

Abstract
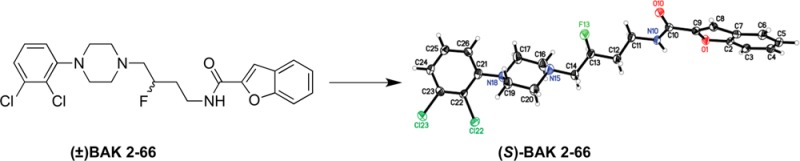
The improved chiral synthesis of the selective dopamine D3 receptor (D3R) antagonist (R)-N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)1H-indole-2-carboxamide ((R)-PG648) is described. The same chiral secondary alcohol intermediate was used to prepare the enantiomers of a 3-F-benzofuranyl analogue, BAK 2-66. The absolute configurations of the 3-F enantiomers were assigned from their X-ray crystal structures that confirmed retention of configuration during fluorination with N,N-diethylaminosulfur trifluoride (DAST). (R)-BAK2-66 showed higher D3R affinity and selectivity than its (S)-enantiomer; however, it had lower D3R affinity and enantioselectivity than (R)-PG648. Further, importance of the 4-atom linker length between the aryl amide and 4-phenylpiperazine was demonstrated with the 4-fluorobutyl-product (8).
Keywords: DAST, asymmetric catalysis, enantioselectivity, dopamine
The neurotransmitter dopamine is synthesized in dopaminergic neurons and released to stimulate G protein-coupled receptors, affecting movement, cognition, and emotion. The five known dopamine receptors (DRs) are divided into two subfamilies, D1-like receptors (D1R and D5R) and D2-like receptors (D2R, D3R, and D4R), on the basis of signaling properties and sequence similarity.1 Dysfunction within the dopamine system can lead to a variety of pathological conditions, including Parkinson’s disease, schizophrenia, ADHD, and drug addiction.1,2
Addictive drugs, such as the psychostimulants cocaine and methamphetamine, enhance dopamine release in mesolimbic pathways. The D3R subtype is expressed primarily in the ventral striatum, particularly in the nucleus accumbens,3 regions in which high levels of dopamine neurotransmission are induced by drug exposure.4 D3R expression is known to be elevated in laboratory animals and humans following exposure to cocaine and methamphetamine.5−7 A significant effort to target the D3R for therapeutics to treat drug abuse and other neuropsychiatric disorders has identified templates for the design of many high-affinity and selective D3R antagonists and partial agonists, which have been studied in numerous animal models of addiction.5,8−11
Extensive investigation of the 4-phenylpiperazine class of D3R antagonists has revealed that a terminal heteroaryl amide attached by a functionalized 4-carbon linking chain can result in highly selective D3R antagonists that have appropriate physicochemical properties to be used as in vivo tools.8−11 The first enantioselective D3R antagonist, (R)-PG648, was discovered to be ∼400-fold more selective for D3R over D2R, with 15-fold higher affinity at D3Rs than its (S) enantiomer; in contrast there was <2-fold enantioselectivity at D2R.12 The recently reported D3R crystal structure provided the coordinates for molecular dynamics studies that suggested an interaction between a tyrosine residue in the seventh transmembrane domain (Y7.43) that may have polar interactions with the 3-OH group on the linking chain of (R)-PG648.13 Note: (R)-PG648 was referred to as R-22 previously.12,13 Using these models and a deconstruction strategy, the molecular determinants of both D3R-selectivity and efficacy were derived that underscored the roles of the primary pharmacophore (2,3-dichloropiperazine) and the secondary pharmacophore (exemplified in (R)-PG648 by the indolylamide function), which binds uniquely to a second binding pocket in D3R.14 Moreover, the length and composition of the linker between these pharmacophoric moieties has also been determined to play a critical role in the affinity and selectivity of these ligands.15 To further explore the linking chain composition and determine if the 3-OH group could be substituted with other functional groups, a highly selective D3R antagonist (BAK2-66) was discovered.16 Note: BAK2-66 was previously referred to as compound 8d.16
Because (R)- and (S)-PG648 showed enantioselectivity at D3R, it was of interest to resolve the enantiomers of BAK2-66 and evaluate these in vitro. Furthermore, in order to complete behavioral studies in rat and nonhuman primate models of addiction, multigram quantities of enantiopure (R)- and (S)-PG648 had to be prepared. Hence in the present study we (1) improved the enantiopure synthesis of (R)- and (S)-PG648 for behavioral studies, (2) used this chiral resolution to synthesize (R)- and (S)-BAK2-66, by converting a 3-OH intermediate to a 3-F analogue using N,N-diethylaminosulfur trifluoride (DAST), wherein retention of stereochemistry resulted, and (3) evaluated the enantiomeric pairs for binding affinities (Ki values) using [3H]N-methylspiperone radioligand binding competition in membranes prepared from HEK293 cells expressing human dopamine D2R, D3R, and D4R (hD2R, hD3R, and hD4R, respectively).
