

Abstract
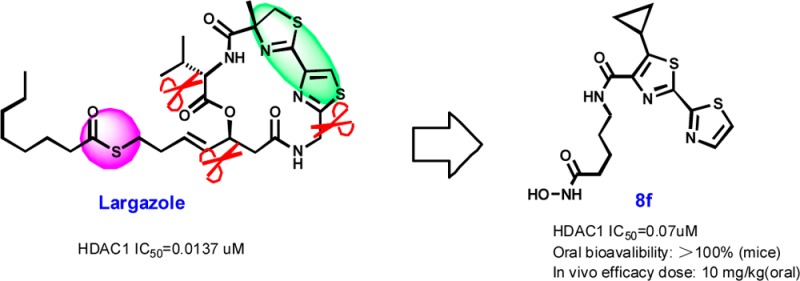
Inspired by the thiazole–thiazoline cap group in natural product largazole, a series of structurally simplified bisthiazole-based histone deacetylase inhibitors were prepared and evaluated. Compound 8f was evaluated in vivo in an experimental autoimmune encephalomyelitis (EAE) model and found to be orally efficacious in ameliorating clinical symptoms of EAE mice.
Keywords: Histone deacetylase inhibitors, largazole, bisthiazole
Precise control of gene expression is important for many cellular processes including cell survival, apoptosis, and proliferation. Histone deacetylases (HDACs) and histone acetyltransferases (HATs) are two functionally opposing enzymes that control gene transcription via regulation of histone lysine acetylation. Histone acetylation neutralizes positive charges on lysine residues and disrupts nucleosome structure, allowing unfolding of associated DNA and binding of transcription factors, resulting in changes of gene expression.1 HDACs have been validated as targets for cancer chemotherapy.2−5 Of the 18 known human HDAC isoforms, which are divided into zinc-dependent classic HDACs (class I, II, and IV enzymes) and NAD+-dependent class III enzymes (sirtuins),6,7 evidence suggests that class I HDACs are most relevant to cancer growth.8,9
HDAC inhibitors (HDACis) block the deacetylation action of HDACs, resulting in hyperacetylation of histones and thereby affecting gene expression.10 Currently reported small-molecule HDACis can be categorized into short-chain fatty acids, hydroxamic acids, cyclic tetrapeptides/depsipeptides, electrophilic ketones, and benzamides.11−13 A typical HDACi has a three-motif pharmacophoric model consisting of a recognition cap group, a hydrophobic linker, and a zinc-binding group (ZBG).14 To date, two HDACis, vorinostat (SAHA) and romidepsin, have been approved by the FDA to treat cutaneous T cell lymphoma.15,16 In addition, the pro-apoptotic activity of HDACis holds potential for treatment of inflammatory and autoimmune diseases.17
Multiple sclerosis (MS), one of the foremost causes of nontraumatic neurological disability in young adults, is a representative autoimmune disease, which induces inflammatory demyelination of the central nervous system (CNS).18−20 Because of the limited understanding of the pathogenesis of MS, there remain many difficulties in treating MS. The development of novel therapeutic targets and agents is in high demand. HDACis have been shown to inhibit proinflammatory cytokine expression.21,22 HDACi trichostatin (TSA) was reported to alleviate clinical symptoms of EAE, an animal model for MS.23 In our previous work, HDACi valproic acid (VPA) was demonstrated to induce apoptosis in pathogenic T cells and ameliorate pathogenesis in EAE mice.24 Accordingly, the idea of repurposing VPA for MS therapy has elicited interest.25
Largazole (1a, Figure 1) is a depsipeptide natural product isolated from the cyanobacterium Symploca sp. by Luesch and co-workers in 2008.26 As a novel natural HDACi with potent antiproliferative activity and selectivity for cancer cells, it has attracted significant interest. To date more than ten groups have reported its total synthesis and SAR studies.27−46
Figure 1.

Structures of synthesized ring-opened analogues.
We previously reported that C7-demethyl bisthiazole largazole analogue 2a (Figure 1) has significantly decreased HDAC inhibitory activity and antiproliferative activity against HCT-116 colon cancer cells compared to largazole.33 de Lera et al. have since reported that C7-demethyl largazole (1c) is equipotent to largazole,34 indicating that the C7-methyl is unimportant for activity; they also reported the potent activity of 2a against NB4 cells. Williams et al. have demonstrated that related thiazole-thiazole analogue 2b displays intermediate HDAC inhibitory activity compared to largazole thiol.31 These results inspired us to design a series of simplified analogues of 2a that retains the bisthiazole cap group while cleaving across the β-hydroxy and thiazole fragments. The synthesis of ring-opened analogues 3a–3d and 4a–4d eventually culminated in the discovery of structurally simple bisthiazole-based HDACis 8a–8t (Figure 1).
