

Abstract
Background
Genetic variants make some contributions to inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC). More than 100 susceptibility loci were identified in Western IBD studies, but susceptibility gene has not been found in Chinese IBD patients till now. Sequencing of individuals with an IBD family history is a powerful approach toward our understanding of the genetics and pathogenesis of IBD. The aim of this study, which focuses on a Han Chinese CD family, is to identify high-risk variants and potentially novel loci using whole exome sequencing technique.
Methods
Exome sequence data from 4 individuals belonging to a same family were analyzed using bioinformatics methods to narrow down the variants associated with CD. The potential risk genes were further analyzed by genotyping and Sanger sequencing in family members, additional 401 healthy controls (HC), 278 sporadic CD patients, 123 UC cases, a pair of monozygotic CD twins and another Chinese CD family.
Results
From the CD family in which the father and daughter were affected, we identified a novel single nucleotide variant (SNV) c.374T>C (p.I125T) in exon 4 of discs large homolog 1 (DLG1), a gene has been reported to play mutiple roles in cell proliferation, T cell polarity and T cell receptor signaling. After genotyping among case and controls, a PLINK analysis showed the variant was of significance (P<0.05). 4 CD patients of the other Chinese family bore another non-synonymous variant c.833G>A (p.R278Q) in exon 9 of DLG1.
Conclusions
We have discovered novel genetic variants in the coding regions of DLG1 gene, the results support that DLG1 is a novel potential susceptibility gene for CD in Chinese patients.
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are classified as chronic, idiopathic inflammatory bowel diseases (IBD) [1], [2]. Familial aggregation, high concordance in twins and a higher prevalence of the disease in a certain ethnic population imply a strong genetic influence on the risk of disease development [3], [4]. Identifying the genetic loci or rare detrimental mutations in different populations or families with the disease will help elucidate the pathogenesis of these complex traits and facilitate the development of more targeted therapy.
It is now widely recognized that common variants shown in GWAS can explain only relatively modest proportions of risk for diseases. Numerous functional and deleterious variants in the population are at frequencies of 0.5 to 5% that are too low to be detected by GWAS [5], [6]. As predisposing variants will present at a much higher frequency in the affected relatives of an index case, family studies may facilitate the detection of the ‘missing heritability’ not identified by GWAS [7]. Exome sequencing, which is a technique that focuses on the protein-coding portion of the genome, is not limited by the detailed and complete pedigree data that are necessary for classical linkage analysis and can be performed on only a few patients for the detection of causal mutations [8], [9].
Researchers have successfully identified a causal hemizygous mutation in the XIAP gene [10] and novel compound heterozygous mutations in interleukin-10 receptor 1 (IL-10R1) [11], using exome sequencing in children presenting with very early-onset and intractable IBD. The sequencing of eight pediatric IBD patients’ exomes revealed various profiles of specific variants with a limited number in each case [12].
Numerous candidate genes for Western IBD patients have been shown, but causality for specific variants in Chinese IBD patients is largely absent. In this study, we applied whole exome sequencing to 4 individuals belonging to a same family (Family A) to discover novel deleterious genetic variants associated with IBD and then validated these findings in other 10 family members of Family A, 401 healthy controls (HC), 278 subjects with sporadic CD, 123 subjects with UC, a pair of monozygotic twins and another Han Chinese CD family (Family B).
Materials and Methods
Patients and Controls
The familial patients included in this study were selected from the Hubei Clinical Center & Key Lab of Intestinal & Colorectal Diseases. Written informed consent was obtained from all subjects and the next of kin on behalf of the children enrolled in the study. This study was approved by the ethics committee of the Zhongnan Hospital of Wuhan University as part of the human subjects’ protocol to study the genetics of IBD in humans. The CD patients and HC were all unrelated subjects of Chinese descent and born to non-consanguineous parents. The ancestry of the patients and control individuals was assessed by self-report and appearance. Phenotypic data were acquired from a review of medical records, phone interviews and photographs. A combination of symptom assessment, laboratory and radiological examinations and endoscopy with histology was applied to make the diagnosis.
