

Abstract
Genetically engineered tumor-selective vaccinia virus (VV) has been demonstrated to be a highly effective oncolytic agent, but immune clearance may limit its therapeutic potential. As previously demonstrated, immunosuppression can lead to significant enhancement of viral recovery and therapeutic effect, but the magnitude of complement-mediated viral inactivation has not been fully elucidated and warrants further investigation. Using fluorescent microscopy and quantitative plaque assays, we have determined complement's key role in viral clearance and its multi-faceted means to pathogen destruction. Complement can lead to direct viral destruction and inhibition of viral uptake into cells, even in the absence of anti-vaccinia antibodies. Our data demonstrate C5 to be integral to the clearance pathway, and its inhibition by Staphylococcal superantigen-like protein (SSL7) leads to a 90-fold and 150-fold enhancement of VV infectivity in both the presence and absence of anti-VV antibodies, respectively. This study suggests that complement inhibition may reduce vaccinia viral neutralization and may be critical to future in vivo work.
Keywords: vaccinia virus, complement, C5, SSL7, compstatin, cobra venom factor
Introduction
Tumor selective, genetically engineered vaccinia virus (VV) represents a highly effective oncolytic agent and serves as a novel approach to cancer therapy.1 VV is a member of the orthopox virus genus and was used as the vaccine for smallpox, formally responsible for its eradication in 1979.1,2 This virus has potential as an efficacious vector for cancer therapy due to its large size, efficiency, and elusiveness.3 In murine models, tumor selective, mutant VV has been shown to infect and express genes specifically in the tumor when delivered systemically, leading to an anti-tumor response.2 Genetic engineering of VV with a single deletion of the thymidine kinase (TK) gene has been effectively performed and tested in a phase I trial, yielding promising results.4,5 Additionally, a high tumor-selective double viral gene deleted VV (vvDD), containing deletions of both the TK and vaccinia growth factor (VGF) genes, has been shown to be more tumor-selective with less pathogenicity than the virus with tk gene deletion and is currently being tested in Phase I trials.6
Despite findings validating VV as an effective biologic agent, the intact immune system in a naïve host eliminates the virus prior to its maximum antitumor effect.7,8 In the immunocompetent host, the virus can replicate for 8 days with high levels of gene activity only lasting for about 4 days.9 VV-immunized people have circulating antibodies directed against viral proteins10 as well as memory T-cells that eliminate VV even faster, thereby increasing host resistance to the virus.1 The body's immune system is comprised of innate and adaptive immunity, both of which work in tandem to eliminate pathogens through non-specific and specific mechanisms.11-15 Ultimately, both components of the immune system enroll the complement system to rid the body of unwanted pathogens.13-16
The complement system can directly lyse viruses and virally infected cells or mediate antibody-directed viral or cell lysis, and its manipulation may be a potential target to enhance VV efficacy.17 Over 25 proteins and protein fragments make up the complement system, including serum proteins, serosal proteins, and cell membrane receptors.17,18 This elegant system is composed of three pathways, which are antibody-mediated, mannose-binding lectin-mediated, and spontaneously activated, respectively.17-19 All three pathways have two major convergence points, which include the cleavage of C3 and C5 to its active compounds.17,20,21
The complement system has previously been found to be particularly important in poxvirus infection.20 Moulton et al demonstrated that by multiple inoculation routes, ectromelia virus caused increased mortality by 7-10 days post-infection in C57BL/6 mice that lacked C3. In the C3 knockout mice, ectromelia virus disseminated earlier to target organs and generated higher peak titers compared to congenic controls as well as increased hepatic inflammation and necrosis.20 In vitro, the complement system in naïve C57BL/6 mouse sera neutralized ectromelia virus through recognition of the virion by natural antibody (Ab) and activation of classical and alternative pathways. Sera deficient in either the classical or alternative pathway components had reduced ability to neutralize viral particles.20 Given the importance of complement in viral clearance, poxviruses have adapted and have developed inherent complement inhibitors. Smallpox has a very potent human complement inhibitor (Smallpox inhibitor of complement enzymes, SPICE), which may be an important factor explaining its virulence compared to VV in human infection.21 In the setting of a tumor selective virus where viral virulence is already inhibited, and its oncolytic function is preserved, a more potent complement inhibitor may be safe and appropriate to increase efficacy.21
Our current investigation was performed to demonstrate the role of complement in VV infectivity/pathogenicity and determine which pathway of the complement system is integral to VV viral clearance and thereby, specifically inhibit this pathway with a monoclonal Ab (mAb) to enhance viral expression. We then sought to examine and compare small protein inhibitors of complement in terms of their effects on VV viral efficacy given their possible clinical use.22 Protein inhibitors of complement include Compstatin, Staphylococcal superantigen-like protein (SSL7), and purified cobra venom factor (CVF). Compstatin is a C3-binding cyclic synthetic peptide, currently being used for macular degeneration.22 Compstatin binds C3 and thereby inhibits binding of C3 convertase, preventing production of C3a and C3b. Without C3b, C5 convertase cannot form, thereby inhibiting production of C5a and C5b. Terminal membrane attack complex formation cannot occur without C5b, thus preventing pathogen clearance. Jayasekera and Caroll et al showed that full activation of the complement pathway can lead to viral lysis by formation of the terminal membrane attack complex, which consists of C5b-C9. Deposition of the membrane attack complex on the surface of an enveloped virus, such as influenza virus, produces membrane-spanning pores that can alter the stability of the virion. 23 SSL7 binds C5 and prevents C5 degradation to its activated components by C5 convertase.24 The final inhibitor, purified CVF, a C3-like molecule, can initiate the alternative pathway by forming an extremely stable C3/C5 convertase by binding to Factor B, which leads to uninhibited C5-consumption and excess C5a production.25 All three inhibitors may serve as possible immune system modulators in future clinical trials, which is our ultimate goal.
Materials and Methods
Cell lines
Mouse colon cancer MC38 cells, African green monkey kidney fibroblasts (CV-1), and human 143B cancer cells have been used frequently in this laboratory. All cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), L-glutamine, penicillin/streptomycin (Invitrogen, Carlsbad, CA) at 37°C under 5% CO2.
Recombinant VV
A WR strain derivative, vvDD-EGFP, a double viral gene-deleted (tk- and vgf-) VV armed with recombinant EGFP gene, was used as the primary virus used in the in vitro assays.6
Human serum
Human sera were obtained from consented donors, IRB approval # REN12110254/ IRB021068. In order to confirm vaccination status, a plaque reduction neutralization assay was performed. All vaccinated serum used was confirmed to have presence of antibody while all nonvaccinated serum used lacked presence of antibody.
Combination of antibody and serum
Vaccinated and nonvaccinated human serum were placed in labeled vials. Heat-inactivated versions of human serum were obtained by placing labeled vials in a 56°C water bath for 45 minutes. 100 μL vaccinated human serum and 200 μL nonvaccinated serum were combined with 1.9 mL and 1.8 mL heat-inactivated DMEM/FBS, respectively, to produce 5% vaccinated human serum and 10% nonvaccinated human serum in labeled vials. 100 μL of these premade serum solutions were added to a pre-labeled 96-well plate of MC38 cells, plated at 1×104 cells/well and used within 24 h of plating. Specific mAb at predetermined doses was then added to the serum samples. The source of all monoclonal antibodies was Abcam. The control IgG used was normal mouse IgG, reactive against human IgG obtained from Santa Cruz Biotechnology. Antibody doses were as follows: C1 mAb (8 μg/mL); C3 mAb (48 μg/mL); C4 mAb (9.6 μg/mL); C5 mAb (3 μg/mL); and C5a mAb (3 μg/mL). Dosing was based on human serum complement factor concentrations found in current literature. The cells were then infected with vvDD-EGFP at an MOI of 1.