To access 3a–3d, the thiazole–thiazole moiety was constructed by a modified Hantzsch procedure (route 1, Scheme 1).47 The thiazoline–thiazole moiety in 4a–4d was accessed via condensation of cyanothiazole with 15.48 Side chain installation via olefin cross-metathesis then afforded ring-opened analogues 3a–3d and 4a–4d.27
Scheme 1. Synthesis of Ring-Opened Analogues.

Reagents and conditions: (a) Lawesson’s reagent, DME, room temperature, 12 h. (b) BrCH2COCOOEt, KHCO3, TFAA, 2,6-lutidine, DME, room temperature, 10 h. (c) LiOH, MeOH/H2O, room temperature, 8 h. (d) EDCI, HOBt, i-Pr2NEt, DMF, room temperature, 12 h. (e) Grubbs’ second generation catalyst, toluene, reflux, 8 h. (f) TFA, Et3SiH, CH2Cl2, room temperature, 2 h. (g) Et3N, TFAA, THF, room temperature, 2 h. (h) Et3N, MeOH, 50 °C, 12 h. (i) NH2OH·HCl, KOH, MeOH, room temperature, 2 h. (j) Zn, dry THF, reflux, 2 h. (k) Pd(OAc)2, PPh3, toluene, reflux, 8 h. (l) NaOH, MeOH/H2O, reflux, 1 h.
With ring-opened analogues 3a–3d and 4a–4d in hand, we assessed their inhibitory activities against human HDAC1, 3, and 6 (Table 1). Unexpectedly, the thiazoline–thiazole analogues (4a–4d) displayed significantly decreased inhibitory activity, while the thiazole–thiazole analogues (3a–3d) retained moderate activity. Compound 3b in particular was nearly equipotent to the thiol form of C7-demethyl bisthiazole largazole 2b, indicating that the thiazole–thiazole moiety might be a suitable cap group for a HDAC inhibitor.
Table 1. Inhibitory Activity against Human HDACs.
| IC50 (μM)d |
|||||
|---|---|---|---|---|---|
| compd | R1 | R2 | HDAC1 | HDAC3 | HDAC6 |
| 1aa | n-C7H15CO | 0.0137 | 0.2450 | 11.50 | |
| 1bb | H | 0.0012 | 0.0034 | 0.049 | |
| 2aa | n-C7H15CO | 2.0 | 16.8 | 14.7 | |
| 2bb | H | 0.077 | 0.085 | >30 | |
| 3a | H | n-C7H15CO | 1.90 ± 0.25 | 2.83 ± 0.34 | NAc |
| 3b | H | H | 0.09 ± 0.02 | 0.06 ± 0.01 | 3.71 ± 0.27 |
| 3c | Me | n-C7H15CO | 2.60 ± 0.45 | 3.56 ± 0.44 | NA |
| 3d | Me | H | 0.12 ± 0.02 | 0.11 ± 0.02 | 4.32 ± 0.33 |
| 4a | H | n-C7H15CO | NAc | NAc | NAc |
| 4b | H | H | 16.82 ± 1.0 | NAc | NAc |
| 4c | Me | n-C7H15CO | NAc | NAc | NAc |
| 4d | Me | H | 13.86 ± 2.2 | NAc | NAc |
A series of HDACis was then synthesized containing the thiazole–thiazole cap group and classic hydroxamic acid ZBG, as found in SAHA (5a–5f, Figure 1). To assess the potential impact of side chain length on activity,30,49 amines 19 with variant linker length were condensed with acids prepared from esters 18a–18b to afford respective amides 20a–20f. Upon treatment with hydroxylamine, compounds 5a–5f were rapidly accessed (route 2, Scheme 1). Disappointingly, most of these hydroxamic acid analogues showed significantly reduced inhibitory activity compared to compound 3b (Table 2). While compounds 5c, 5e, and 5f showed moderate inhibition to HDAC1 and 3, compounds 5a and 5d displayed no inhibitory activity. These results were consistent with previous reports regarding the importance of the trans double bond linker in largazole analogues.30
Table 2. Inhibitory Activity against Human HDACs.