For whole exome sequencing, we selected a Han Chinese family (Family A) including a daughter and a father both affected with CD from Hubei province. In this family, the father is the proband, the proband’s unaffected mother and wife were taken as exome sequencing controls. The father, who was diagnosed with CD at the age of 31 years in 1999 with terminal ileitis and proctitis, was treated with oral prednisolone and aminosalicylic acid (5-ASA). Small intestine computed tomography enterography (CTE) showed a thickened ileum wall in 2012.
The affected daughter developed CD at the age of 16 years in 2012, with high fever, diarrhea, oral ulcers and an anal fistula. Endoscopy showed upper digestive tract ulcers, aphthous ulcers at the ileocecal junction and colitis involving the rectum, sigmoid colon, descending colon and transverse colon. A biopsy showed non-specific granulomatous inflammation and staining was negative for acid-fast bacilli. She was finally diagnosed with CD and was treated with an intravenous injection of corticosteroids, 5-ASA, immunosuppressants and infliximab for severe refractory disease. She is now in remission with azathioprine and 5-ASA (the supporting data are provided in Fig. 1).
Figure 1. Clinical characteristics of two patients in Chinese CD family A.
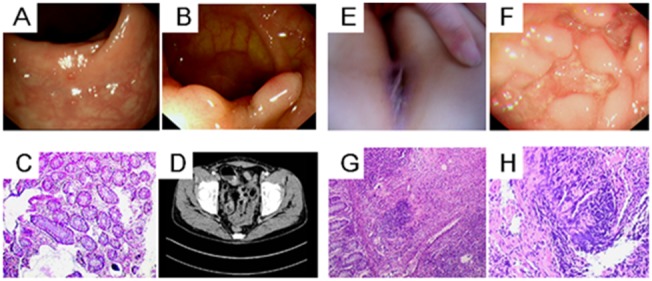
The index patient in the family, the father (Panels A to D) had evidence of mucous membrane granulation, polypoid proliferation and hyperemia in his colonoscopy, as shown in Panels A and B. Panel C shows the patient’s pathological findings of chronic intestinal inflammation. Panel D shows the thickening of the ileum wall by small intestine computed tomography enterography (CTE). The daughter in the family is another Patient (Panels E to H). Panel E shows her anal fistula at disease onset. Endoscopy showed intestinal poly-ulcers in Panel F. A biopsy showed non-specific granulomatous inflammation, as shown in Panel G, and the higher magnification of the pathology shown in Panel H reveals negative acid-fast staining granulomas. All of the images were collected in March 2012 in Zhongnan Hospital of Wuhan University.
An additional 10 healthy members’ blood DNA samples from family A were also taken as Sanger sequencing controls to validate the co-segregation of the mutations in the CD family.
In addition, 278 sporadic CD and 123 sporadic UC patients were enrolled from the Inflammatory Bowel Disease Center of Zhongnan Hospital of Wuhan University (131 CD, 76 UC), Renji Hospital of Shanghai Jiaotong University School of Medicine (40 CD) and the Institute of Digestive Disease of The Chinese University of Hong Kong (107 CD, 47 UC) from January 2001 to December 2012; 401 HC were from Wuhan, China (Table 1).
Table 1. Characteristics of 401 sporadic inflammatory bowel disease (IBD) patients and 401 healthy controls.
| Index | CD 278 | UC 123 | HC |
| Male (n = ) | 176 | 72 | 239 |
| Female (n = ) | 102 | 51 | 162 |
| Average age (years) | 32.25±13.38 | 35.40±10.63 | 36.42±12.42 |
Moreover, we collected 25 young and intractable CD cases (Table 2), including a pair of monozygotic twins (Patient ID in Table 2: 24 and 25) and 23 cases selected from 131 sporadic CD patients of Hubei province. Another CD family (family B) was also from Wuhan city.