Fluorescent microscopy
A fluorescent microscope was used to image the expression of EGFP in cells infected by vvDD-EGFP. As in conventional epifluorescence photomicroscopy, GFP expressed in cells was distinguished as a bright green signal over a yellow-green autofluorescence background. Images were obtained via live automated camera within IDM viewer computer program. The magnification used was 10X, and the images all represent multiple foci of multiple plaques.
Purification of SSL7
Recombinant SSL7 was kindly provided by the Fraser Group, University of Auckland, New Zealand. Dose of 0.4 mg/mL SSL7 was used in all experiments including this protein inhibitor. MC38 cells were plated at 1×104 cells/well in a 96-well plate were again incubated in 10% nonvaccinated and 5% vaccinated human serum. 0.4 mg/mL SSL7 was added to predesignated wells. All wells were then infected with vvDD-EGFP at an MOI of 1. Fluorescent microscopy and a plaque assay were then performed following a 48 h infection period.
Compstatin
It was obtained from Tocris Bioscience, of R&D Systems, Inc. (Minneapolis MN). Soluble to 2 mg/mL in 30% aceto-nitrile/water. A dose of 65 μg/mL compstatin was used in all experiments including this complement inhibitor. Similar to the use of SSL7, predesignated wells of MC38 cells were incubated with 65 μg/mL compstatin, and then all wells were infected with vvDD-EGFP. Following a 48 h infection period, fluorescent microscopy and a plaque assay were performed.
Cobra venom factor, purified
It was obtained from Quidel Corporation (San Diego, CA), from the Naja naja kaouthia species. Dose of CVF used in all in vitro experiments was 40 μg/mL. MC38 cells, incubated in human serum, were treated with CVF and were then infected with vvDD-EGFP. Following a 48 h infection period, fluorescent microscopy and a plaque assay were performed.
Plaque assays
CV-1 cells were plated in 6-well plates 24 to 72 h prior to use in assay at 1x106 cells/well. The MC38 cells that had been infected by virus under different serum conditions were harvested and used to infect the CV1 cell line. Each sample was diluted in 1× DMEM, supplemented with 2% FBS (10 mL FBS/500 mL DMEM), in a 10-fold fashion according to the experimental design with dilution factors of 2, 3 and 4. The culture medium in the 6-well plate was then removed by either swift decanting or aspiration by pipette and virus inocula of various concentrations were added to replicate wells in volumes of 2mL. Inoculum fluid was distributed by gentle rocking, manually. The plates were incubated at 37 °C for about 1 hour after which the samples were removed, and 10% DMEM/FBS was added to the CV1 cells. After 48 h, the media was removed, and 1 mL 0.1% crystal violet in 10% ethanol was added to each well. The number of plaques was counted for each sample.
Plaque counting and titer calculation
Plates were inverted on a light box and plaques were counted by hand with the aid of a marker by one operator. If there were wells at two different serial dilutions with counts within this range, then the counts from wells with the more concentrated dilution were chosen for calculating titer. Counts from replicate wells were averaged, and that number was multiplied by the dilution factor of the inoculum which produced that count, and the volume of inoculum plated to calculate the plaque forming units (PFU) per ml of the original stock virus preparation. The calculation is: average value of plaques in replicate wells × mL of virus inoculum × dilution factor = titer in PFU/mL.
Measurement of plaque size
For the experiment designed to elicit the effect of complement inhibition on viral spread, determination of plaque size was more critical than counting the number of plaques. For each sample again titred on CV1 cells in a serially diluted manner, the diameters of 10 plaques were measured using a standard ruler and averaged.
Cell viability assay
For the viral cytotoxicity assay, cancer cells were plated at 1.0 × 104 cells per well in 96-well plates for growth over night, and then subsequently incubated in human serum (vaccinated, nonvaccinated, and heat-inactivated versions of both). Predesignated wells of cells were then treated with specific complement inhibitors (C5 mAb, Compstatin, SSL7, and CVF) and were then infected with vvDD-EGFP at an MOI of 1.0 following a half-hour incubation period with the treatment regimen. Cell viability was determined at 48 h after infection by first harvesting the infected MC38 cells. Each sample of cells was then centrifuged, and the cell pellet was then resuspended in 10% DMEM/FBS. An equal volume of trypan blue was added to each sample, and viable cells were then counted using microscopy.
Statistical analysis
Raw data were recorded electronically and statistical analyses were performed with Microsoft Excel or the SPSS Software version 18 (IBM, NY, USA). Central tendencies were expressed either as means +/- SD with Student's t-test or medians with Kruskal-Wallis as appropriate. An alpha value (p) of 0.05 was considered as statistically significant.
Results
Complement inhibition enhances VV gene expression/infectivity
We grew MC38 cells in 96 well plates and incubated the cells with different samples of human serum: 5% vaccinated human serum (vacc. HS), 10% non-vaccinated human serum (nonvacc. HS), and heat-inactivated versions of both. These concentrations of human serum were determined previously, where 5% and 10% were found to be the minimal concentrations of vaccinated and nonvaccinated human serum, respectively, needed to effectively inhibit VV infection of rapidly dividing cells. MC38 cancer cells were then infected with vvDD-EGFP at an MOI of 1, and viral expression of EGFP was examined under the fluorescent microscope at 24 and 48 h. Viral titers were then performed using a plaque assay. Inhibition of the complement system via heat-inactivation resulted in enhanced viral expression (Figure 1a). Additionally, the number of viral plaques was significantly higher than the samples incubated with non-heat-inactivated human serum. One hundred seventy PFU/mL virus was recovered with heat-inactivated vaccinated serum (HI vacc. HS) as opposed to 50 PFU/mL virus with the non-heat inactivated vaccinated serum. In the nonvaccinated group, the heat-inactivated serum produced 400 PFU/mL virus as opposed to 70 PFU/mL obtained in the non-heat inactivated serum (Figure 1b). Even in presence of preformed Ab to VV, complement inhibition led to a 3-4 fold increase in viral recovery.
Figure 1.
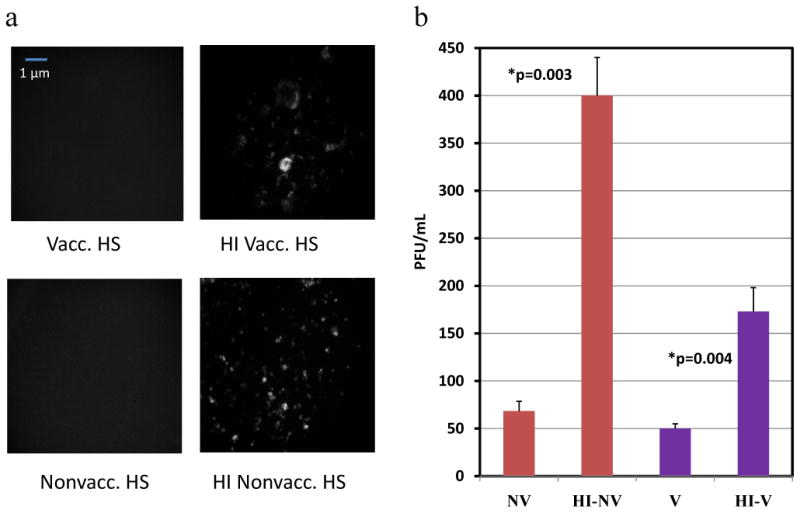
Effect of complement on viral infectivity. (a) Viral expression following 48 h incubation period. Fluorescent microscopy images of MC38 cells incubated in Vacc. HS/Heat-Inactivated (HI) Vacc. HS as well as Nonvacc. HS/HI Nonvacc. HS following vvDD-EGFP infection at MOI of 1; (b) Plaque assay illustrating differing number of plaques between vaccinated and nonvaccinated serum groups and heat-inactivated serum groups. p-value < 0.05 was considered statistically significant, and used to describe differences between heat-inactivated and non-heat inactivated serum groups.