Scheme 1 outlines the synthetic strategy used for the chiral intermediate (R)-4, used for both (R)-BAK2-66 and (R)-PG648. As previously described, compound (R)-3 was synthesized by asymmetric hydrolysis of the terminal epoxide 2 using Jacobsen’s catalyst.12,17 This hydrolytic kinetic resolution provided the terminal epoxide with ≥99% ee. (S)-2-(Bromoethyl) oxirane ((S)-1) is commercially available, and thus, chiral resolution to obtain (S)-3 was not required. The rest of the synthetic strategy to obtain (S)-BAK2-66 was identical and is not depicted. The purity and ee of both (R)- and (S)-enantiomers of compounds 3, 5, and BAK2-66 were determined using chiral HPLC analysis (experimental details and HPLC spectra in Supporting Information).
Scheme 1. Synthesis of (R)-BAK2-66.

Reagents and conditions: (a) potassium phthalimide, DMF, RT, 12 h; (b) (Salen)Co(III)(OAc) complex, H2O, THF, 72 h; (c) 1-(2,3-dichlorophenyl)piperazine, isopropanol, reflux, 12 h; (d) DAST, CH2Cl2, −78 °C to RT, 12 h; (e) hydrazine, EtOH, reflux, 3 h; (f) benzofuran-2-carboxylic acid, SOCl2, 3 h.
Interestingly, when the alcohol intermediate 4 was converted to its fluoro analogue 5 using DAST, no change in the configuration at C-3 was observed, where inversion of stereochemistry was expected.18 Typically DAST reactions undergo an SN2 nucleophilic displacement of the OH group with F, leading to an inversion of stereochemistry; however, inversion was not observed. This was confirmed via extensive HPLC and X-ray analysis of (S)-4, (S)-5, and (S)-BAK2-66 that established the absolute configuration (S) at C-14 as shown in Figure 1. (Note: C-14 as depicted in the X-ray structures corresponds to C3, based on IUPAC nomenclature.) To our knowledge, there has only been one report of stereochemistry retention in a DAST-mediated fluorination reaction, with a double inversion mechanism suggested.19
Figure 1.
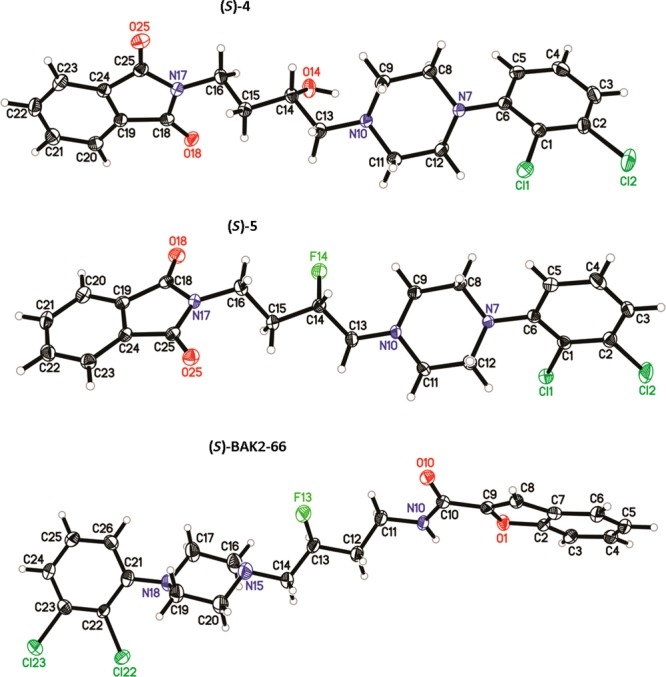
X-ray crystal structures of (S)-4, (S)-5, and (S)-BAK2-66.
In Scheme 2, a side reaction of our synthetic strategy is described. Fluorination of the alcohol intermediate (4) gave the expected intermediate 5a and an additional intermediate 5b in a ratio of ∼2.5:1, which were separated by column chromatography. Deprotection of 5b using hydrazine gave the free amine 7, which was reacted with benzofuran-2-carboxylic acid to afford compound 8, a structural isomer of BAK2-66.