Table 2. Data of 25 young and intractable CD patients.
| PatientID | Age atdiagnosis(year) | Sex | Diseaselocation | Treatment | |||||
| Diagnosticanti-TB | Aminosalicylates | Corticosteroids | Immunosuppressiveagentes | Biologicalagentes | Surgery | ||||
| 1 | 25 | Female | Terminal ileum | NO | YES | NO | NO | NO | YES |
| 2 | 15 | Male | Terminal ileum | NO | YES | YES | NO | YES | NO |
| 3 | 24 | Male | Terminal ileum | NO | YES | NO | YES | NO | NO |
| 4 | 34 | Male | Terminal ileum and ascendingcolon | YES | YES | NO | YES | NO | YES |
| 5 | 16 | Male | Terminal ileumand sigmoid colon | YES | YES | YES | YES | YES | NO |
| 6 | 14 | Male | Terminal ileum | YES | YES | NO | YES | YES | NO |
| 7 | 19 | Female | Terminal ileum and right sidedcolon | NO | YES | YES | YES | YES | YES |
| 8 | 17 | Male | Terminal ileum and descendingcolon | YES | YES | YES | YES | NO | NO |
| 9 | 21 | Male | Small intestine | NO | YES | YES | YES | NO | NO |
| 10 | 20 | Male | Terminal ileum and right sidedcolon | YES | YES | YES | YES | YES | YES |
| 11 | 24 | Female | Terminal ileum | NO | YES | NO | NO | YES | NO |
| 12 | 21 | Male | Terminal ileum | YES | YES | YES | YES | YES | NO |
| 13 | 22 | Female | Terminal ileumand sigmoid colon | YES | YES | YES | YES | YES | NO |
| 14 | 23 | Male | Terminal ileum and right sidedcolon | NO | YES | YES | YES | YES | NO |
| 15 | 23 | Male | Small intestine | NO | YES | YES | YES | YES | NO |
| 16 | 17 | Male | Terminal ileum and right sidedcolon | NO | YES | YES | YES | YES | NO |
| 17 | 13 | Female | Terminal ileum and ascendingcolon | YES | YES | YES | YES | YES | YES |
| 18 | 24 | Female | Terminal ileum | NO | YES | YES | YES | YES | NO |
| 19 | 25 | Male | Terminal ileum and ascendingcolon | NO | YES | YES | YES | YES | YES |
| 20 | 11 | Male | Terminal ileum and right sidedcolon | YES | YES | YES | YES | YES | YES |
| 21 | 21 | Female | Terminal ileum and ascendingcolon | YES | YES | YES | YES | NO | NO |
| 22 | 14 | Female | Terminal ileum and right sidedcolon | NO | YES | YES | YES | NO | YES |
| 23 | 11 | Male | Terminal ileum and right sidedcolon | YES | YES | YES | YES | YES | YES |
| 24 | 26 | Male | Terminal ileum | NO | YES | YES | YES | NO | YES |
| 25 | 29 | Male | Terminal ileum | NO | YES | YES | YES | YES | YES |
TB: tuberculosis.
DNA Extraction
Genomic DNA was extracted from EDTA-anticoagulated peripheral venous blood samples using a QIAamp DNA Blood Midi Kit (Qiagen, Germany) according to the manufacturer’s instructions.
Whole Exome Sequencing and Variant Detection
Using an E210 ultrasonicator (Covaris, MA, USA), the genomic DNA samples were randomly fragmented into 250–300 bp fragments and subjected to library preparation according to NimbleGen’s standard protocol. Target region enrichment was performed for the shotgun libraries using the NimbleGen SeqCap EZ custom design kit (NimbleGen, Madison, WI, USA), which consisted of SeqCap EZ Human Exome Library v2.0 and a continuous region covering the MHC genes. The enriched shotgun libraries were sequenced using the Hiseq2000 platform, and 90-bp paired-end reads were generated. Raw image data and base calling were processed by Illumina Pipeline software version 1.7 with the default parameters. Quality control for the reads was performed by discarding adaptor-containing reads and low-quality reads. For SNP calling, SOAP aligner [13] was used to align the reads to the human reference genome (hg19), and SOAP snp [14] was then used to assemble the consensus sequence and call SNPs. As another quality control, low-quality SNPs satisfying one of the four following criteria were discarded: (i) genotype quality<20; (ii) total reads covering the variant site<4; (iii) estimated copy number >2; (iv) distance from the nearest SNP<5 bp (except for SNPs present in dbSNP). For indel calling, high-quality reads were aligned to the human reference genome using BWA (version 0.5.9-r16) [15]. GATK Indel Realigner was used to realign reads around insertion/deletion sites, and then small indels were called using the IndelGenotyperV2 tool from GATK (version v1.0.4705) [16], [17]. Indels were called as heterozygous and homozygous if indel-supporting reads consisted of 30–70% and >70% of the total reads, respectively. SNP and indel detection was performed only for the targeted regions and flanking regions within 200 bp of the targeted regions.