Inhibition of C5 of the complement system using a monoclonal antibody enhances vaccinia viral expression
After the initial study illustrated the importance of the complement system in VV viral expression and infectivity, we next hoped to identify the specific pathway of the complement system involved in viral clearance. We obtained monoclonal antibodies against specific components of the complement system: C1q, C3, C4, C5, and C5a. It has been shown in several in vitro and in vivo assays that monoclonal antibodies directed against complement components reduce the complement activity of serum. Functional assays have determined that mAbs for C1q, C3, C5, and C5a can deplete circulating complement serum protein levels in vivo and in vitro. The C1q functional assay is well established and has illustrated a depletion of C1 with the addition of the mAb. Alsenz et al illustrated the inhibitory ability of C3 mAb in serum in his article entitled “Structural and Functional analysis of C3 using monoclonal antibodies. Additionally, C3 mAb function had been verified by Whitehead et al, illustrating its ability to inhibit a C3 hemolytic assay. Anti-C5 mAb was used in Wang et al's article entitled “anti-C5 monoclonal antibody therapy prevents collagen induced arthritis and ameliorates established disease” and was shown to diminish terminal complement activation.26-29 C5 mAb binds specifically to the C5 beta chain between residues Tyr334 and Lys41, while C5a mAb binds to a neo-epitope on human C5a/C5a des-Arg. The monoclonal antibody directed against C5b reacts with an epitope present on C5b as well as on C5. C5a acts as an anaphylatoxin and is a chemotactic attractant for neutrophils. C5b serves as the anchor for the formation of the terminal membrane attack complex.30 MC38 cells were again incubated in 5% vaccinated human serum, and 10% non-vaccinated human serum +/- each individual monoclonal antibody. The cells were then infected with vvDD-EGFP at an MOI of 1. Viral expression was determined by fluorescent microscopy at 48 h (Figure 2a). As seen in the aforementioned figure, viral gene expression was markedly enhanced in both the vaccinated and nonvaccinated serum settings when mAb against C5 was added. We then quantified viral recovery via plaque assay on CV1 cells. The number of viral plaques was highest in the vaccinated and nonvaccinated serum groups where C5 mAb had been added. The addition of C5 mAb increased viral recovery 20-fold and 50-fold in the vaccinated and nonvaccinated serum settings, respectively (Figure 2b, 2c). Though the addition of C3 mAb appeared to be effective in enhancing viral expression/recovery in the presence of vaccinated human serum, in the presence of nonvacc. HS, C3 inhibition did not appear to be entirely successful. Therefore, of all the complement system components targeted, C5 inhibition led to the greatest significant upregulation of viral infectivity and efficacy in both vacc. and nonvacc. HS.
Figure 2.
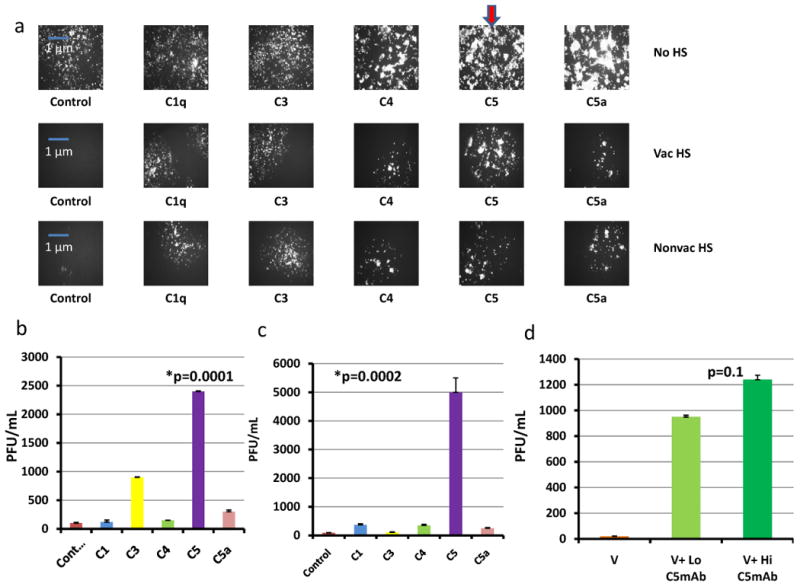
Inhibition of specific components of the complement pathway and viral expression/infectivity. (a) Inhibition of specific components of the complement pathway and viral expression. MC38 cells incubated in no human serum or human serum (vaccinated, nonvaccinated) and added monoclonal antibody against C1 (8 μg/mL), C3 (48 μg/mL), C4 (9.6 μg/mL), C5 (3 μg/mL), or C5a (3 μg/mL) were then infected by vvDD-EGFP at an MOI of 1. Fluorescent microscopy images of infected cells after 48 h are shown here. The bold red arrow indicates marked enhancement of viral expression in both nonvaccinated and vaccinated serum settings with the addition of C5 mAb. (b) mAb inhibition of specific components of the complement system in the setting of vaccinated serum. (c) Monoclonal Antibody inhibition of specific components of the complement system (plaque assay) in the setting of nonvaccinated serum. P-values were noted to compare the serum groups with the addition of C5 mAb to the control IgG groups. The addition of all monoclonal antibodies, except C4, resulted in significant increases in viral recovery in both settings, designated by the asterisks. C4 mAb addition in the vaccinated serum group did not significantly enhance viral recovery. As expected, C5 mAb had a more drastic effect on viral recovery than C5a mAb. (d) Dose Effectiveness of C5 mAb. C5 mAb was administered to human serum-incubated MC38 cells at high (0.1 mg/mL) and low (0.01 mg/mL) concentrations. Cells were then infected with vvDD-EGFP at an MOI of 1. p-value noted in the figure compared the high and low concentration C5 mAb groups. The addition of both high concentration and low concentration C5 mAb resulted in significant increases in viral recovery compared to the control group.
An experiment designed to determine dose-effectiveness of C5 mAb was performed and revealed high dose C5 mAb (0.1 mg/mL) to be more effective in enhancing viral gene expression and viral recovery than low dose (0.01 mg/mL solution), but this difference was not statistically significant (Figure 2d).
Complement inhibition enhances vaccinia viral oncolysis
In order to determine whether complement inhibition enhances VV-mediated oncolysis, MC38 cells incubated in vaccinated, nonvaccinated, and heat-inactivated versions of both +/- C5 mAb were infected with vvDD-EGFP at an MOI of 1. Following a 48 h infection, viability of the tumor cells was assessed by trypan blue exclusion and manual counting of viable cells. The results revealed markedly fewer viable tumor cells in vaccinated and nonvaccinated serum where C5mAb was added (Figure 3). In the vaccinated serum setting, the addition of C5 mAb decreased cell viability by 74%, and in the nonvaccinated serum group, cell viability fell by 80% with the addition of C5 mAb.
Figure 3.
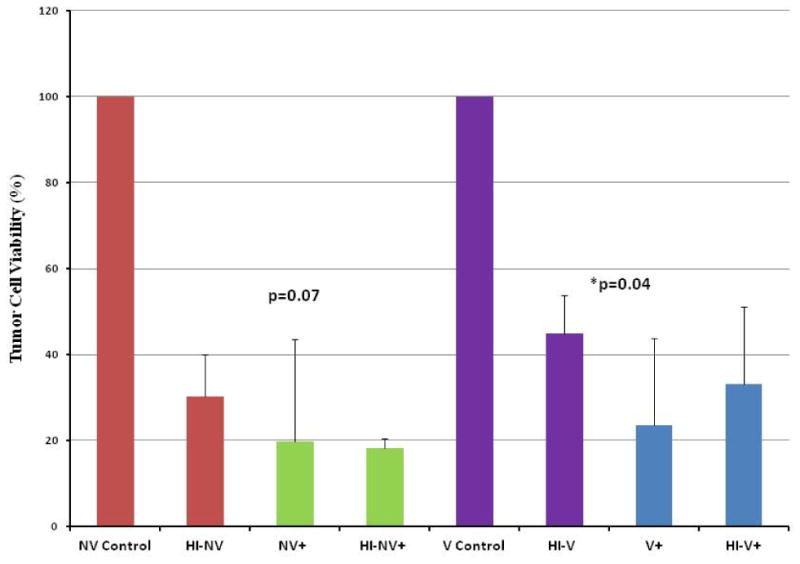
Effect of complement inhibition via mAb on VV oncolysis. MC38 cells incubated in serum (vacc., nonvacc., and heat-inactivated versions) +/- C5 mAb were then infected by vvDD at an MOI of 1.0. After a 48 h infection period, the cells were harvested and counted. Complement inhibition with addition of C5 monoclonal antibody resulted in a marked decrease in the % tumor cell viability in both the vaccinated and nonvaccinated serum settings. The addition of C5 mAb resulted in a significant decrease in % viable cells in the vaccinated setting (designated by the asterisk) and resulted in a trend towards a significant decrement in % viable cells in the nonvaccinated serum setting.
