

Abstract

As part of our program to study neurotransmitter releasers, we report herein a class of hybrid dopamine reuptake inhibitors that display serotonin releasing activity. Hybrid compounds are interesting since they increase the design potential of transporter related compounds and hence represent a novel and unexplored strategy for therapeutic drug discovery. A series of N-alkylpropiophenones was synthesized and assessed for uptake inhibition and release activity using rat brain synaptosomes. Substitution on the aromatic ring yielded compounds that maintained hybrid activity, with the two disubstituted analogues (PAL-787 and PAL-820) having the most potent hybrid activity.
Keywords: Dopamine uptake inhibitors, serotonin releasers, serotonin receptor, cocaine, methamphetamine
Plasma membrane biogenic amine transporters (BATs) terminate aminergic signaling by transporting previously released neurotransmitters, dopamine, norepinephrine, and serotonin (DA, NE, and 5-HT transported via DAT, NET, and SERT, respectively), from the synaptic cleft back into the neuronal cytoplasm. One of the most successful strategies for drug discovery over the past 30 years has been the development of BAT ligands, which block the reuptake of amine neurotransmitters, thereby elevating synaptic concentrations of these neurotransmitters. At the molecular level, BAT ligands block reuptake by either inhibiting endogenous transmitter binding or preventing the transporter protein from undergoing obligatory conformational changes. From a therapeutic perspective, these compounds are known as reuptake inhibitors, an example of which is the DA/NE reuptake inhibitor bupropion 1 (Figure 1).1 Alternatively, other BAT ligands can act as transporter substrates that induce neurotransmitter release. These compounds are called neurotransmitter releasers, such as the DA/NE releaser methcathinone 2a.2 Reuptake inhibition and release are the primary modes of action for most BAT ligands.
Figure 1.

Propiophenone compounds.
Both types of BAT compounds can elevate synaptic concentrations of neurotransmitters, and this enhancement of neurotransmission can have profound in vivo effects. For instance, the stimulatory in vivo effects of DAT ligands are well-known as demonstrated by the ability of DA reuptake blockers and releasers to induce locomotor activity via elevating synaptic DA.2−4 Despite their shared acute physiological effects, mechanistic differences between reuptake inhibitors and releasers lead to dissimilar kinetics, downstream effects, and feedback control.5 In particular, a reuptake inhibitor depends on ongoing exocytotic release of neurotransmitter to achieve its effects, while a releaser causes rapid efflux of a cytoplasmic pool of neurotransmitter that is independent from exocytosis. These differences in mechanism can lead to opposing changes in biological regulation. As a specific example, the administration of the DA uptake inhibitor cocaine upregulates striatal DATs,6 as does bupropion,7 while administration of the DA releaser methamphetamine downregulates striatal DATs.8 Therefore, while uptake inhibitors and releasers can both elevate synaptic concentrations of neurotransmitter, the mechanisms and pharmacological consequences of their use are different.5
Transporter ligand discovery and development efforts have traditionally focused on the development of a drug within a given mechanistic class of BAT ligand, either a compound that inhibits reuptake or one that induces neurotransmitter release. Most transporter ligands recruit one mechanism or the other. This pattern suggests that BAT ligands possess general structural features that cause them to act as either releasers or uptake inhibitors, but not both. However, given the relatively recent confirmation that a compound must be a substrate in order to induce transporter-mediated release,9 the possibility exists that a BAT ligand might be a substrate of one transporter but not of another and instead act as a reuptake inhibitor. In turn, this implies that it might be possible to find compounds with mixed (i.e., hybrid) transporter activity, whereby certain ligands induce release at one transporter but block reuptake at another.
