely resembled those in control mice with larger diameters and increased spacing between the vessels (Fig. 4F). Thus, HIF1α stabilization in SP7-positive cells alters vascular organization in the bone marrow, and VEGFA appears to be a principle mediator.
Suppression of Glycolysis Blocks HIF1α-Induced Bone Formation in Vivo.
The lack of a VEGFA effect on HIF1α-induced bone formation prompted us to examine the potential role of metabolic regulation, as HIF1α is known to stimulate glycolysis through direct regulation of multiple key enzymes (26). We confirmed that pyruvate dehydrogenase kinase 1 (PDK1), lactate dehydrogenase A (LDHA), and hexokinase II (HK2) were all more abundant in the bone protein extracts of SH−dox versus the control mice (Fig. 5A). Also consistent with a metabolic shift toward lactate-producing glycolysis, we detected a significant increase in circulating lactate levels in the SH−dox mice compared with control (Fig. 5B). Thus, HIF1α stabilization stimulates glycolytic metabolism in bone.
Fig. 5.
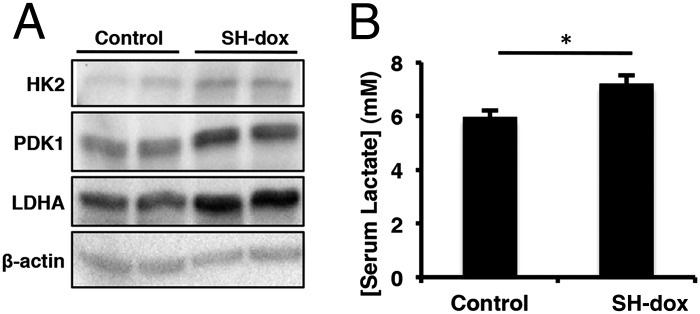
SH-dox bones have increased levels of glycolytic enzymes. (A) Western blot analysis of protein extracts from whole bones of control and SH-dox mice. The protein levels of HK2, PDK1, and LDHA were all elevated in bone from SH-dox mice. For these experiments, dox was removed for 21 d. β-actin is included as a loading control. (B) Serum lactate concentration measurements. SH-dox mice had significantly higher levels of circulating lactate. P = 0.0358, n = 5 (control); n = 6 (SH-dox). Control mice were sex-matched littermates without SP7-tTA, subjected to same dox regime as experimental groups.
To test whether the glycolytic shift in SP7-positive cells is important for increased bone mass in SH−dox mice, we treated the mice with sodium dichloroactetate (DCA), a potent PDK1 inhibitor that suppresses glycolysis (27). SH or control mice were exposed to DCA through the drinking water during the 15-d period of HIF induction. Strikingly, µCT analyses of the tibia showed that DCA essentially abolished the increase in cancellous bone mass caused by HIF1-PPN, but did not cause a significant change in the control mice (Fig. 6A). Similarly, DCA corrected the trabecular number and thickness in the SH−dox mice, without a significant effect on trabecular separation (Fig. 6 B–D). Thus, PDK1 inhibition by DCA effectively overcomes the bone-inducing effect of HIF1α and restores normal bone mass in the mouse.
Fig. 6.

PDK1 inhibition blocks the bone anabolic effects of HIF1-PPN. (A–D) Quantification of µCT results. DCA treatment significantly reduced the BV/TV, trabecular number, and trabecular thickness in SH-dox mice (P = 0.0005, P = 0.0232, and P = 0.0018, respectively). BV/TV in control mice was also reduced with DCA treatment, but this number did not reach significance (P = 0.0921). n = 4 (control, SH-dox, SH-dox+DCA); n = 5 (control+DCA). (E–H) Histological sections of the tibia with higher magnification views of the boxed regions shown in the lower panels. (I) Osteoblast numbers per bone surface, as evaluated by histomorphometry. DCA treatment significantly lowered osteoblast numbers in SH-dox mice (P = 0.0327). (J) Osteoclast numbers per bone surface, as evaluated by histomorphometry. n = 3 (control, SH-dox+DCA) and n = 4 (control+DCA, SH-dox) for cell counting. (K) Mineral apposition rate (MAR) as determined by calcein double labeling. No significant changes were observed. Control mice were sex-matched littermates without SP7-tTA, subjected to same dox regime as experimental groups.