Inhibition of complement during the initial viral infection enhances VV expression/infectivity
To determine the mechanisms of action of complement in clearance of VV, a series of experiments was performed. The first of which was to determine whether the complement system is critical in the initial tumor cell infection rather than the actual spread of VV amongst the cells. This was assessed by placing vaccinated human serum and nonvaccinated human serum in vials +/- C5 mAb. To each of the vials, vvDD-EGFP was added at an MOI of 1. Following a 3 h incubation period, each of these samples was added to MC38 cells in a 96-well plate for a 1-2 h infection period. Viral gene expression was again assessed via fluorescent microscopy at 24 and 48 h. At 48 h, the highest EGFP expression was seen in both the vaccinated and nonvaccinated serum settings when C5 mAb was added. The infected MC38 cells were then harvested, and a plaque assay was performed. Viral recovery was enhanced four-fold in the nonvaccinated serum setting and increased from 0 PFU/mL to 90 PFU/mL in the vaccinated serum setting when C5 mAb was added (Figure 4). The data suggest that complement can directly lyse vaccinia virions prior to infection of tumor cells and may play a major role in the initial VV infection.
Figure 4.
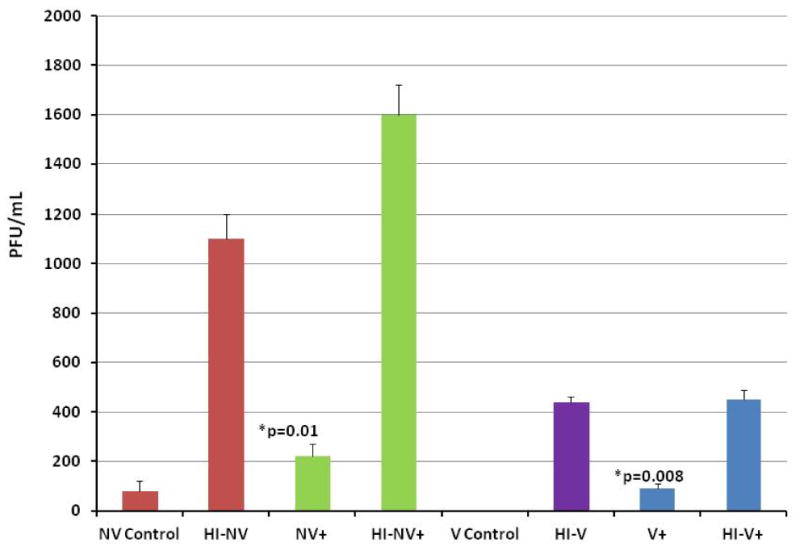
Effect of complement system on initial VV infectivity. vvDD at an MOI of 1 was added to samples of human serum (vaccinated/non-vaccinated/heat-inactivated versions) +/- C5a mAb, C5 mAb, or C5b mAb. After a 3-hour incubation period, each sample was added to a 96-well plate of MC38 cells. Following a 48 hour period, the MC38 cells were harvested and used to perform a plaque assay. The addition of C5 mAb resulted in a significant increase in viral recovery in both serum settings, designated by the asterisks and p-values.
Inhibition of complement does not affect cell-to-cell VV viral spread
To determine whether complement also inhibits the cell-to-cell spread of vaccinia virus, another experiment was performed in which CV1 cells were infected with vvDD-EGFP at an MOI of 0.001. Vaccinated, nonvaccinated, and heat-inactivated versions of both serums +/- C5 mAb were added to the infected cells after a 6 h infection period, and viral expression was noted after 24 h followed by addition of crystal violet to all wells to determine plaque size (Figure 5a). The size of the viral plaques did not vary in the different groups, illustrating the ability of cell-to-cell VV viral spread to proceed regardless of complement inhibition (Figure 5b). This experiment was repeated using 143B cells, an osteosarcoma cell line, and again viral expression was not significantly enhanced when the infected cells were incubated with vaccinated or nonvaccinated serum where C5 mAb had been added compared to their control counterparts (Figure 5c). Additionally, the sizes of the plaques were not significantly different between the groups, illustrating that complement inhibition does not affect VV viral spread (Figure 5d). VV spreads in a cell-to-cell manner, unexposed to serum components. Therefore, viral spread is unaffected by complement and is unhindered by its inhibition.
Figure 5.
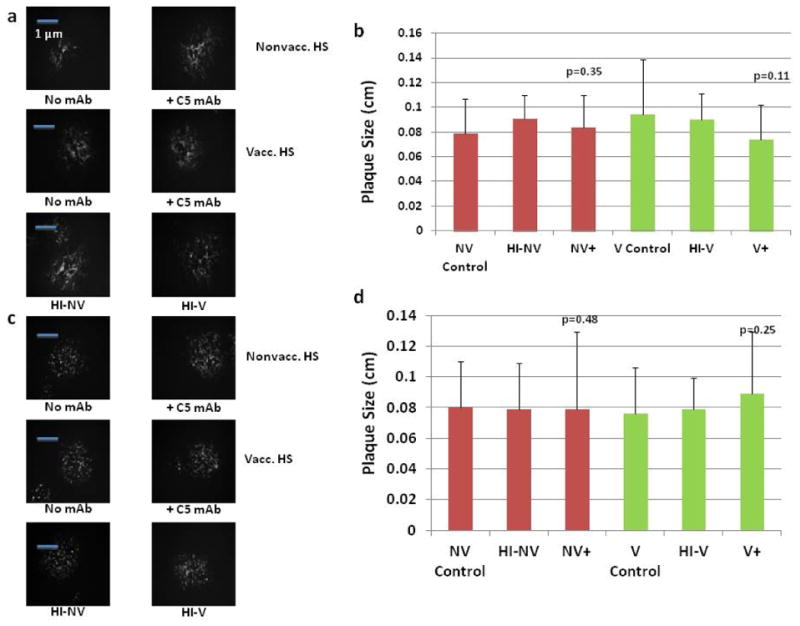
Effect of complement inhibition on vaccinia viral spread. (a) CV1 cells infected with vvDD at an MOI of .1. After a 6 h infection period, samples of human serum (vacc., and nonvacc) +/- C5 mAb were added, fluorescent microscope images were obtained after 48hr incubation period. (b) For each sample again titred on CV1 cells and following administration of crystal violet to each well, the diameters of 10 plaques were measured in cm using a standard ruler and averaged. (c) The previous experiment was performed using 143B cells, fluorescent microscopy was performed after a 48 h incubation period. (d) Sizes of plaques were measured, and the average of 10 plaques was taken. The averages of all treatment groups were not significantly different from the control groups, as shown by the marked p-values.