| IC50 (μM)b |
||||
|---|---|---|---|---|
| compd | R1 | HDAC1 | HDAC3 | HDAC6 |
| 5a | H | NAa | NAa | NAa |
| 5b | H | 9.69 ± 2.5 | 3.17 ± 0.45 | NAa |
| 5c | H | 2.59 ± 0.49 | 1.64 ± 0.13 | NAa |
| 5d | Me | NAa | NAa | NAa |
| 5e | Me | 2.66 ± 0.71 | 2.22 ± 0.41 | NAa |
| 5f | Me | 2.45 ± 0.31 | 1.58 ± 0.30 | NAa |
| 6a | H | 0.45 ± 0.11 | 0.27 ± 0.06 | 2.24 ± 0.32 |
| 6b | Me | 0.24 ± 0.03 | 0.15 ± 0.04 | 1.19 ± 0.25 |
| 7 | H | 0.21 ± 0.03 | 0.11 ± 0.02 | 1.10 ± 0.06 |
No inhibitory activity observed at 20 μg/mL.
Values are expressed as the mean ± SD, and the test was performed in triplicate.
Further structural simplification, featuring replacement of the chiral l-valine moiety with a chained carbon linker, was achieved by condensation of amine 21 with 2,4′-bisthiazole-4-carboxylic acids obtained by hydrolysis of 10a,10b to give intermediates 22a,22b. Upon treatment with hydroxylamine, hydoxamic acids 6a,6b were afforded (route 3, Scheme 1). Negishi coupling of methyl 2-bromothiazole-4-carboxylates 24 with 2-thiazole-zinc bromide, prepared by treating 2-bromothiazole 23 with activated zinc dust, afforded 2,2′-bisthiazole 25,50 which was hydrolyzed to the corresponding acid (route 4, Scheme 1). Condensation of amine 21 with this acid and subsequent treatment with hydroxylamine afforded compound 7 directly. Analogues 6a,6b and 7 displayed improved inhibitory activity compared with 5a–5f (Table 2). Interestingly, compound 7, which contains a 2,2′-bisthiazole moiety instead of a 2,4′-bisthiazole moiety, showed a 2-fold increase in activity compared to 6a.
Encouraged by the comparable inhibitory activity of compound 7 and its facile synthetic accessibility, a series of 2,2′-bisthiazole-based hydroxamic acids 8a–8n were synthesized (route 4, Scheme 1). The preparation of compounds 8o–8t is additionally described in the Supporting Information. This approach allowed for rapid diversification of the substituent on the left thiazole ring (alkyl, aryl, and alkenyl, Table 3) and efficient synthesis of a small focused 2,2′-bisthiazole-based library.
Table 3. Inhibitory Activity against Human HDACs.