The DCA rescue of bone mass was confirmed and elaborated by histomorphometry. Histology of tibial sections indicated that the overall area of cancellous bone in SH−dox+DCA was similar to the control level, but much lower than that in SH−dox (Fig. 6 E–H, Upper). Interestingly, the fibrosis phenotype was also eliminated in the SH−dox+DCA mice (Fig. 6 E–H, Lower). Quantitative histomorphometry revealed that DCA prevented the increase in osteoblast number in SH−dox mice and returned it to the normal level, but did not change the parameter in the control mice (Fig. 6I). On the other hand, DCA did not change osteoclast numbers (Fig. 6J). Similarly, DCA did not alter osteoblast activity in either control or SH−dox mice, as indicated by MAR (Fig. 6K). Thus, glycolytic suppression in bone prevents a HIF1α-related increase in osteoblast number, thereby precluding the increase in bone formation.
Discussion
We have investigated the role of HIF1α stabilization in bone formation in vivo. Despite close proximity to the marrow vasculature, osteoblast precursors in the cancellous bone region of long bones experience low oxygen tension expected to stabilize HIF proteins. Brief expression of a constitutively active form of HIF1α in SP7-positive cells in postnatal mice markedly enhanced cancellous bone formation through increased osteoblast numbers. HIF1α stabilization also stimulated fibrosis and altered the vascular architecture in the cancellous bone region. Importantly, although VEGFA is necessary for the fibrotic and vascular phenotype, it is dispensable for the increase in bone mass in these mice. Instead, increased glycolysis is the main driver for HIF1α-induced bone formation. This study therefore uncovers both VEGFA-dependent and -independent functions of HIF1α in bone, and identifies the stimulatory role of enhanced glycolysis in bone formation.
It is somewhat unexpected that VEGFA is dispensable for HIF1α-induced bone formation despite its clear influence on the bone marrow vasculature. Angiogenesis has been long linked with bone formation. For example, mouse embryonic studies have indicated that blood vessels invading the hypertrophic cartilage of developing long bones may carry osteogenic signals (28). Moreover, stabilization of HIFα proteins by deletion of VHL in mature osteoblasts with OC-Cre greatly increased cancellous bone mass in postnatal mice, and this effect was attributed to increased angiogenesis driven by high levels of VEGF expression (14). Similarly, pharmacological stabilization of HIFα proteins greatly improved bone regeneration coupled with a marked increase in angiogenesis in a mouse distraction osteogenesis model (29). However, when HIF1α and HIF2α were deleted individually with OC-Cre, only HIF1α deletion caused a marked decrease in cancellous bone formation, even though both deletions similarly impaired skeletal angiogenesis, indicating that HIF1α may function independently of angiogenesis (15). The present study provides direct evidence that HIF1α promotes cancellous bone formation through stimulation of glycolysis. SP7-tTA directs HIF1-PPN expression in the osteoblast precursors within the cancellous bone region, but did not change cell proliferation or apoptosis among the population, as determined by BrdU labeling or TUNEL staining, respectively. Nonetheless, HIF1-PPN increased osteoblast number. Thus, we conclude that HIF1α likely enhances osteoblast differentiation from the SP7-positive preosteoblasts.