Protein inhibitors of complement also increase vaccinia viral infectivity
Given previous findings illustrating the importance of C5 in the clearance of VV in both the vaccinated and nonvaccinated serum settings, further experiments were performed to determine whether protein inhibitors of the central portions of the complement system are as effective as mAb for the purposes of future in vivo experiments. Three protein inhibitors of complement: Compstatin, SSL7, and CVF were obtained. An experiment was designed similar to the experiments performed with mAb against components of the complement system. MC38 cells were treated with vaccinated, nonvaccinated, and heat-inactivated versions of both serums with or without compstatin at its appropriate dose of 10 μl/well (at 2 mg/mL concentration). The cells were then infected with vvDD-EGFP at an MOI of 1. Viral marker gene expression was assessed via fluorescent microscopy at 24 and 48 h, followed by plaque assay. As figure 6a demonstrates, the addition of compstatin increased viral EGFP expression at 48 h in both the vaccinated and nonvaccinated serum settings. The plaque assay confirmed these findings (Figure 6b). In the nonvaccinated serum setting, the addition of compstatin doubled viral recovery, and in the vaccinated serum group, viral recovery was enhanced almost four-fold.
Figure 6.
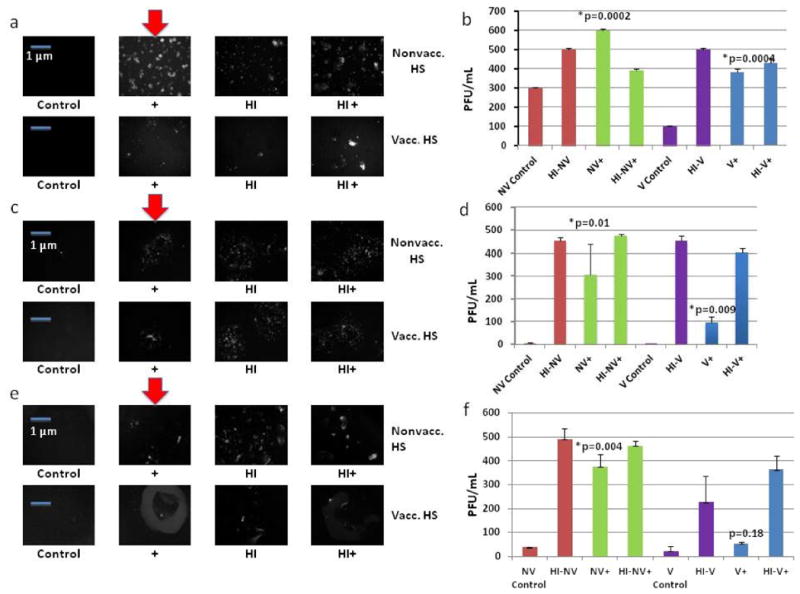
Small molecule/protein inhibitors of complement. (a) MC38 cells incubated in serum (vacc., nonvacc., and heat-inactivated versions) +/- Compstatin were then infected by vvDD at an MOI of 1.0. (b) Compstatin plaque assay. (c) MC38 cells incubated in serum(vacc., nonvacc., and heat-inactivated versions) +/- SSL7 were then infected by vvDD at MOI of 1.0. (d) SSL7 Plaque Assay. (e) MC38 cells incubated in serum(vacc., nonvacc. and heat-inactivated versions) +/- CVF were then infected by vvDD at MOI of 1; f) CVF Plaque Assay. Bold red arrows indicate addition of complement inhibitor and marked enhancement of viral expression. p-values were noted to compare treatment groups to control groups, indicating statistically significant results with the addition of all complement inhibitors in both serum settings except the addition of CVF to vaccinated serum.
This same experiment was performed using SSL7 and similar findings were obtained.24 At 48 h, viral expression under fluorescent microscopy was enhanced in the vaccinated and nonvaccinated groups where SSL7 was added compared to their control counterparts (Figure 6c). A plaque assay was again performed, which resulted in a 90-fold increase in vvDD recovery in the vaccinated serum group when SSL7 was added and a 150-fold upregulation in viral recovery in the nonvaccinated serum setting with the addition of SSL7 (Figure 6d).
Lastly, an in vitro experiment was performed in which MC38 cells were incubated with vaccinated, nonvaccinated, and heat-inactivated serum with or without CVF. The cells were then infected with vvDD-EGFP at an MOI of 1. Viral expression in both vaccinated and nonvaccinated serum groups were enhanced with the addition of CVF (Figure 6e). A plaque assay was once again performed. In the vaccinated group, vvDD recovery was almost tripled with the addition of CVF. In the nonvaccinated serum group, viral recovery was enhanced more than 10-fold with the addition of CVF (Figure 6f).
Though all three inhibitors did enhance VV viral gene expression and viral recovery, the addition of SSL7 to serum had the greatest impact on viral infectivity, indicating that direct C5 inhibition using a complement inhibitor can indeed increase VV efficacy and may have potential use in future clinical trials.
Protein inhibitors of complement enhance VV oncolysis
In order to determine whether complement inhibition via protein inhibitors also enhances VV oncolysis, MC38 cells were once again incubated in vaccinated, nonvaccinated, and heat-inactivated versions of both. Each sample was with or without SSL7, compstatin, or purified CVF. All wells of cells were infected with vvDD-EGP at an MOI of 1. Following a 48 h infection, viability of the tumor cells was assessed by trypan blue exclusion and manual counting of viable cells. The results showed that markedly fewer viable tumor cells were found in vacc. and nonvacc. HS with the addition of SSL7, compstatin, or CVF. The addition of SSL7, compstatin, and CVF decreased the number of viable MC38 cells by 75%, 74%, and 73% in the vaccinated group, respectively and by 77%, 66%, and 71%, respectively in the nonvaccinated serum setting (Figure 7).
Figure 7.
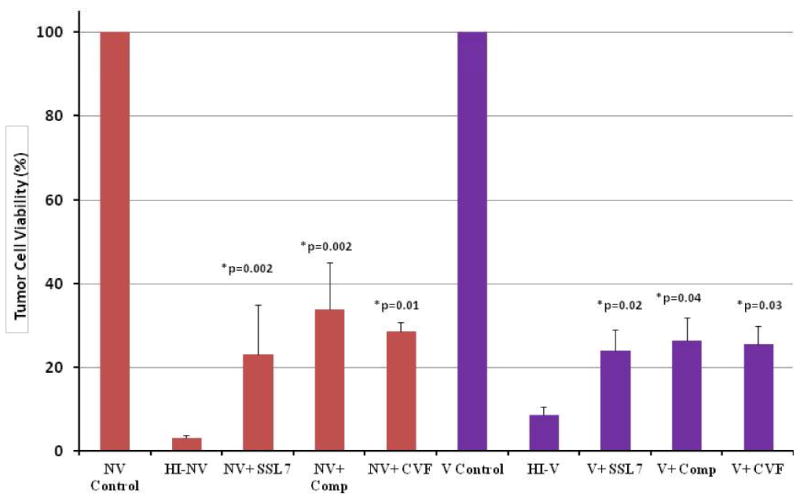
Effect of complement inhibition via small molecule/protein inhibitors on VV oncolysis. MC38 cells incubated in serum (vacc., nonvacc., and heat-inactivated versions) +/- SSL7 or Compstatin were then infected by vvDD at an MOI of 1. After a 48 h infection period, the cells were harvested, and viable cells were counted following addition of trypan blue to the cells. The addition of both SSL7 and Compstatin significantly decreased the % tumor cell viability in both the vaccinated and nonvaccinated serum settings, while the addition of CVF resulted in a significant reduction of % tumor cell viability in the nonvaccinated setting only. p-values <0.05 were obtained for all treatment groups in both serum settings when compared to control groups.
Discussion
VV has been shown to be an effective cancer therapeutic agent, but its effects are limited by immune system clearance.1 The immune response is a complex mixture of effector molecules and cells that interact with one another to eliminate the virus.10 Recent years have seen tremendous advances in the development of tumor-targeting virotherapeutics that can safely and selectively destroy malignancies.7 Though it holds great potential, significant implementation of this effective oncolytic virus may be hindered by vulnerability to immune system barriers and early systemic clearance.