Scheme 2. Synthesis of Compound 8.

Reagents and conditions: (a) DAST, CH2Cl2, −78 °C to RT, 12 h; (b) hydrazine, EtOH, reflux, 3 h; (c) benzofuran-2-carboxylic acid, SOCl2, 3 h.
A proposed mechanism is shown in Figure 2 for the retention of configuration in Scheme 1 and the formation of the additional minor intermediate in Scheme 2, during the fluorination reactions. We propose that an aziridinium ion intermediate is formed via neighboring group participation to give the resulting products.20 The liberated F– can attack the aziridinium ion either at C-1 or C-2 to produce (S)-5 or (S)-5b (via (±)-4), respectively. To our knowledge this is the first time this mechanism has been suggested for a fluorination reaction using DAST and reflects the unique juxtaposition of the piperazine nitrogen to the 3-position −OH group to enable ultimate retention of configuration.
Figure 2.

Proposed mechanism for fluorination reactions. Step 1: The 3-OH is converted to the leaving group −O–SF2–NEt2 with DAST. Step 2: The piperazine N attacks from the backside via neighboring group participation (SN2). Step 3: F– attacks the vulnerable aziridinium ion intermediate at C-1 by SN2 nucleophilic displacement resulting in double inversion with overall retention of stereochemistry. Step 4: In the case of (±)4, nucleophilic attack by F– occurs at the C-2 giving intermediate 5b.
The synthesis of (R)-PG648, presented in Scheme 3, has previously been reported on a small scale.12 However, modification of the original method resulted in the synthesis of multigram quantities of this compound as the pure single enantiomer in ≥99% ee in comparison to the earlier reported method (>90% ee).12(S)-PG648 was also synthesized from the commercially available starting material (S)-2-(bromoethyl) oxirane to give (S)-4 (not shown) using the same procedure as described for the (R)-enantiomer (experimental details in Supporting Information). We have previously reported the X-ray analysis of (S)-PG648.12
Scheme 3. Synthesis of (R)-PG648.
Reagents and conditions: (a) hydazine, EtOH, reflux, 3 h; (b) indole-2-carboxylic acid, CDI, 3 h.
Evaluation of the analogues described herein for binding at hD2R, hD3R, and hD4R revealed enantioselectivity similar to previously results for (R)- and (S)-PG648, although the absolute Ki values are not identical, due to different binding assay conditions and radioligands used (Table 1 and Supporting Information).12(±)-BAK2-66 showed a similar affinity for D3R (10 vs 5.4 nM) as previously reported, but a significantly higher affinity for D2R than previously reported (960 vs 9160 nM), resulting in a 96-fold selectivity for D3R over D2R, in contrast to our previously published selectivity ratio of >1000.16(R)-BAK2-66 (Ki = 6.9 nM) was similarly D3R-selective (∼115-fold) compared to its racemate (∼86-fold). This contrasts to the more dramatic D3R enantioselectivity observed with the (R)- and (S)-enantiomers of PG648 (Table 1). These data suggest that the polar interaction between the Y7.43 residue of the D3R and the 3-OH group in (R)-PG648 is more prominent than with the 3-F group of (R)-BAK2-66, although enantioselectivity was retained. It must also be noted that PG648 is an indolylamide, whereas BAK2-66 is a benzofuranylamide. Therefore, the structure of this secondary pharmacophore likely affects the binding pocket interactions of these molecules.15 Of note, (R)-PG648 has ∼13-fold higher affinity for D3R than (R)-BAK2-66; in contrast, (R)-BAK2-66 has only 3-fold lower affinity at D2R than (R)-PG648.
Table 1. Human D2-Like Family Receptor Subtype Binding Results.
| compd | Ki D3R (nM)a | Ki D2R (nM)a | D2/D3 | Ki D4R (nM)a |
|---|---|---|---|---|
| (±)-PG648 | 1.9 ± 0.11 | 750 ± 120 | 395 | 2600 ± 660 |
| (R)-PG648 | 0.53 ± 0.091 | 300 ± 65 | 566 | 4000 ± 880 |
| (S)-PG648 | 3.9 ± 0.36 | 790 ± 160 | 233 | 1900 ± 490 |
| (±)-BAK2-66 | 10 ± 2.0 | 960 ± 270 | 96 | NT |
| (R)-BAK2-66 | 6.9 ± 1.5 | 800 ± 100 | 116 | >9000 |
| (S)-BAK2-66 | 29 ± 8.6 | 2500 ± 790 | 86 | >14000 |
| 8 | 560 ± 80 | 7400 ± 1400 | 13 | >3000 |
Ki values determined by competitive inhibition of [3H]N-methylpsiperone binding in membranes harvested from HEK 293 cells stably expressing hD2R, hD3R, or hD4R. NT, not tested. Further details in the Supporting Information.