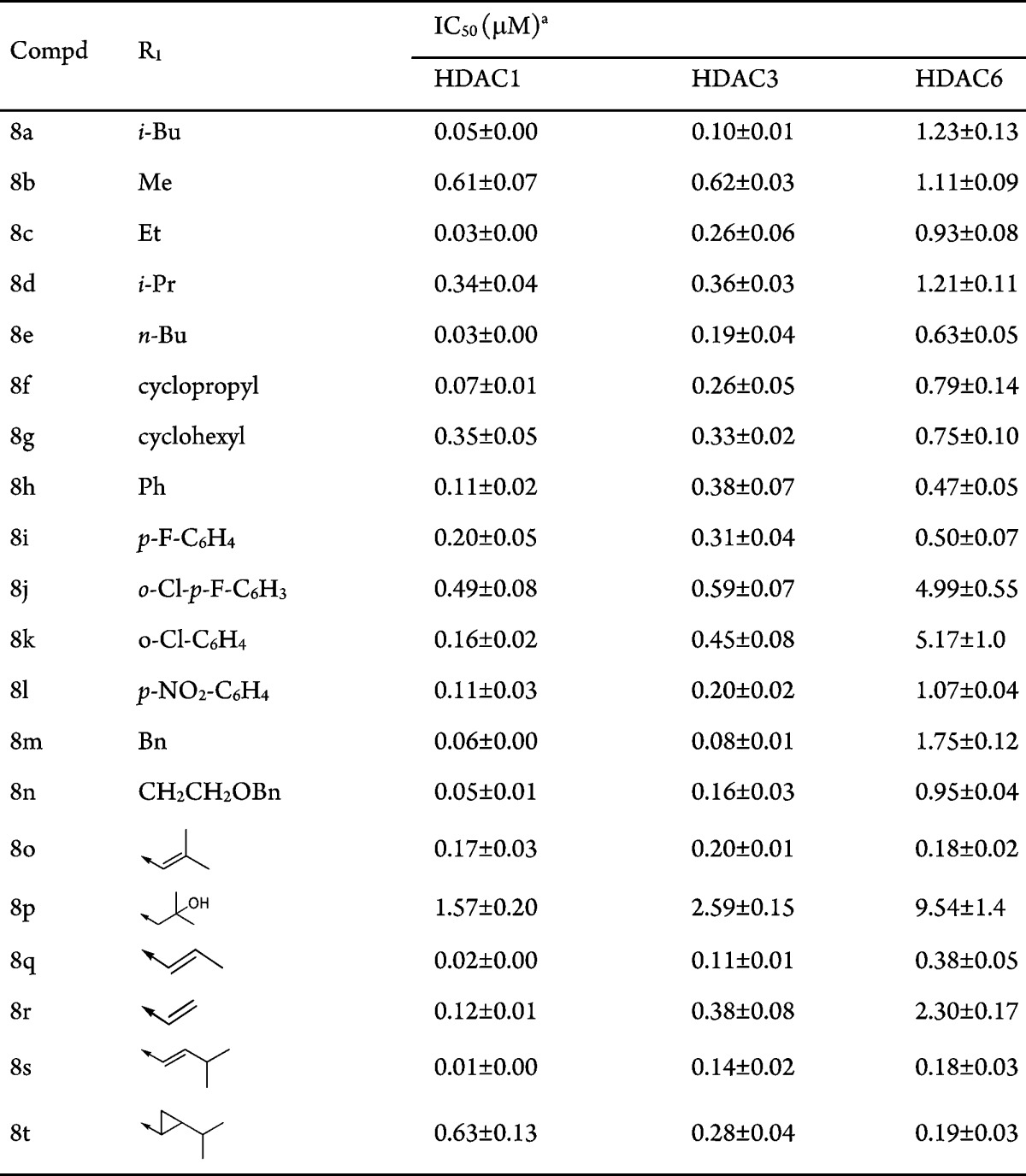
Values are expressed as the mean ± SD, and the test was performed in triplicate.
Pleasingly, most of the R1 substituents were well tolerated (Table 3). Among compounds 8a–8t, 8s was found to be the most potent HDAC1 inhibitor with IC50 as low as 0.01 μM. The isopropyl group in 8s might contribute to key hydrophobic interactions with the protein surface. In contrast, compound 8p, which lacks a hydrophobic substitute on the left thiazole, showed much reduced activity. Moreover, compound 8f not only displayed potent activity against nine HDACs except HDAC7 and HDAC9 (Table S1, Supporting Information) but also showed excellent pharmacokinetic profiles characterized by high Cmax (7.04 μg/mL) and AUC0-t (12.07 μg·h/mL) and high absolute bioavailability (F% = 118.7%) in mice (20 mg/kg, P.O., Table S2, Supporting Information). We thus went on to evaluate its efficacy in vivo.
Compound 8f induced hyperdeacetylation of histone H3 and H4 and apoptosis of T cells in vitro. The cellular histone acetylation levels after treatment with 8f were determined using mouse embryonic fibroblast (MEF) cells. The cells were cultured with 8f (0.3, 1, or 3 μM) for 6 h, and cell lysates were subjected to Western blot analysis. As shown in Figure 2a,b, resting levels of acetylated histone H3 and H4 were low in MEF cells. When treated with 8f, acetylation levels of histone H3 and H4 increased significantly in a dose-dependent manner. Notably, at a concentration of 3 μM, 8f displayed better inhibitory effects than VPA at 4 mM.
Figure 2.

Compound 8f induces histone acetylation in MEF cells and apoptosis in T cells. (a,b) MEF cells were treated with 8f or VPA for 6 h and then lysed for Western blot analysis to detect the acetylation level of histone H3 and H4. Quantification of histone acetylation was analyzed with Quantity One software (Bio-Rad). (c,d) Mouse lymphocytes were stimulated in vitro and treated with 8f for 36 h. Apoptosis was detected by Annexin V and PI staining, and the percentages of apoptotic cells (Annexin V single positive) were analyzed by flow cytometry.