Pre-existing antipoxvirus immunity in cancer patients presents a severe impediment to oncolytic virotherapy.10 We have learned that both innate and adaptive immunity are critical to viral clearance16 and have previously examined the combination of immunosuppression with carrier cell delivery of an oncolytic VV.31 Using an MC38 peritoneal carcinomatosis model, immunosuppression alone did not enhance viral recovery from tumor after viral delivery, but the combination of immunosuppression and carrier cells led to the most effective viral delivery, viral replication, and viral spread in tumor.31 We found that this combination significantly enhanced the efficacy of the oncolytic poxvirus in the pre-immunized host.31 However, immunosuppression is not without side effects and may manifest as increased susceptibility to opportunistic infections and decreased cancer immunosurveillance.32-34 Therefore, caution must be heralded when employing immunosuppressants. Other modes of immune system modulation may be as effective as the immunosuppressants used previously without the same side effect profiles and must be further investigated.
Though some earlier studies have shown enhancement of oncolysis by combining VV with immunosuppression of cellular responses,35 our lab has demonstrated that immunosuppressive drug therapy alone does not significantly enhance infectivity or prolong VV infection in the setting of pre-formed anti-vaccinia antibodies.32,36,37 In this previously mentioned study, the mice were preimmunized to VV, and suppression of the T cell response was not enough to overcome the barriers set by the immune system.31 Though the use of carrier cells to circumvent pre-formed antibodies did enhance viral infectivity in the preimmunized host, this technique of virus delivery may be impractical in the human subject. Additionally, carrier cell delivery of virus may not be potent enough to evade the complement system. Therefore, other means of immune system inhibition are sought to prevent early clearance of VV.
Complement inhibition has been implemented in studies using herpes simplex virus (HSV) oncolytic therapy and has demonstrated promising results. Ikeda et al demonstrated that in immunocompetent hosts, cell transduction by replication-conditional HSV administered intraarterially was inhibited.38,39 They found that complement depletion improved initial infection of intracerebral brain tumors with HSV using CVF. However, complement inhibition was insufficient in enhancing subsequent viral propagation within tumor tissues.38
Complement has been shown to be critical in pathogen clearance and plays a central role in poxviral pathogenicity in particular.17-19 Moulton et al demonstrated the role of complement in ectromelia viral infection. Complement-deficient mice, when infected by ectromelia virus, had much higher morbidity and mortality when compared to control mice.20
Our study identifies complement as a potent player in immune system surveillance and VV clearance, and a major hindrance to effective virotherapeutics. Our findings demonstrate that complement inhibition markedly enhanced VV efficacy, and that C5 is the most critical target for inhibition. In the setting of non-heat-inactivated serum and presence of the complement system, vaccinia viral expression and recovery was inhibited entirely, a quite surprising and remarkable finding. Our lab has never previously performed in vitro experiments using complement-intact serum. Therefore, this finding was the crux of our further experimentation. We believe that the presence of neutralizing antibodies still present in the immune serum continues to play a large and persistent role in diminishing viral infection. Though one component (the complement system) is eliminated with heat inactivation, the neutralizing antibodies are still present in the immune serum and can still have an effect on viral efficacy. C5 inhibition resulted in the most amplified VV infectivity, recovery, and oncolytic activity in both the nonvaccinated and vaccinated settings. This implies that complement inhibition can aid VV in evading the preformed antibody response as well as the innate immune response. C5 most likely plays a critical role because it serves as the final convergence point for all three complement pathways, and its breakdown products form the terminal membrane complex responsible for pathogen clearance. As expected, C5 mAb is more effective than C5a as noted in figure 2 because C5a serves as a byproduct of C5 breakdown and acts as an anaphylatoxin and chemoattractant for neutrophils and is not involved in formation of the terminal membrane attack complex. Therefore, C5a inhibition should not drastically affect viral neutralization.
We conclude that C3 mAb is effective in the presence of vaccinated human serum. However, in the presence of nonvaccinated human serum, C3 inhibition does not appear to be extremely effective. This experiment had been repeated several times with the same findings. A possibility for this difference is that 10% nonvaccinated human serum was used for these in vitro experiments while only 5% vaccinated human serum was used concurrently. Given that a higher concentration of nonvaccinated human serum was used, it may be that the quantity of C3 mAb added may have not been adequate to completely neutralize the higher levels of C3 present in the nonimmune serum. Though C3 mAb was more effective in the vaccinated setting, its addition to nonvaccinated serum did result in a significant increase in viral recovery compared to the control group. Since this experiment was repeated with the same findings, we can only surmise to why C3 mAb did not work more effectively in the nonvaccinated setting and will require further experimentation.
Protein inhibitors against central components of the complement system, particularly C5, were used to demonstrate their impact on viral efficacy for possible future in vivo experiments and potential use in human trials. All three available inhibitors--SSL7, compstatin, and CVF--enhanced viral marker gene expression and increased viral recovery, with SSL7 achieving the best results. These findings all illustrate complement's key role in VV viral clearance and purport inhibition of complement as a method to enhance viral infectivity in both the presence and absence of preformed neutralizing antibodies against vaccinia virus.
The mechanism of VV viral clearance by the complement system is not completely understood. A set of experiments was performed to define whether complement targets the virus directly, the initial infection phase, or further viral spread. These experiments showed that complement plays a role in inhibition of the initial cell infection of vaccinia virus, but does not appear to affect oncolytic VV spread. This can be explained due to the inherent complexity of VV viral structure and adaptations to immune system barriers. VV is purified and delivered to patients in the IMV form, which is susceptible to antibody and complement clearance. VV spreads in a cell-to-cell manner via the cell-associated enveloped virion (CEV) form of VV. Cell-to-cell spread can occur without the viral internal membrane ever being exposed to antibodies or complement.40 To aid virus dissemination within a host, the outer membrane of the EEV particle is obtained from the host cell and acquires host complement factors which protect EEV particles from destruction by complement for distant spread.40 Poxviruses will also produce complement control proteins after infection to inhibit the complement-mediated clearance. Though complement's mechanism of action in pathogen clearance is multi-faceted due to complement's three pathways of clearance, the alternative and spontaneous pathways result in direct destruction of microorganisms. Therefore, complement can directly clear the IMV form of VV, but once infection occurs, the virus is protected via production of its endogenous complement inhibitor, thereby leading to no change in viral spread when an exogenous complement inhibitor is added.
Emerging combination treatments using oncolytic virus therapy and immune therapy have been shown for years to be effective tumor-targeting regimens and safe.41 Recently, genetically-engineered VV expressing a specific chemokine to enhance tumor infiltration with cytotoxic T cells was shown to enhance VV infectivity and prolong viral persistence.42 However, a gentle balance between immune system stimulation and suppression must be sought in order to allow successful VV infection and proliferation without early clearance.41,42 In addition to enhancing viral infection, complement inhibition has also been shown to inhibit suppressor cells in the tumor microenvironment leading to an enhanced antitumor immune response. Complement inhibition may be another form of immunotherapy, which may enhance VV delivery while improving the actions of activated chemokines in the tumor microenvironment.
VV holds great potential for effective oncolytic therapy. However, overcoming immune system barriers may significantly enhance its antitumor effects prior to clearance by preformed antibodies, cytotoxic cells, and ultimately, the complement system.43-45 This study provides a clinically feasible means of inhibiting complement with known drugs in combination with VV therapy. Further pre-clinical animal studies need to be performed to validate these in vitro results.
Supplementary Material
Supplemental Figure 1. Determination of minimal concentrations of vaccinated and nonvaccinated serum required to inhibit viral expression. 1×104 MC38 cells were incubated in various concentrations of immune and nonimmune sera, ranging from 20% to 0.5%. The cells were then infected with vvDD-EGFP at an MOI of 1. Fluorescent microscope images were obtained following a 48 hour infection period.