It is not clear at present why osteoblast differentiation requires active glycolysis. Such metabolic reprogramming may alter the levels of key metabolites (e.g., NAD+, acetyl-coA, α-ketoglutarate) necessary for epigenetic modifications as suggested in other experimental settings (30–32). The glycolytic shift may also change the consumption of other nutrients such as amino acids or fatty acids, which may in turn be important for driving osteoblast differentiation. Future studies are warranted to explore these potential mechanisms. Finally, in addition to hypoxia and HIFα stabilization, WNT signaling, a well-known mechanism for stimulating bone anabolism, also induces aerobic glycolysis during osteoblast differentiation (33). Thus, increased glycolysis may represent a general mechanism for osteoblast differentiation in response to various physiologic stimuli. A better understanding of the metabolic regulation of osteoblast formation and function may open avenues for developing a new class of bone anabolic therapeutics.
Materials and Methods
Mice.
The Animal Studies Committee at Washington University reviewed and approved all mouse procedures used in this study. The SP7-tTA,tetO-EGFP/cre (Jax strain 006361), HIF1-PPN, and VEGF-flox mouse lines have all been previously described (20, 21, 25). Analyses were performed on sex-matched, paired littermates. Mice treated with dox received drinking water containing 2% (wt/vol) sucrose and 50 mg/L dox hyclate (Sigma). Sodium dichloroacetate (Sigma) was dissolved in drinking water at a concentration of 2 g/L, and the pH was adjusted to 7 with NaOH. All drinking water was provided ad libitum. At the time of sacrifice, mice were anesthetized with a ketamine/xylazine mixture and whole blood was collected retro-orbitally. Ten milliliters of heparinized saline were then perfused through the left ventricle of the heart, followed by 10 mL of 4% (wt/vol) paraformaldehyde in PBS, pH 7.4. For protein isolation, mice were euthanized by CO2 inhalation.
Hypoxyprobe Labeling.
Six-week-old wild-type mice were injected intraperitoneally with 60 mg/kg Hypoxyprobe-1 (pimonidazole HCl) in PBS. After 75 min, mice were perfusion-fixed and bones were isolated and fixed overnight at 4 °C before processing. Mice that did not receive hypoxyprobe were analyzed in parallel to serve as a negative control for hypoxyprobe antibody specificity.
Immunostaining.
Perfusion-fixed bones were incubated overnight at 4 °C in 4% (wt/vol) paraformaldehyde followed by 3 d of decalcification in 14% (wt/vol) EDTA, pH 7.4. Bones were then rinsed, equilibrated in 20% (wt/vol) sucrose, embedded in optimum cutting temperature (OCT) compound (Tissue-Tek), and frozen in liquid nitrogen. Sections at 8 μm in thickness were cut using the Cryo-Jane Tape-Transfer system (Leica). Sections were rinsed, incubated briefly in 0.1% Triton X-100, and blocked with 5% (vol/vol) normal serum, followed by overnight incubation in primary antibody at 4 °C. Following secondary detection at room temperature, sections were rinsed and mounted with Vectashield containing DAPI (Vector Laboratories). Primary antibodies included hypoxyprobe (1:20; Hypoxyprobe Inc.), osteocalcin (1:50; Santa Cruz sc-30045), and GFP (1:1,000; abcam ab13970). Biotinylated isolectin B4 (Vector Laboratories) was used at 10 µg/mL in PBS. Alexa Fluor-conjugated secondary antibodies (Life Technologies) were used at 1:500. Immunofluorescence images were captured using a Nikon Eclipse 80i equipped with d-EclipseC1 confocal system and processed with Adobe Photoshop and Image J.
μCT.
Tibias were embedded in 1% agarose and scanned by µCT (µCT 40; Scanco Medical AG) with the following settings: 55 kVp, 145 µA, 16 µm voxel size, and 150 ms integration time. Cancellous bone immediately beneath the growth plate was manually selected, and 100 slices (1.6 mm total) were analyzed. Gaussian filter parameters were a support of 2 and a sigma of 1.2. The threshold was set at 240.
Histology and Histomorphometry.