We next examined the effect of 8f on apoptosis induction in lymphocytes; HDAC enzymes play important roles in cell survival and proliferation, and HDACis have been shown to induce apoptosis in several cell types.51−53 Naive lymphocytes were isolated from mouse spleen and activated with anti-CD3 antibody or LPS, respectively, and cultured with various concentrations of 8f for 36 h. Activated splenocytes displayed a relatively high apoptosis level (∼40% for T cells) when cultured in vitro. Compound 8f treatment further enhanced apoptosis in a dose-dependent way in T cells (∼75%), but not in B cells (Figure 2c). Subpopulation analysis with cell surface markers revealed that 8f enhanced apoptosis in both CD4+ and CD8+ T cells after activation in vitro (Figure 2d).
Compound 8f Ameliorates Clinical Symptoms of EAE Mice
HDACis have been shown to ameliorate inflammation in several autoimmune animal models,54,55 including EAE.23 To examine the in vivo effect of 8f, EAE was induced in C57BL/6 mice by immunization with MOG35–55 peptide. Compound 8f (10 mg/kg) was given b.i.d. via oral administration from day 3 or day 15 following immunization until the end of the experiment. PBS was used as a vehicle control. Compound 8f treatment led to decreased incidence, reduced peak severity, and decreased cumulative clinical score of EAE when given from day 3 (Figure 3a and Table S2, Supporting Information). More interestingly, when given after the onset of the disease (day 15), 8f (10 mg/kg) was still able to reduce the severity of EAE (Figure 3b and Table S2, Supporting Information), indicating both a therapeutic benefit and preventative effect of this drug.
Figure 3.
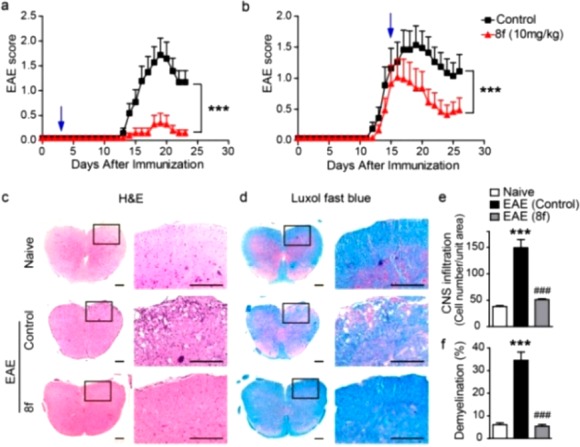
Compound 8f alleviates clinical symptoms of EAE. (a,b) Clinical score of EAE mice treated with 8f (n = 10) or PBS (n = 10) b.i.d. by oral administration on the day indicated by the arrow. Data are mean ± SEM, ***p < 0.001 (two-way ANOVA test). (c,d) Representative H&E or Luxol fast blue stained spinal cord sections isolated from naive, 8f, or vehicle-treated EAE mice on day 22 postimmunization. Boxed areas in left columns are presented enlarged on the right. (e,f) Quantitative analysis of the number of spinal cord infiltrates in the sections presented in panel c and the amount of demyelination presented in panel d. Data are mean ± SEM; ***p < 0.001 versus naive group; ###p < 0.001 versus vehicle control (Student’s t-test).
Compound 8f Ameliorates CNS Pathology and Leukocytes Infiltration in EAE Mice
Histological examination of spinal cords was performed at day 22 postimmunization. A dramatic decrease in leukocyte infiltration in spinal cord was observed with 8f treatment (Figures 3c,e). In vehicle-treated EAE mice, Luxol fast blue staining showed multiple widespread areas of myelin damage in white matter regions; demyelination was greatly ameliorated in 8f-treated EAE mice (Figures 3d,f). Leukocyte infiltration was further assessed by immunofluorescent staining of the spinal cord sections with anti-CD45 antibody; 8f treatment significantly reduced the number of CD45+ cells in the spinal cords (Figure S1a, Supporting Information).
Leukocyte infiltration into the CNS was isolated and quantified by flow cytometry at day 18 postimmunization. Results confirmed that the total CNS infiltrates (Figure S1b, Supporting Information) and CD4+ T cells accumulated in CNS (Figure S1c, Supporting Information) both decreased with 8f treatment. IL-17-producing Th17 and IFN-γ-producing Th1 are the main pathogenic CD4+ T effector cells in EAE. With surface staining of CD4 and intracellular staining of IL-17 and IFN-γ, the number of both Th17 and Th1 cells was observed to decrease significantly in the CNS of 8f-treated EAE mice (Figures S1c,d, Supporting Information).