{kind=link}
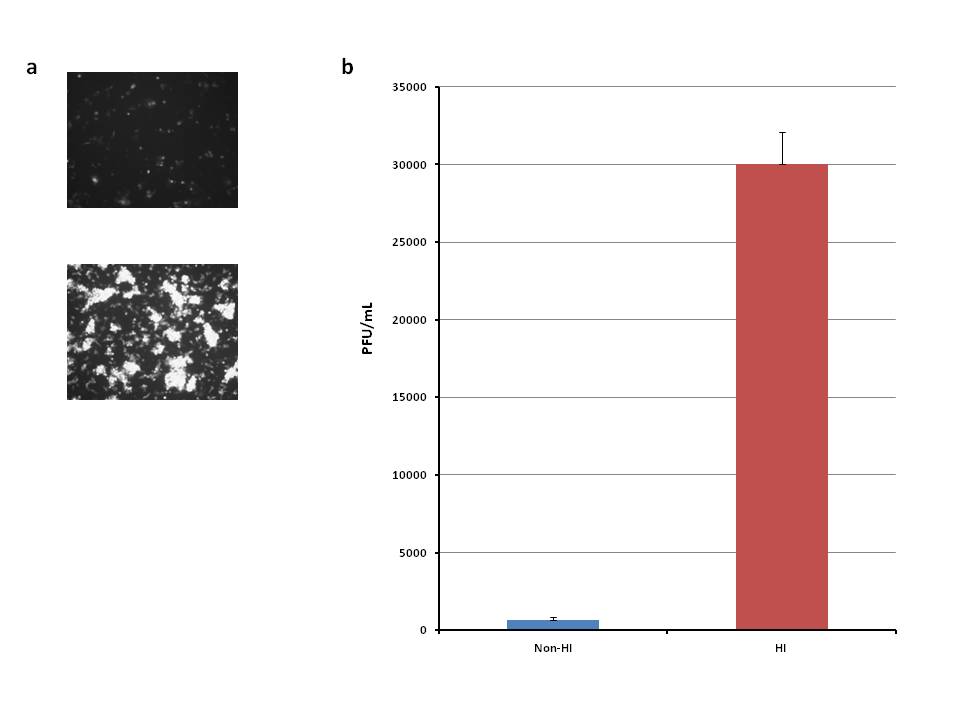
Supplemental Figure 2. The effect of complement inhibition via heat inactivation on vvDD expression and recovery using a more efficient model. (a) MC38s were plated in a 6-well plate at 1×106 cells/well and were then incubated in 10% nonvaccinated and 10% heat-inactivated nonvaccinated serum. The cells were then infected with vvDD-EGFP at an MOI of 1, and following a 48 hour infection period, fluorescent images were obtained. (b) A plaque assay was then performed. p<0.05 was considered statistically significant, determined via a t-test.
{kind=link}
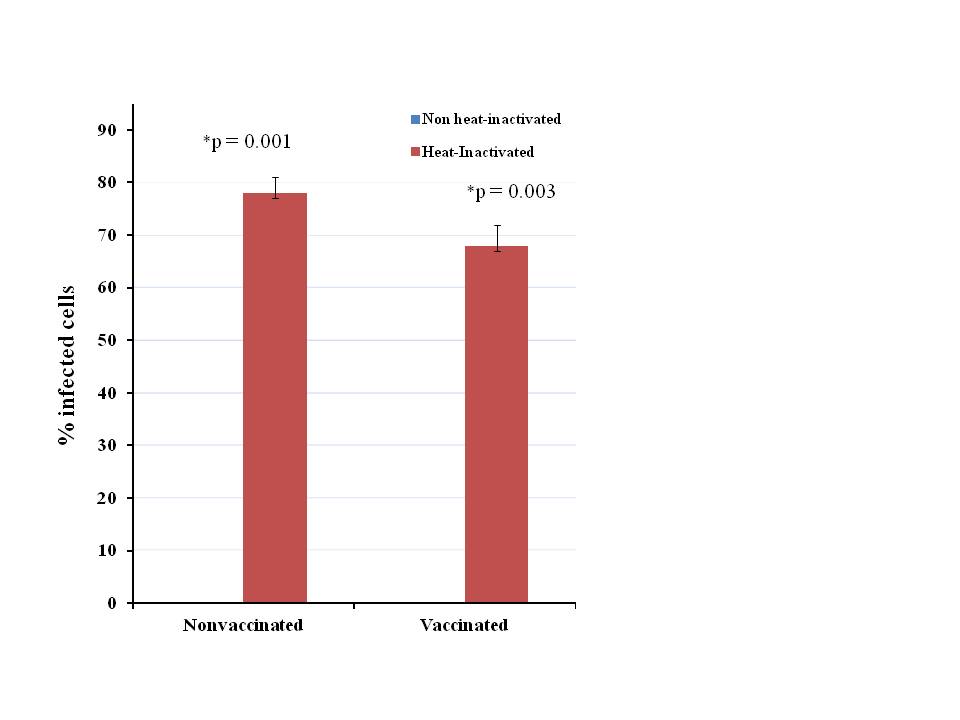
Supplemental Figure 3. Viability assay. Following incubation in either 10% nonvaccinated or 5% vaccinated serum or heat-inactivated versions of both, 1×104 MC38 cells were infected with vvDD at an MOI of 1. Following a 48 hour infection period, rather than harvesting cells in this repeated assay, samples in the 96 well plate were observed under the fluorescent microscope to determine an accurate measure of the number of infected cells, given as a percentage of total cells in the field. In the nonvaccinated serum setting, where the serum was heat-inactivated, we observed an increase in the percentage of infected cells from 0% to 78% while in the vaccinated serum setting, heat-inactivation resulted in an increase from 0% to 68% infected cells. The differences between control groups and heat-inactivated groups were statistically significant, p<0.05.
{kind=link}
Acknowledgments
This work is supported by NIH grant R01CA155925 (to DLB) and by David C. Koch Regional Therapy Cancer Center. DM is supported by training grant T32CA113263 from the National Cancer Institute. This project used University of Pittsburgh Cancer Institute (UPCI) shared resources that are supported in part by NIH grant award P30CA047904. Special thanks to Dr.John Fraser's group in Aukland, New Zealand for their collaboration and with providing SSL7 protein for our study.
Footnotes
Disclosure/Conflict of interests: DBL serves as a member of the Scientific Advisory Board, of Jennerex BioTherapeutics, a company focusing on development of oncolytic viruses.