Variant Annotation and Prioritization
The detected variants were annotated based on four databases, including NCBI CCDS, RefSeq, Ensembl and Encode (http://genome.ucsc.edu/ENCODE/). Exclusion steps were taken to help identify candidate mutations. Variants falling within intergenic, intronic and untranslated regions and synonymous substitutions were excluded; variants documented in 4 public genetic variant databases, including dbSNP132, 1000 Genomes, HapMap and YH (http://yh.genomics.org.cn/), with an allele frequency >0.5% (except for YH) were rejected; and variants shared by 2 exome sequenced cases and absent from 2 exome sequenced controls were kept. Additionally, we used the following criteria to evaluate and prioritize the candidate genes: (i) SIFT (http://sift.bii.a-star.edu.sg/), MutationTaster (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) and PMut (http://mmb.pcb.ub.es/PMut/) were used to predict whether single amino acid changes in genes would alter the protein function; (ii) the conservation of candidate mutations was analyzed by evaluating the GERP score (http://snp.gs.washington.edu/SeattleSeqAnnotation137/); (iii) the candidate mutations’ total frequency of occurrence in the EVS (http://evs.gs.washington.edu/EVS/) and the BGI in-house database was analyzed and (iiii) the tissue distributions and functions were analyzed using the online tools BioGPS (http://biogps.org/), Entrez Gene (http://www.ncbi.nlm.nih.gov/gene) and Proteinatlas (http://www.proteinatlas.org/).
As a final step, we compared and prioritized the remaining candidate genes using 4 internet tools: GeneDistiller2 (http://www.genedistiller.org/), SUSPECTS (http://www.genetics.med.ed.ac.uk/suspects/index.shtml), ToppGene (http://toppgene.cchmc.org/) and Endeavour (http://www.esat.kuleuven.be/endeavour). To prioritize the SNVs and genes, we chose 20 reported CD susceptibility genes (NOD2, ATG16L1, STAT3, IL23R, IL10R2, IL10R1, JAK2, ICOSLG, CDKAL1, MST1, PTGER4, IRGM, TNFSF15, ZNF365, NKX2-3, PTPN2, PTPN22, IL12B, XIAP and ITLN1) as the training set.
Validation Phase
All shared SNVs of the two affected individuals were verified for all members acquired from family A to detect co-segregation, by direct polymerase chain reaction (PCR) amplification followed by Sanger sequencing (PCR primers are listed in Table 3, Invitrogen). The sequencing reactions were conducted on an ABI 3730XL DNA Analyzer.
Table 3. The PCR primers of 22 candidate SNPs by Sanger Sequencing.