In conclusion, a series of bisthiazole-based hydroxamic acids evolved from natural product largazole were synthesized as potent HDAC inhibitors. Several compounds exhibited low micromolar to midnanomolar IC50 values in in vitro HDAC inhibition assay. Compound 8f was evaluated in vivo in an EAE model and showed efficacy in ameliorating clinical symptoms of EAE mice when administered orally. These bisthiazole-based HDACis are under further investigation for treatment of autoimmune diseases and cancer, and the results will be reported in due course.
Glossary
ABBREVIATIONS
- DME
ethylene glycol dimethyl ether
- DMF
N, N-dimethylformamide
- EAE
experimental autoimmune encephalomyelitis
- EDCI
1-ethyl-3-(3-dimethyl-aminopropyl)-carbodiimide hydrochloride
- IC50
half maximal inhibitory concentration
- TFA
trifluoroacetic acid
- TFAA
trifluoroacetic anhydride
- HOBt
1-hydroxybenztriazole
- THF
tetrahydrofuran
- Trt
triphenylmethyl
Supporting Information Available
Synthetic details and characterization data for all compounds reported in this letter. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† F.C., H.C. and M.-B.S. contributed equally to this work.
This work was supported by grants from The National Natural Science Foundation of China (30725049, 81021062, and 81072652), Chinese Academy of Sciences (XDA01040301), Ministry of Science and Technology of China (2009CB940900), and Shanghai Science and Technology Committee (No. 11431921102).
The authors declare no competing financial interest.
Supplementary Material
References
- Thiagalingam S.; Cheng K. H.; Lee H. J.; Mineva N.; Thiagalingam A.; Ponte J. F. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann. N.Y. Acad. Sci. 2003, 983, 84–100. [DOI] [PubMed] [Google Scholar]
- Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes. Dev. 1998, 125599–606. [DOI] [PubMed] [Google Scholar]
- Johnstone R. W. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug. Discovery 2002, 14287–99. [DOI] [PubMed] [Google Scholar]
- Somech R.; Izraeli S.; A J. S. Histone deacetylase inhibitors--a new tool to treat cancer. Cancer Treat. Rev. 2004, 305461–72. [DOI] [PubMed] [Google Scholar]
- Bolden J. E.; Peart M. J.; Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug. Discovery 2006, 59769–84. [DOI] [PubMed] [Google Scholar]
- Yang X. J.; Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell. Biol. 2008, 93206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S.; Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol. Sci. 2010, 315212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis T. C.; El-Osta A. Will broad-spectrum histone deacetylase inhibitors be superseded by more specific compounds?. Leukemia 2007, 21161–5. [DOI] [PubMed] [Google Scholar]
- Sweet M. J.; Shakespear M. R.; Kamal N. A.; Fairlie D. P. HDAC inhibitors: modulating leukocyte differentiation, survival, proliferation and inflammation. Immunol. Cell Biol. 2012, 90114–22. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M.; Clarke C.; Marks P. A. Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer. Res. 2007, 510981–9. [DOI] [PubMed] [Google Scholar]
- Miller T. A.; Witter D. J.; Belvedere S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46245097–116. [DOI] [PubMed] [Google Scholar]
- Paris M.; Porcelloni M.; Binaschi M.; Fattori D. Histone deacetylase inhibitors: from bench to clinic. J. Med. Chem. 2008, 5161505–29. [DOI] [PubMed] [Google Scholar]
- Newkirk T. L.; Bowers A. A.; Williams R. M. Discovery, biological activity, synthesis and potential therapeutic utility of naturally occurring histone deacetylase inhibitors. Nat. Prod. Rep. 2009, 26101293–320. [DOI] [PubMed] [Google Scholar]
- Sternson S. M.; Wong J. C.; Grozinger C. M.; Schreiber S. L. Synthesis of 7200 small molecules based on a substructural analysis of the histone deacetylase inhibitors trichostatin and trapoxin. Org. Lett. 2001, 3264239–42. [DOI] [PubMed] [Google Scholar]
- Mann B. S.; Johnson J. R.; Cohen M. H.; Justice R.; Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12101247–52. [DOI] [PubMed] [Google Scholar]
- Campas-Moya C. Romidepsin for the treatment of cutaneous T-cell lymphoma. Drugs Today 2009, 4511787–95. [DOI] [PubMed] [Google Scholar]