References
- 1.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 2.Smith GL, Vanderplasschen A. Extracellular enveloped vaccinia virus. Entry, egress, and evasion. Adv Exp Med Biol. 1998;440:395–414. [PubMed] [Google Scholar]
- 3.Zeh HJ, Bartlett DL. Development of a replication-selective, oncolytic poxvirus for the treatment of human cancers. Cancer Gene Ther. 2002;9:1001–1012. doi: 10.1038/sj.cgt.7700549. [DOI] [PubMed] [Google Scholar]
- 4.Breitback CJ, Burk J, Jonker D, Stephenson J, Haas AR, Kirn DH, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 5.Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kirn DH, et al. Use of a targeted oncolytic virus, JX-594 in patients with refractory primary or metastatic liver cancer: A Phase I trial. Lancet Oncol. 2008;9:533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 6.McCart JA, Ward JM, Lee J, Hun Y, Alexander R, Libutti S, Bartlett DL, et al. Systemic Cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- 7.Stanford MM, Bell JC, Vaha-Koskela MJ. Novel oncolytic viruses: riding high on the next wave? Cytokine Growth Factor Rev. 2011;21:177–183. doi: 10.1016/j.cytogfr.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Bourke MG, Salwa S, Harrington KJ, Kucharczyk MJ, Forde PF, de Kruijf M, et al. The emerging role of viruses in the treatment of solid tumours. Cancer Treat Rev. 2011;37:618–632. doi: 10.1016/j.ctrv.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Moss B. Regulation of vaccinia virus transcription. Ann Rev Biochem. 2005;59:661–668. doi: 10.1146/annurev.bi.59.070190.003305. [DOI] [PubMed] [Google Scholar]
- 10.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tracey KJ. Reflex control of immunity. Nat Revs Immunol. 2009;9:418–28. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wakimoto H, Johnson PR, Knipe DM, Chiocca EA. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther. 2003;10:983–990. doi: 10.1038/sj.gt.3302038. [DOI] [PubMed] [Google Scholar]
- 13.Janeway CA, Travers P, Walport M, Capra JD, Shlomchik MJ. Garland Science. 5th. New York: 2001. Immunobiology: The immune system in health and disease; pp. 509–511. [Google Scholar]
- 14.Pier GB, Lyczak JB, Wetzler LM. Immunology, Infection and Immunity. Washington, DC: ASM Press; 2004. pp. 85–109. [Google Scholar]
- 15.Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol. 2004;5:971–974. doi: 10.1038/ni1004-971. [DOI] [PubMed] [Google Scholar]
- 16.Carrol MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–986. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 17.Lambris J, Ricklin D, Geisbrecht B. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodruff T, Nandakumar K, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol. 2011;48:1631–1642. doi: 10.1016/j.molimm.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Moulton EA, Atkinson JP, Buller RML. Surviving mousepox infection requires the complement system. PLOS Pathog. 2008;4:e1000249. doi: 10.1371/journal.ppat.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liszewski MK, Leung MK, Hauhart R, Fang CJ, Bertram P, Atkinson JP. Smallpox inhibitor of complement enzymes (SPICE): dissecting functional sites and abrogating activity. J Immunol. 2009;183:3150–3159. doi: 10.4049/jimmunol.0901366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernet J, Mullick J, Singh A, Sahu A. Viral mimicry of the complement system. J Biosci. 2003;28:249–264. doi: 10.1007/BF02970145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambris J, Ricklin D. Compstatin: A complement inhibitor on its way to clinical application. Adv Exp Med Biol. 2008;632:273–292. doi: 10.1007/978-0-387-78952-1_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jayasekera J, Moseman E, Caroll M. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J Virol. 2007;81:3487–3494. doi: 10.1128/JVI.02128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laursen N, Gordon N, Hermans S, Lorenz N, Jackson N, Anderson GR. Structural basis for inhibition of complement C5 by the SSL7 protein from Staphylococcus aureus. Proc Natl Acad Sci U S A. 2010;107:3681–3686. doi: 10.1073/pnas.0910565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma S, Jabeen T, Singh RK, Bredhorst R, Vogel CW, Singh TP. Structural studies on the cobra venom factor: isolation, purification, crystallization and preliminary crystallographic analysis. Acta Crystallogr D Biol Crystallogr. 2001;57:596–598. doi: 10.1107/s0907444901001342. [DOI] [PubMed] [Google Scholar]
- 26.Kirschfink M, Mollnes TE. Modern complement analysis. Clin Vaccine Immunol. 2003;10:982–989. doi: 10.1128/CDLI.10.6.982-989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alsenz J, Becherer JD, Nilsson B, Lambris JD. Structural and functional analysis of C3 using monoclonal antibodies. Curr Topics Microbiol Immunol. 1989;153:235–245. doi: 10.1007/978-3-642-74977-3_13. [DOI] [PubMed] [Google Scholar]
- 28.Whitehead AS, Sim RB, Bodmer WF. A monoclonal antibody against human complement component C3: the production of C3 by human cells in vitro. Eur J Immunol. 1981;11:140–146. doi: 10.1002/eji.1830110215. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci U S A. 1995;92:8955–8959. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas TC, Rollins SA, Rothers RP, Giannoni MA, Hartman SL, Elliott EA, Nye SH, Matis LA, Squinto SP, Evans MJ. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996;33:1389–1401. doi: 10.1016/s0161-5890(96)00078-8. [DOI] [PubMed] [Google Scholar]
- 31.Guo ZS, Parimi V, O'Malley ME, Thirunavukasaru P, Sathaiah M, Bartlett DL. The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene Ther. 2010;17:1465–1475. doi: 10.1038/gt.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srinivas TR, Meier-Kriesche HU. Minimizing immunosuppression, an alternative approach to reducing side effects: objectives and interim Result. Clin J Am Soc Nephrol. 2008;3:101–116. doi: 10.2215/CJN.03510807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Penn I. Chemical immunosuppression and cancer. Cancer. 1974;34:1474–1480. doi: 10.1002/1097-0142(197410)34:8+<1474::aid-cncr2820340820>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 34.Newstead CG. Assessment of risk of cancer after renal transplantation. Lancet. 1998;351:610–611. doi: 10.1016/s0140-6736(98)22009-5. [DOI] [PubMed] [Google Scholar]
- 35.Lun XQ, Jang JH, Tang N, Deng H, Head R, McCart A. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res. 2009;15:2777–2788. doi: 10.1158/1078-0432.CCR-08-2342. [DOI] [PubMed] [Google Scholar]
- 36.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Power A, Wang J, Falls T, Paterson J, Parato KA, Bell J, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15:123–130. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 38.Ikeda K, Wakimoto H, Ichikawa T, Jhung S, Hochberg F, Chiocca E, et al. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J Virol. 2000;74:4765–4775. doi: 10.1128/jvi.74.10.4765-4775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Diesboeck TS, Chiocca EA, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 40.Law M, Hollinshead R, Smith GL. Antibody-sensitive and antibody-resistant cell-to-cell spread by vaccinia virus: role of the A33R protein in antibody-resistant spread. J Gen Virol. 2002;83:209–222. doi: 10.1099/0022-1317-83-1-209. [DOI] [PubMed] [Google Scholar]
- 41.Bernards R, Destree A, McKensie S, Gordon E, Weinberg RA, Panicali D. Effective tumor immunotherapy directed against an oncogene-encoded product using a vaccinia virus vector. Proc Natl Acad Sci U S A. 1987;84:6854–6858. doi: 10.1073/pnas.84.19.6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, O'Malley ME, Urban J, Sampath P, Guo ZS, Kalinski P, et al. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther. 2011;19:650–657. doi: 10.1038/mt.2010.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirn DH, Wang Y, Liang W, Contag C, Thorne SH. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 2008;68:2071–2075. doi: 10.1158/0008-5472.CAN-07-6515. [DOI] [PubMed] [Google Scholar]
- 44.Isaacs S, Kotwal G, Moss B. Vaccinia virus complement-control protein prevents antibody-dependent complement-enhanced neutralization of infectivity and contributes to virulence. Immunol. 1992;89:628–632. doi: 10.1073/pnas.89.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Purushottam J, Kotwal G. Vaccinia complement control protein: Multi-functional protein and a potential wonder drug. J Biosci. 2003;28:265–271. doi: 10.1007/BF02970146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Determination of minimal concentrations of vaccinated and nonvaccinated serum required to inhibit viral expression. 1×104 MC38 cells were incubated in various concentrations of immune and nonimmune sera, ranging from 20% to 0.5%. The cells were then infected with vvDD-EGFP at an MOI of 1. Fluorescent microscope images were obtained following a 48 hour infection period.
Supplemental Figure 2. The effect of complement inhibition via heat inactivation on vvDD expression and recovery using a more efficient model. (a) MC38s were plated in a 6-well plate at 1×106 cells/well and were then incubated in 10% nonvaccinated and 10% heat-inactivated nonvaccinated serum. The cells were then infected with vvDD-EGFP at an MOI of 1, and following a 48 hour infection period, fluorescent images were obtained. (b) A plaque assay was then performed. p<0.05 was considered statistically significant, determined via a t-test.
Supplemental Figure 3. Viability assay. Following incubation in either 10% nonvaccinated or 5% vaccinated serum or heat-inactivated versions of both, 1×104 MC38 cells were infected with vvDD at an MOI of 1. Following a 48 hour infection period, rather than harvesting cells in this repeated assay, samples in the 96 well plate were observed under the fluorescent microscope to determine an accurate measure of the number of infected cells, given as a percentage of total cells in the field. In the nonvaccinated serum setting, where the serum was heat-inactivated, we observed an increase in the percentage of infected cells from 0% to 78% while in the vaccinated serum setting, heat-inactivation resulted in an increase from 0% to 68% infected cells. The differences between control groups and heat-inactivated groups were statistically significant, p<0.05.