Hybrid compounds are intriguing for several reasons. First, the kinetics of ligand–transporter interaction may be different for releasers versus reuptake blockers. A releaser mimics an endogenous neurotransmitter and acts quickly at the target transporter, possibly because it interacts at a well-defined domain suited for neurotransmitter translocation. However, an uptake inhibitor can have much slower on- and off-rates of interaction with transporter proteins. Second, the profile of synaptic neurotransmitter change should differ according to molecular mechanism since an uptake inhibitor relies on ongoing exocytotic release to achieve its effects, while a releaser overrides the system and triggers neurotransmitter efflux regardless of the state of the system. Third, the effects of chronic use may also be different since neuroadaptive changes are related to acute drug mechanism, and in some cases may have the opposite effects, an example of which is the long-term effect of BAT ligands on DATs described above. Thus, the in vivo properties of a dual DA-5-HT uptake inhibitor may be different than a dual DA-5-HT releaser, which in turn may be different than a DA uptake inhibitor-5-HT releaser or a DA releaser-5-HT uptake inhibitor. These hybrid possibilities increase the drug design potential for transport related compounds and represent a novel strategy for therapeutic discovery.
Indeed, a compound with hybrid transporter activity has already been identified in the Rothman laboratory: 2-(N-ethylamino)propiophenone (2b), the bioactive metabolite of the clinically available appetite suppressant diethylpropion (2c).10 This analogue of both 1 and 2a was found to be a DA reuptake inhibitor with a Ki value of 1014 nM and a 5-HT releaser with an EC50 value of 2118 nM. To our knowledge this was the first compound identified to have hybrid activity, albeit weak and structural analogues did not have the same hybrid activity. As part of our program to study neurotransmitter releasers, other hybrid compounds have been identified from our screening data. For example, we recently reported that the (−)-isomer of the abused street drug 3,4-methylenedioxy-N-ethylamphetamine also has hybrid transporter activity.11
Here we describe the first example of a family of related compounds with hybrid uptake inhibitor-releaser activity: substituted 2-(N-cyclopropylamino)propiophenones (3a–h). These BAT ligands are DA reuptake inhibitors with 5-HT releasing activity. The lead structure, 2-(N-cyclopropylamino)-3′-chloropropiophenone (3a) was originally synthesized as part of a set of bupropion related compounds and was found to be a DA reuptake inhibitor with an IC50 of 265 nM and a 5-HT uptake inhibitor with an IC50 of 3180 nM.1 This compound elicited unique in vivo behavioral activity related to drug abuse, specifically a slower onset and longer duration of action in generalization to cocaine. Such a profile is well suited for a DA agonist pharmacotherapy for treating addiction to cocaine, methamphetamine, nicotine, and other drugs of abuse. Compound 3a was originally thought to be a DA/5-HT uptake inhibitor, but a more detailed analysis of the transporter activity uncovered the 5-HT releasing activity. This suggests 3a was originally mischaracterized as a SERT blocker but is in reality a SERT substrate that induces 5-HT release. All of the other compounds except for the cyclobutyl compound (data not shown) in the original bupropion series were found to be uptake blockers as originally described. A series of additional analogues was then synthesized and tested for transporter activity to validate the activity across the structural class, all of which were found to have the same unique transporter profile.
The synthesis of 3a–3h is shown in Scheme 1. Commercially available propiophenones 4a–h were brominated using bromine in methanol or dichloromethane followed by amination with cyclopropylamine to form compounds 3a–h with good yields in both steps. The N-propyl and N-ethyl bupropion analogues (6 and 7, respectively) were synthesized as previously described.1 Compounds 8, 9, 10, and 12 were synthesized as described in the Supporting Information. Compound 11 was synthesized as described in the literature.12 The optical isomers of 3a were synthesized using a modified procedure used to synthesize the optical isomers of bupropion, but using cyclopropylamine instead of tert-butylamine.13,14
Scheme 1. Synthesis of Hybrid Analogues.

(a) Br2, MeOH; (b) cyclopropylamine.