- Adcock I. M. HDAC inhibitors as anti-inflammatory agents. Br. J. Pharmacol. 2007, 1507829–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sospedra M.; Martin R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [DOI] [PubMed] [Google Scholar]
- John G. R.; Shankar S. L.; Shafit-Zagardo B.; Massimi A.; Lee S. C.; Raine C. S.; Brosnan C. F. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med. 2002, 8101115–21. [DOI] [PubMed] [Google Scholar]
- Hemmer B.; Nessler S.; Zhou D.; Kieseier B.; Hartung H. P. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat. Clin. Pract. Neurol. 2006, 24201–11. [DOI] [PubMed] [Google Scholar]
- Matsuoka H.; Fujimura T.; Mori H.; Aramori I.; Mutoh S. Mechanism of HDAC inhibitor FR235222-mediated IL-2 transcriptional repression in Jurkat cells. Int. Immunopharmacol. 2007, 7111422–32. [DOI] [PubMed] [Google Scholar]
- Takahashi I.; Miyaji H.; Yoshida T.; Sato S.; Mizukami T. Selective inhibition of IL-2 gene expression by trichostatin A, a potent inhibitor of mammalian histone deacetylase. J. Antibiot. 1996, 495453–7. [DOI] [PubMed] [Google Scholar]
- Camelo S.; Iglesias A. H.; Hwang D.; Due B.; Ryu H.; Smith K.; Gray S. G.; Imitola J.; Duran G.; Assaf B.; Langley B.; Khoury S. J.; Stephanopoulos G.; De Girolami U.; Ratan R. R.; Ferrante R. J.; Dangond F. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 1641–210–21. [DOI] [PubMed] [Google Scholar]
- Lv J.; Du C.; Wei W.; Wu Z.; Zhao G.; Li Z.; Xie X. The antiepileptic drug valproic acid restores T cell homeostasis and ameliorates pathogenesis of experimental autoimmune encephalomyelitis. J. Biol. Chem. 2012, 2873428656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough N. R. Repurposing antiepileptic drugs for multiple sclerosis. Science Signalling 2012, 5239ec223. [Google Scholar]
- Taori K.; Paul V. J.; Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 13061806–7. [DOI] [PubMed] [Google Scholar]
- Ying Y.; Taori K.; Kim H.; Hong J.; Luesch H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130268455–9. [DOI] [PubMed] [Google Scholar]
- Bowers A.; West N.; Taunton J.; Schreiber S. L.; Bradner J. E.; Williams R. M. Total synthesis and biological mode of action of largazole: a potent class I histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 1303311219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiser T.; Kamena F.; Cramer N. Synthesis and biological activity of largazole and derivatives. Angew. Chem., Int. Ed. 2008, 47346483–5. [DOI] [PubMed] [Google Scholar]
- Ying Y.; Liu Y.; Byeon S. R.; Kim H.; Luesch H.; Hong J. Synthesis and activity of largazole analogues with linker and macrocycle modification. Org. Lett. 2008, 10184021–4. [DOI] [PubMed] [Google Scholar]
- Bowers A. A.; West N.; Newkirk T. L.; Troutman-Youngman A. E.; Schreiber S. L.; Wiest O.; Bradner J. E.; Williams R. M. Synthesis and histone deacetylase inhibitory activity of largazole analogs: alteration of the zinc-binding domain and macrocyclic scaffold. Org. Lett. 2009, 1161301–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers A. A.; Greshock T. J.; West N.; Estiu G.; Schreiber S. L.; Wiest O.; Williams R. M.; Bradner J. E. Synthesis and conformation–activity relationships of the peptide isosteres of FK228 and largazole. J. Am. Chem. Soc. 2009, 13182900–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Gao A. H.; Li J.; Nan F. J. Synthesis and biological evaluation of c7-demethyl largazole analogues. ChemMedChem 2009, 481269–72. [DOI] [PubMed] [Google Scholar]
- Souto J. A.; Vaz E.; Lepore I.; Poppler A. C.; Franci G.; Alvarez R.; Altucci L.; de Lera A. R. Synthesis and biological characterization of the histone deacetylase inhibitor largazole and C7- modified analogues. J. Med. Chem. 2010, 53124654–67. [DOI] [PubMed] [Google Scholar]
- Nasveschuk C. G.; Ungermannova D.; Liu X.; Phillips A. J. A concise total synthesis of largazole, solution structure, and some preliminary structure activity relationships. Org. Lett. 2008, 10163595–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Kulkarni S. Enantioselective total synthesis of (+)-largazole, a potent inhibitor of histone deacetylase. Org. Lett. 2008, 10173907–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q.; Dai L.; Zhang H.; Tan W. F.; Xu Z. S.; Ye T. Total synthesis of largazole. Synlett 2008, 15, 2379–2383. [Google Scholar]