As noted above, a minor product (5b) with a shorter tether was obtained in the fluorination reaction (Scheme 2). Decreasing the length of the linking chain between the primary pharmacophore (4-phenylpiperazine) and the secondary pharmacophore (benzofuranylamide), from the optimum 4-carbon linker to a 3-carbon linker, and increasing the steric bulk on the linker with the fluoromethylene group significantly decreased the binding affinities of compound 8 for both D3R and D2R. This was not unexpected, as previous structure–activity relationships (SAR) have exemplified the importance of the 4-carbon linker length and the lack of tolerance for functional groups that are larger than OH.12,21,22 Recently, molecular dynamic studies demonstrated that a 3-carbon linker prevents optimal placement of the secondary pharmacophore in the ptm 23 subpocket of both D3R and D2R, which is likely responsible for the lower binding affinity.15 In the D3R, the shorter linker results in the secondary pharmacophore being positioned away from the critical Gly94 in the EL1 loop.
In summary, we report herein an improved chiral synthesis of (R)- and (S)-PG648, for in vivo studies, and the chiral resolution of its analogue BAK2-66. In the DAST-mediated fluorination reaction, retention of configuration was confirmed by both HPLC and X-ray analysis. In addition, we report the product of a side reaction during fluorination that yielded a structural isomer of BAK2-66. Binding experiments at hD2R, hD3R, and hD4R confirmed enantioselectivity at D3R for (R)- and (S)-PG648 as well as for the enantiomers of BAK2-66. However, less significant enantioselectivity was noted between the (R)- and (S)-BAK2-66 pair, likely due to less pronounced polar interactions with a Y7.43 residue previously reported to interact with the 3-OH group of (R)-PG648.13−15 On the basis of the previously reported off-target binding profiles for both PG648(12) and BAK2-66(16) and the lower enantioselectivity for the (R)- and (S)-enantiomers of BAK2-66, only PG648 and its enantiomers have been selected for further evaluation in vivo and will be reported in due course. The present findings serve to extend SAR in the 4-phenylpiperazine class of D3R ligands, especially highlighting the critical role of the length and functionalization of the linker between the primary and secondary pharmacophores in producing high D3R affinity and favorable D2R-like subtype selectivity of these compounds. We also highlight serendipitous reactions with DAST, likely via neighboring group participation to form an aziridinium intermediate with the piperazine N, that led to the final products described.
Acknowledgments
HEK cells expressing hD2R, hD3R, or hD4R were provided by Dr. David Sibley of NINDS. The authors thank Caitlin Burzynski and Catherine Schweppe for assistance in radioligand binding, Dr. Joshua Antoline for his insight on the fluorination mechanisms, and Dr. Rachel Slack for her careful reading of this manuscript and important suggestions for improvement.
Glossary
ABBREVIATIONS
- SAR
structure–activity relationship
- CDI
1,1′-carbonyldiimidazole
- DAST
N,N-diethylaminosulfur trifluoride
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- D4R
dopamine D4 receptor
- ee
enantiomeric excess
- RT
room temperature
- PTSA
para-toluenesulfonic acid
- EL
extracellular loop
- ptm 23
pocket between the second and third transmembrane segments
Supporting Information Available
Experimental details for the synthesis and purification and the in vitro pharmacological characterizations of the compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was funded by the NIDA Intramural Research Program, National Institutes of Health, Department of Health and Human Services. V.K. was supported by an NIH Visiting Fellowship, and T.M.K. was supported by an NIH Postdoctoral Intramural Research Training Award (IRTA) Fellowship. The X-ray crystallographic studies were supported by NIDA through Interagency Agreement #Y1-DA1101 with the Naval Research Laboratory (NRL).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Missale C.; Nash S. R.; Robinson S. W.; Jaber M.; Caron M. G. Dopamine receptors: from structure to function. Physiol. Rev. 1998, 78, 189–225. [DOI] [PubMed] [Google Scholar]
- Iversen S. D.; Iversen L. L. Dopamine: 50 years in perspective. Trends Neurosci. 2007, 30, 188–93. [DOI] [PubMed] [Google Scholar]