The transporter activity of all compounds was assessed using the previously described protocol for identifying releasers and uptake inhibitors, using synaptosomes generated from rat brain homogenate.15,16 Compounds were binned as releasing substrates or uptake inhibitors by assessment in both synaptosomal release and uptake inhibition assays. Once the functional activity of a compound was determined, dose–response curves were completed for the binned activity.15
The activity of these compounds is shown in Table 1. Contrary to the original report by Carroll et al.,1 in our SERT rat synaptosome assays, the lead compound 3a was found to be a 5-HT releaser with an EC50 of 1328 nM. Potencies and mechanism of activity at the other two transporters were roughly the same compared to the original report. The optical isomers of 3a were examined in order to assess enantiospecific effects, but the potencies were found to be the same. It is uncertain if this observation is a real effect of the two enantiomers, or due to epimerization to the racemic mixture under the assay conditions. Bupropion has been reported to epimerize rapidly, but more detailed analysis suggests it does so slowly under physiological conditions.17 Stirring the enantiomers in water or alcohol failed to induce much epimerization (by optical rotation) but could occur under the assay conditions. Other compounds from the original set of bupropion analogues from Carroll et al. were tested, two of which are included in Table 1. The N-propyl analogue (6) was found to be a selective DA uptake inhibitor with an IC50 of 793 nM. The N-ethyl analogue (7) was found to have much weaker activity than 3a and did not show hybrid activity as did the originally reported hybrid compound (unsubstituted analogue 2b).
Table 1. Comparison of Dopamine, Serotonin, and Norepinephrine Transporter Uptake Inhibition and Release Data for a Series of N-Alkylpropiophenones Using Rat Synaptosomesa.
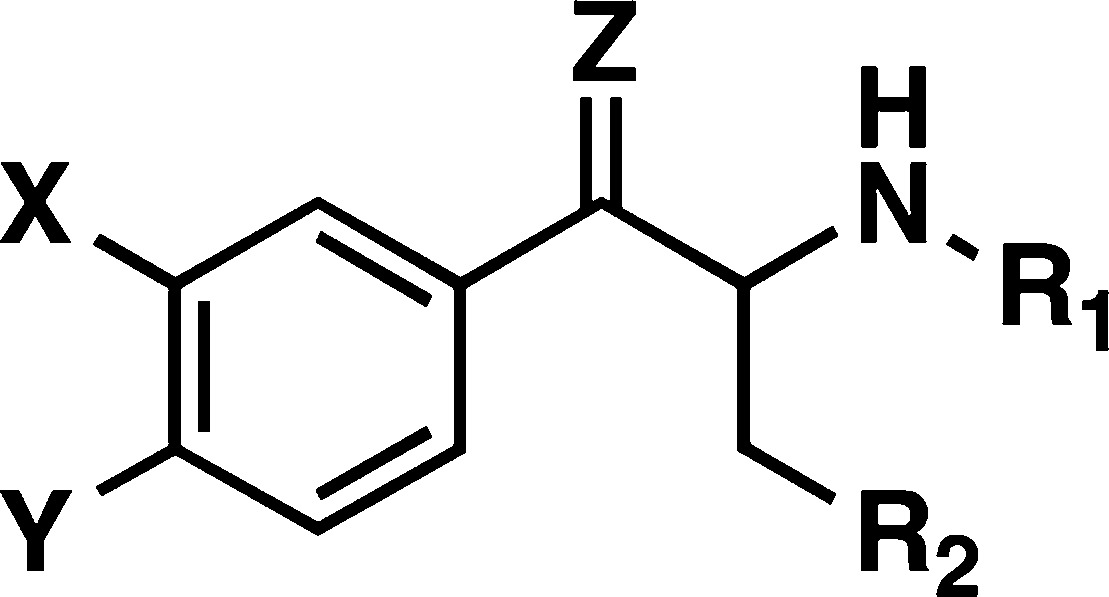
| DATb |
SERTb |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PAL # | compd | Z | R1 | R2 | X | Y | inhibition IC50 (nM) | release EC50 (nM) | inhibition IC50 (nM) | release EC50 (nM) |
| 433 | 3a | O | CH(CH2CH2) | H | Cl | H | 533 ± 61 | 1328 ± 148 | ||
| 265 ± 94c | 3180 ± 170c | |||||||||
| 1122 | (−)-3a | O | CH(CH2CH2) | H | Cl | H | 294 ± 24 | 562 ± 152 | ||
| 1123 | (+)-3a | O | CH(CH2CH2) | H | Cl | H | 369 ± 40 | 733 ± 163 | ||
| 363 | 6 | O | nC3H7 | H | Cl | H | 793 ± 66 | >10k | ||
| 361 | 7 | O | C2H5 | H | Cl | H | 5920 ± 621 | >10k | ||
| 586 | 3b | O | CH(CH2CH2) | H | Br | H | 386 ± 29 | 621 ± 141 | ||
| 588 | 3c | O | CH(CH2CH2) | H | CH3 | H | 1104 ± 69 | 1067 ± 203 | ||
| 591 | 3d | O | CH(CH2CH2) | H | OCH3 | H | 2372 ± 175 | 1014 ± 118 | ||
| 743 | 3e | O | CH(CH2CH2) | H | H | Cl | 2011 ± 178 | 1632 ± 161 | ||
| 744 | 3f | O | CH(CH2CH2) | H | H | CH3 | 1103 ± 884 | 667 ± 92 | ||