Perfusion-fixed bones were incubated overnight in 10% (vol/vol) neutral-buffered formalin and then decalcified for 2 wk in 14% (wt/vol) EDTA, pH 7.4. Paraffin sections at 6 μm in thickness were stained according to standard hematoxylin and eosin or tartrate-resistant acid phosphatase (TRAP) protocols. Reticulin and Masson’s trichrome staining were performed according to the manufacturers’ instructions (Polysciences, Inc.). For dynamic histomorphometry, calcein (Sigma) was injected intraperitoneally 6 and 2 d before euthanasia at 20 mg/kg. Femurs were fixed in 70% (vol/vol) ethanol and embedded in methyl-methacrylate for sectioning. Measurements of double-labeled cortical bone were taken on the medial and lateral endocortical surfaces at the metaphysis/diaphysis border. Measurements and calculations for static and dynamic histomorphometry were done with the computer software Bioquant II (Bioquant Image Analysis Corp.).
BrdU Labeling.
Mice were injected peritoneally with a BrdU (10 mg/mL, Sigma B5002)/FdU (1.2 mg/mL, Sigmal F0503) solution at 0.1 mL/10 g, 4 h before sacrifice. Bones were collected and fixed in 10% (vol/vol) neutral-buffered formalin overnight at room temperature and decalcified with 14% EDTA (pH 7.4) for 2 wk before paraffin embedding and sectioning. Sections were deparaffinized and stained with a BrdU staining kit (Invitrogen 93–3943) according to the manufacturer’s instructions. BrdU labeling indices were calculated as percentage of positive over total cells within similar areas under the growth plate across the different mice. Three mice per group were analyzed.
TUNEL Staining.
Mouse bones were fixed in 4% (wt/vol) paraformaldehyde overnight and decalcified with 14% (wt/vol) EDTA (pH 7.4) for 2 d at 4 °C. Bones were incubated sequentially in 15% and 30% (wt/vol) sucrose before OCT embedding and cryostat sectioning. Sections were incubated in 0.1% triton X-100 for 10 min before proceeding with TUNEL staining with the In Situ Cell Death Detection Kit, TMR red (Roche 12 156 792 910), according to the manufacturer’s instructions.
Serum Biochemistry.
Whole blood collected at the time of sacrifice was centrifuged and the serum retained. Serum collagen I C-telopeptide (CTX-I) assays were performed with the RatLaps ELISA Kit (Immunodiagnostic Systems, Ltd.). Serum osteocalcin measurements were done with the mouse osteocalcin EIA Kit (Biomedical Technologies). For serum lactate measurements, the serum was first precipitated with two volumes of ice-cold ethanol, and lactate levels in the supernatant were measured with the l-lactate assay kit (Eton Bioscience).
Vascular Casting.
Mice were anesthetized and perfused with heparinized saline followed by 10% (vol/vol) neutral-buffered formalin and then again with heparinized saline. Microfil compound (MV-122, Flow Tech, Inc.) prepared according to the manufacturer’s instructions was perfused transcardially. The body was stored at 4 °C overnight to allow full polymerization and then bones were isolated. The epiphyses and all excess tissue were removed and the bones were decalcified for 48 h in Cal-Ex II (Fisher). Tibias were held in styrofoam and scanned by µCT (µCT 40; Scanco Medical AG) with the following settings: 70 kVp, 114 µA, and 6 µm voxel size. 3D reconstructions of the vessels of the cancellous region immediately beneath the growth plate were generated with the following parameters: Gaussian support of 1 and sigma of 2 with the threshold at 120.
Protein Extraction and Western Blotting.