- Murray A. M.; Ryoo H. L.; Gurevich E.; Joyce J. N. Localization of dopamine D3 receptors to mesolimbic and D2 receptors to mesostriatal regions of human forebrain. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 11271–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G.; Bassareo V.; Fenu S.; De Luca M. A.; Spina L.; Cadoni C.; Acquas E.; Carboni E.; Valentini V.; Lecca D. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology 2004, 47Suppl 1227–41. [DOI] [PubMed] [Google Scholar]
- Heidbreder C. A.; Newman A. H. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N.Y. Acad. Sci. 2010, 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley J. K.; Mash D. C. Adaptive increase in D3 dopamine receptors in the brain reward circuits of human cocaine fatalities. J. Neurosci. 1996, 16, 6100–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau I.; Payer D.; Houle S.; Behzadi A.; Rusjan P. M.; Tong J.; Wilkins D.; Selby P.; George T. P.; Zack Z.; Furukawa Y.; McCluskey T.; Wilson A. A.; Kish S. J. Higher binding of the dopamine D3 receptor-preferring ligand [11C]-(+)-propyl-hexahydro-naphtho-oxazin in methamphetamine polydrug users: a positron emission tomography study. J. Neurosci. 2012, 32, 1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T. M.; Burzynski C.; Shi L.; Newman A. H. Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv. Pharmacol. 2014, 69, 267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A. H.; Blaylock B. L.; Nader M. A.; Bergman J.; Sibley D. R.; Skolnick P. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem. Pharmacol. 2012, 84882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F. Recent advances in the development of dopamine D3 receptor antagonists: a medicinal chemistry perspective. ChemMedChem 2011, 6, 1152–1162. [DOI] [PubMed] [Google Scholar]
- Micheli F.; Heidbreder C. Dopamine D3 receptor antagonists: a patent review (2007–2012). Expert Opin. Ther. Pat. 2013, 23, 363–381. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Grundt P.; Cyriac G.; Deschamps J. R.; Taylor M.; Kumar R.; Ho D.; Luedtke R. R. N-(4-(4-(2,3-Dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem. 2009, 52, 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E. Y. T.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. Structure of the human dopamine d3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A. H.; Beuming T.; Banala A. K.; Donthamsetti P.; Pongetti K.; LaBounty A.; Levy B.; Cao J.; Michino M.; Luedtke R. R.; Javitch J. A.; Shi L. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012, 55, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M.; Donthamsetti P.; Beuming T.; Banala A.; Duan L.; Roux T.; Han Y.; Trinquet E.; Newman A. H.; Javitch J. A.; Shi L. A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol. Pharmacol. 2013, 84, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banala A. K.; Levy B. A.; Khatri S. S.; Furman C. A.; Roof R. A.; Mishra Y.; Griffin S. A.; Sibley D. R.; Luedtke R. R.; Newman A. H. N-(3-Fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J. Med. Chem. 2011, 54, 3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M.; Larrow J. F.; Kakiuchi F.; Jacobsen E. N. Asymmetric catalysis with water: efficient kinetic resolution of terminal epoxides by means of catalytic hydrolysis. Science 1997, 277, 936–938. [DOI] [PubMed] [Google Scholar]
- Middleton W. J. New fluorinating reagents. dialkylaminosulfur fluorides. J. Org. Chem. 1975, 40, 574–578. [Google Scholar]
- Jeong L. S.; Nicklaus M. C.; George C.; Marquezt V. E. Unanticipated retention of configuration in the DAST fluorination of deoxy-4′-thiopyrimidine nucleosides with “up” hydroxyl groups. Tetrahedron Lett. 1994, 35, 7569–7572. [Google Scholar]
- Métro T. X.; Duthion B.; Pardo D. G.; Cossy J. Rearrangement of β-amino alcohols via aziridiniums: a review. Chem. Soc. Rev. 2010, 39, 89–102. [DOI] [PubMed] [Google Scholar]
- Robarge M. J.; Husbands S. M.; Kieltyka A.; Brodbeck R.; Thurkauf A.; Newman A. H. Design and synthesis of [(2,3-dichlorophenyl)piperazin-1-yl]alkylfluorenylcarboxamides as novel ligands selective for the dopamine D3 receptor subtype. J. Med. Chem. 2001, 44, 3175–3186. [DOI] [PubMed] [Google Scholar]
- Ehrlich K.; Götz A.; Bollinger S.; Tschammer N.; Bettinetti L.; Härterich S.; Hübner H.; Lanig H.; Gmeiner P. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J. Med. Chem. 2009, 52, 4923–4952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.