| 787 | 3g | O | CH(CH2CH2) | H | Cl | Cl | 425 ± 22 | 356 ± 96 | ||
| 820 | 3h | O | CH(CH2CH2) | H | Cl | CH3 | 592 ± 31 | 181 ± 31 | ||
| 304 | 8 | H,H | H | H | Cl | H | 11.8 ± 0.7 | 120 ± 6 | ||
| 434 | 9 | O | CH3 | H | Cl | H | 46.8 ± 4.0 | 410 ± 38 | ||
| 426 | 10 | H,H | CH3 | CH3 | H | H | 225 ± 9 | 4698 ± 479 | ||
| 429 | 11 | O | CH3 | CH3 | H | H | 411 ± 98 | >10K | ||
| 550 | 12 | H,H | H | C2H5 | H | H | 2596 ± 408 | >10K | ||
ND = not determined; PAL = phenyl amine library.
Data are represented as means ± SD and are the result of N = 3 performed in triplicate.
Data from Carroll et al.1
Substitution on the aromatic ring yielded compounds with a variety of potencies, but all were found to maintain hybrid DA reuptake/5-HT releaser activity. The site of ring substitution did not significantly alter activity, with 3a and 3e, as well as 3c and 3f, having approximately the same activity despite being substituted in the 3- and 4-positions, respectively. The disubstituted compounds 3g and 3h were the most potent in the series. The 3,4-dichloro analogue 3g and 3-chloro-4-methyl analogue 3h had roughly the same potencies for DA reuptake inhibition as for 5-HT release. These compounds had the most potent 5-HT release EC50 values of 356 and 181 nM, respectively, an order of magnitude more potent than the lead compound 3a. All of the compounds were also active at NET, either as weak uptake inhibitors or as partial releasers (data not shown).
Hybrid activity was preserved with aromatic substitution, but other changes resulted in the compounds either becoming DA/5-HT releasers or DAT/SERT uptake inhibitors. Smaller structures such as 3-chloroamphetamine (8), 3′-chloro-2-methylaminopropiophenone (9), and N-methyl-1-phenyl-2-butanamine (10) were found to be DA and 5-HT releasers. The addition of an oxygen to 10 to form 2-methylaminobutyrophenone (11) maintained DA release activity but eliminated 5-HT releasing activity. Alkyl extension alpha to the nitrogen to form phenylpentanamine (12) eliminated both DA and 5-HT release. Interestingly, the release potencies weaken for compounds 8–12 as the overall structures get larger. 3-Chloroamphetamine (8) has a 5-HT release EC50 value of 120 nM and the EC50 values weaken to 410 nM and then 4698 nM for 9 and 10, respectively. Increasing the size further makes the compound inactive. Similarly, 3-chloroamphetamine (8) has a DA release EC50 value of 11.8 nM, and the EC50 values weaken progressively to an EC50 value of 411 nM for 11. Increasing the size of the ligand molecule past a critical point serves to flip the activity to a DAT uptake inhibitor. Since release activity depends on transportability, these trends suggest that the larger the structure, the less likely the compound can be transported by either DAT or SERT and thus be a releaser. The N-cyclopropyl and N-cyclobutyl analogues retain their ability to be transported through SERT but not DAT thus leading to hybrid activity.