- Numajiri Y.; Takahashi T.; Takagi M.; Shin-Ya K.; Doi T. Total synthesis of largazole and its biological evaluation. Synlett 2008, 16, 2483–2486. [Google Scholar]
- Wang B.; Forsyth C. J. Total synthesis of largazole: devolution of a novel synthetic strategy. Synthesis-Stuttgart 2009, 17, 2873–2880. [Google Scholar]
- Zeng X.; Yin B.; Hu Z.; Liao C.; Liu J.; Li S.; Li Z.; Nicklaus M. C.; Zhou G.; Jiang S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010, 1261368–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q.; Wang L. P.; Jiao X. Z.; Liu X. Y.; Wu Q.; Xie P. Concise total synthesis of largazole. J. Asian Nat. Prod. Res. 2010, 1211940–9. [DOI] [PubMed] [Google Scholar]
- Benelkebir H.; Marie S.; Hayden A. L.; Lyle J.; Loadman P. M.; Crabb S. J.; Packham G.; Ganesan A. Total synthesis of largazole and analogues: HDAC inhibition, antiproliferative activity and metabolic stability. Bioorg. Med. Chem. 2011, 19123650–8. [DOI] [PubMed] [Google Scholar]
- Wang B.; Huang P. H.; Chen C. S.; Forsyth C. J. Total syntheses of the histone deacetylase inhibitors largazole and 2-epi-largazole: application of N-heterocyclic carbene mediated acylations in complex molecule synthesis. J. Org. Chem. 2011, 7641140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole K. E.; Dowling D. P.; Boone M. A.; Phillips A. J.; Christianson D. W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 1333212474–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhansali P.; Hanigan C. L.; Casero R. A.; Tillekeratne L. M. Largazole and analogues with modified metal-binding motifs targeting histone deacetylases: synthesis and biological evaluation. J. Med. Chem. 2011, 54217453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Tu Z.; Li H.; Liu C.; Li Z.; Sun Q.; Yao Y.; Liu J.; Jiang S. Biological evaluation of new largazole analogues: alteration of macrocyclic scaffold with click chemistry. ACS Med. Chem. Lett. 2012, 41132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diness F.; Nielsen D. S.; Fairlie D. P. Synthesis of the thiazole–thiazoline fragment of largazole analogues. J. Org. Chem. 2011, 76239845–51. [DOI] [PubMed] [Google Scholar]
- Pattenden G.; Thom S. M.; Jones M. F. Enantioselective synthesis of 2-alkyl substituted cysteines. Tetrahedron 1993, 49102131–2138. [Google Scholar]
- Salmi-Smail C.; Fabre A. L.; Dequiedt F.; Restouin A.; Castellano R. M.; Garbit S.; Roche P.; Morelli X.; Brunel J. M.; Collette Y. Modified CAP group suberoylanilide hydroxamic acid histone deacetylase inhibitor derivatives reveal improved selective antileukemic activity. J. Med. Chem. 2010, 5383038–3047. [DOI] [PubMed] [Google Scholar]
- Chen H. J.; Wang W. L.; Wang G. F.; Shi L. P.; Gu M.; Ren Y. D.; Hou L. F.; He P. L.; Zhu F. H.; Zhong X. G.; Tang W.; Zuo J. P.; Nan F. J. Rational design and synthesis of 2,2-bisheterocycle tandem derivatives as non-nucleoside hepatitis B virus inhibitors. ChemMedChem 2008, 391316–21. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Richon V.; Ni X.; Talpur R.; Duvic M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J. Invest. Dermatol. 2005, 12551045–52. [DOI] [PubMed] [Google Scholar]
- Ruefli A. A.; Ausserlechner M. J.; Bernhard D.; Sutton V. R.; Tainton K. M.; Kofler R.; Smyth M. J.; Johnstone R. W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 2001, 981910833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrana J. A.; Decker R. H.; Johnson C. R.; Wang Z.; Jarvis W. D.; Richon V. M.; Ehinger M.; Fisher P. B.; Grant S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 1999, 18507016–25. [DOI] [PubMed] [Google Scholar]
- Lin H. S.; Hu C. Y.; Chan H. Y.; Liew Y. Y.; Huang H. P.; Lepescheux L.; Bastianelli E.; Baron R.; Rawadi G.; Clement-Lacroix P. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br. J. Pharmacol. 2007, 1507862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C. A.; Fossati G.; Mascagni P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol. Med. 2011, 175–6333–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.