For protein extraction from bone, femurs and tibias were isolated, and the epiphyses and all excess tissue were removed. Bone marrow was removed by centrifugation, and bones were snap-frozen in liquid nitrogen, followed by pulverization in a Mikro Dismembrator U (Sartorius). Protein was extracted with 150 µL radioimmunoprecipitation assay buffer containing protease inhibitors on ice for 30 min. Protein concentrations were measured by BCA assay (Thermo Scientific), and Western blotting was performed according to standard protocols. HK2 (2867S), LDHA (2012S), and β-actin (4970S) antibodies were all from Cell Signaling Technologies. The PDK1 (ADI-KAP-PK112) antibody was purchased from Enzo.
Statistics.
Quantitative data are presented as mean ± SD. Sample numbers are as indicated in the figure legends. Statistical significance was determined by Student t test. Asterisks indicate P ≤ 0.05.
Supplementary Material
Acknowledgments
The work is supported by National Institutes of Health Grants R01 DK065789 and R01 AR055923 (to F.L.), T32 HL007873 (to J.N.R.), and P30 AR057235 (Washington University Center for Musculoskeletal Research).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1324290111/-/DCSupplemental.
References
- 1.Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 2008;9(4):285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lendahl U, Lee KL, Yang H, Poellinger L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat Rev Genet. 2009;10(12):821–832. doi: 10.1038/nrg2665. [DOI] [PubMed] [Google Scholar]
- 3.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998;95(14):7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maxwell PH, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 5.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 6.Semenza GL, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271(51):32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 7.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92(12):5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benita Y, et al. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37(14):4587–4602. doi: 10.1093/nar/gkp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mole DR, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284(25):16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schödel J, et al. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117(23):e207–e217. doi: 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 12.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117(6):1616–1626. doi: 10.1172/JCI31581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shomento SH, et al. Hypoxia-inducible factors 1alpha and 2alpha exert both distinct and overlapping functions in long bone development. J Cell Biochem. 2010;109(1):196–204. doi: 10.1002/jcb.22396. [DOI] [PubMed] [Google Scholar]
- 16.Maes C, et al. VEGF-independent cell-autonomous functions of HIF-1alpha regulating oxygen consumption in fetal cartilage are critical for chondrocyte survival. J Bone Miner Res. 2012;27(3):596–609. doi: 10.1002/jbmr.1487. [DOI] [PubMed] [Google Scholar]
- 17.Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys J. 2001;81(2):685–696. doi: 10.1016/S0006-3495(01)75733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arteel GE, Thurman RG, Yates JM, Raleigh JA. Evidence that hypoxia markers detect oxygen gradients in liver: Pimonidazole and retrograde perfusion of rat liver. Br J Cancer. 1995;72(4):889–895. doi: 10.1038/bjc.1995.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long F, et al. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development. 2004;131(6):1309–1318. doi: 10.1242/dev.01006. [DOI] [PubMed] [Google Scholar]
- 20.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–3244. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 21.Bekeredjian R, et al. Conditional HIF-1alpha expression produces a reversible cardiomyopathy. PLoS ONE. 2010;5(7):e11693. doi: 10.1371/journal.pone.0011693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsythe JA, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maes C, et al. Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J. 2010;29(2):424–441. doi: 10.1038/emboj.2009.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerber HP, et al. VEGF is required for growth and survival in neonatal mice. Development. 1999;126(6):1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- 26.Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347–353. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- 27.Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974;141(3):761–774. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132(19):4339–4351. doi: 10.1242/dev.02025. [DOI] [PubMed] [Google Scholar]
- 29.Wan C, et al. Activation of the hypoxia-inducible factor-1alpha pathway accelerates bone regeneration. Proc Natl Acad Sci USA. 2008;105(2):686–691. doi: 10.1073/pnas.0708474105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imai S, et al. Sir2: An NAD-dependent histone deacetylase that connects chromatin silencing, metabolism, and aging. Cold Spring Harb Symp Quant Biol. 2000;65:297–302. doi: 10.1101/sqb.2000.65.297. [DOI] [PubMed] [Google Scholar]
- 31.Wellen KE, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324(5930):1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esen E, et al. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 2013;17(5):745–755. doi: 10.1016/j.cmet.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.