The agonist activity of the compounds at the 5-HT2A, 5-HT2B, 5-HT2C, and 5-HT1A G-protein-coupled receptors (GPCRs) was determined in calcium mobilization assays and in a 5-HT2A β-arrestin assay. These GPCRs were chosen due to the long-standing association of these receptors with the pharmacology of abused drugs, in particular 5-HT releasers. Agonists of these receptors would be considered off-target liabilities. The 10 μM agonist screens revealed that all of the hybrid compounds were inactive at the 5-HT1A, 5-HT2A, 5-HT2B, and 5-HT2C receptors. Taken together, the screening data indicate that these hybrid compounds may not elicit the adverse effects often associated with drug abuse. Thus, the hybrid compounds synthesized in this report could represent a new structural class that could be examined as treatments for drug addiction.
While the potency of these compounds may not appear substantial (i.e., midnanomolar to micromolar range), this potency range agrees with that of clinically used compounds exhibiting similar structures. For example, the clinically used antidepressant and smoking cessation compound bupropion has about the same level of potency.1 The optimal selectivity for DAT reuptake inhibition versus SERT release is unknown due to the differences in kinetics, mechanisms of neurotransmitter elevation, and effects after chronic use. The mixed mechanistic profile of these compounds makes such an assessment more complicated, compared to developing a DA/5-HT releaser or a DA/5-HT reuptake inhibitor, but the differences in mechanistic profile make the study of these compounds intriguing, especially regarding chronic use.
The structure–activity data presented here characterize the first class of novel compounds with hybrid DA reuptake/5-HT releaser activity. To our knowledge, compounds with this type of hybrid activity have never been developed or even considered as treatment medications. The lead structure 3a is currently being evaluated in advanced in vivo studies by the National Institute on Drug Abuse (NIDA) as a treatment medication for cocaine abuse. The recharacterization of 3a from being a SERT uptake inhibitor to a SERT releaser demonstrates the need to carefully assess compound activity using both uptake inhibition and transporter release assays. We consider compounds 3g and 3h to be lead candidates for further development. Additional compounds have been synthesized and are being evaluated, including the optical isomers and N-cyclobutyl analogues.
Acknowledgments
We would like to thank Dr. Ivy Carroll for the original samples of 3a, 6, and 7.
Glossary
ABBREVIATIONS
- BAT
biogenic amine transporter
- DA
dopamine
- 5-HT
serotonin
- DAT
dopamine transporter
- SERT
serotonin transporter
- 5-HT2A
serotonin 2A receptor
- 5-HT2B
serotonin 2B receptor
- 5-HT2C
serotonin 2C receptor
- 5-HT1A
serotonin 1A receptor
Supporting Information Available
Selected experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ (R.B.R.) Department of Psychiatry, MedStar St. Mary’s Hospital, 25500 Point Lookout Road, Leonardtown, Maryland 20650, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding was provided by the National Institute on Drug Abuse (DA12970). Aspects of this work were supported by the Intramural Research Program, National Institute on Drug Abuse, NIH.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Carroll F. I.; Blough B. E.; Abraham P.; Mills A. C.; Holleman J. A.; Wolckenhauer S. A.; Decker A. M.; Landavazo A.; McElroy K. T.; Navarro H. A.; Gatch M. B.; Forster M. J. Synthesis and biological evaluation of bupropion analogues as potential pharmacotherapies for cocaine addiction. J. Med. Chem. 2009, 52, 6768–81. [DOI] [PubMed] [Google Scholar]
- Glennon R. A.; Yousif M.; Naiman N.; Kalix P. Methcathinone: A new and potent amphetamine-like agent. Pharmacol., Biochem. Behav. 1987, 26, 547–551. [DOI] [PubMed] [Google Scholar]
- Nielsen J. A.; Shannon N. J.; Bero L.; Moore K. E. Effects of acute and chronic bupropion on locomotor activity and dopaminergic neurons. Pharmacol., Biochem. Behav. 1986, 24, 795–9. [DOI] [PubMed] [Google Scholar]
- Sparago M.; Wlos J.; Yuan J.; Hatzidimitriou G.; Tolliver J.; Dal Cason T. A.; Katz J.; Ricaurte G. Neurotoxic and pharmacologic studies on enantiomers of the N-methylated analog of cathinone (methcathinone): a new drug of abuse. J. Pharmacol. Exp. Ther. 1996, 279, 1043–52. [PubMed] [Google Scholar]
- Gowrishankar R.; Hahn M. K.; Blakely R. D. Neurochem. Int. 2013, 10.1016/j.neuint.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Little K. Y.; Kirkman J. A.; Carroll F. I.; Clark T. B.; Duncan G. E. Cocaine use increases [3H]WIN 35428 binding sites in human striatum. Brain Res. 1993, 628, 17–25. [DOI] [PubMed] [Google Scholar]
- Tella S. R.; Ladenheim B.; Cadet J. L. Differential regulation of dopamine transporter after chronic self-administration of bupropion and nomifensine. J. Pharmacol. Exp. Ther. 1997, 281, 508–13. [PubMed] [Google Scholar]
- Sandoval V.; Riddle E. L.; Ugarte Y. V.; Hanson G. R.; Fleckenstein A. E. Methamphetamine-induced rapid and reversible changes in dopamine transporter function: an in vitro model. J. Neurosci. 2001, 21, 1413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D.; Sonders M. S.; Poulsen N. W.; Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog. Neurobiol. 2005, 75, 406–33. [DOI] [PubMed] [Google Scholar]
- Yu H.; Rothman R. B.; Dersch C. M.; Partilla J. S.; Rice K. C. Uptake and release effects of diethylpropion and its metabolites with biogenic amine transporters. Bioorg. Med. Chem. 2000, 8, 2689–92. [DOI] [PubMed] [Google Scholar]
- Rothman R. B.; Partilla J. S.; Baumann M. H.; Lightfoot-Siordia C.; Blough B. E. Studies of the biogenic amine transporters. 14. Identification of low-efficacy ″partial″ substrates for the biogenic amine transporters. J. Pharmacol. Exp. Ther. 2012, 341, 251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power J. D.; McDermott S. D.; Talbot B.; O’Brien J. E.; Kavanagh P. The analysis of amphetamine-like cathinone derivatives using positive electrospray ionization with in-source collision-induced dissociation. Rapid Commun. Mass Spectrom. 2012, 26, 2601–11. [DOI] [PubMed] [Google Scholar]
- Fang Q. K.; Han Z.; Grover P.; Kessler D.; Senanayake C. H.; Wald S. A. Rapid access to enantiopure bupropion and its major metabolite by stereospecific nucleophilic substitution on an a-ketotriflate. Tetrahedron: Asymmetry 2000, 11, 3659–63. [Google Scholar]
- Lukas R. J.; Muresan A. Z.; Damaj M. I.; Blough B. E.; Huang X.; Navarro H. A.; Mascarella S. W.; Eaton J. B.; Marxer-Miller S. K.; Carroll F. I. Synthesis and characterization of in vitro and in vivo profiles of hydroxybupropion analogues: aids to smoking cessation. J. Med. Chem. 2010, 53, 4731–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman R. B.; Baumann M. H.; Dersch C. M.; Romero D. V.; Rice K. C.; Carroll F. I.; Partilla J. S. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 2001, 39, 32–41. [DOI] [PubMed] [Google Scholar]
- Rothman R. B.; Katsnelson M.; Vu N.; Partilla J. S.; Dersch C. M.; Blough B. E.; Baumann M. H. Interaction of the anorectic medication, phendimetrazine, and its metabolites with monoamine transporters in rat brain. Eur. J. Pharmacol. 2002, 447, 51–7. [DOI] [PubMed] [Google Scholar]
- Coles R.; Kharasch E. D. Stereoselective analysis of bupropion and hydroxybupropion in human plasma and urine by LC/MS/MS. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007, 857, 67–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.