| SNP_ID | Gene | F-primer sequences (5′-3′) | R-primer sequences (5′-3′) |
| chr17_55183450 | AKAP1 | TCAGAGTCCTCGGGCATT | CTGCTACATACTCTTCCTCC |
| chr20_30232655 | COX4I2 | ACAGTCCTTGGGGTCTAA | CCACTGCTTCTTCTCATAG |
| chr9_110249480 | KLF4 | AGTCCCGCCGCTCCATTA | TCTTTGGCTTGGGCTCCT |
| chr1_26368197 | SLC30A2 | ACTGCCTTATTCTGAACTGT | GAAGCATAATCCTCACCC |
| chr7_73279329 | WBSCR28 | GAGAATCGCCCGAAACC | CCAGGCACTGAGCAAGG |
| chr9_125582872 | PDCL | GATTCTTGTTGTGCCTCAG | TTCCTGGTGAACTGACTGC |
| chr6_90418252 | MDN1 | AACCTCTTCCCCATCAT | TCCAACACCCCACAACT |
| chr20_420894 | TBC1D20 | GACCTGACACCTGCCTTTC | ACCCAGCATTTCCCAACT |
| chr9_2717768 | KCNV2 | CCACAGCCAGGAGGAAA | CTCGTAGTCGTCGCACA |
| chr16_20492206 | ACSM2A | CAGGGCAGGGGATTTAG | TTGCTGGATCGTATGGTAGTT |
| chr15_41275952 | INO80 | AGCCAAAGCAGCCTCAAC | GGAATCAGGACCTTACCC |
| chr6_168366533 | MLLT4 | GAAGCAGGAGGCTGAGAA | TTGAGGTAGGAGGCGTTT |
| chr3_196921405 | DLG1 | GGTAAGAAATGAGCAATCAATATTCAG | GGGCGAACCTACATGAAAGAATA |
| chr1_1470881 | TMEM240 | GACGCCTCCGAGAACTACTTTG | ACAGCTTGGGCAGCCAGGTC |
| chr16_16170185 | ABCC1 | AACCCGTGGCTGATGTC | TGTCCAAGGCTGCTGTA |
| chr15_39885853 | THBS1 | TGGGTGCTGAGGATGTC | TGGTGATGCTGGGAACT |
| chr10_116225553 | ABLIM1 | TTCCTTGGCAGTGTTTG | GGAAATGTTTAGTCGTTGA |
| chr7_98602860 | TRRAP | TTTCCCGTGACAGTTCG | CTCTTGGTGGTCTCCTTT |
| chr6_152536152 | SYNE1 | TTGGCTTTTCGCTATTC | ACCTTGACTGCGGACTT |
| chr14_21860964 | CHD8 | GCCCAAGGTAACAAACAG | CCAGGAGTCAATGAGGGA |
| chr7_99160120 | ZNF655 | TATGGGCTTTATTCCGTAG | CGGAGAAGACGATGTGAA |
| chr7_87051466 | ABCB4 | ATCCAAGTGGGCGTTTT | TGAATGTCTGCTGAGGG |
Genotyping was conducted by the MassARRAY (MALDI-TOF MS) method using the SEQUENOM System (Sequenom, Inc.) to screen the candidate genes in an additional 401 HC individuals (278 sporadic CD patients and 123 UC cases), and the data were analyzed using TYPER 4.0 software. The primer sequences for genotyping were designed and synthesized using Primer 5.0 software (PCR primer sequences are listed in Table 4, and the primers were synthesized by Invitrogen). To further study the genes (DLG1 and PDCL) that we identified through the series of steps listed above, we applied PCR amplification followed by Sanger sequencing to examine all of the exons of DLG1 and PDCL in 25 young and intractable CD cases (the PCR primer sequences are listed in Table 5).
Table 4. The primers of the 22 candidate SNPs for the MassARRAY method.
| SNP_ID | Gene | F-primer sequences (5′-3′) | R-primer sequences (5′-3′) |
| chr17_55183450 | AKAP1 | ACGTTGGATGAGAGGGCAAGAGAGACAGGT | ACGTTGGATGACAGAGCTTCTTCAAGCACC |
| chr20_30232655 | COX4I2 | ACGTTGGATGAGCGCATGCTGGACATGAAG | ACGTTGGATGCTGCTTCTTCTCATAGTCCC |
| chr9_110249480 | KLF4 | ACGTTGGATGTCTTTGGCTTGGGCTCCTCT | ACGTTGGATGATGATGCTCACCCCACCT |
| chr1_26368197 | SLC30A2 | ACGTTGGATGAACCTTGACCATCCTGAGAG | ACGTTGGATGAAGAGCAAAAAGGGAGCCAC |
| chr7_73279329 | WBSCR28 | ACGTTGGATGTGATGGCTGACGGTTGTCTC | ACGTTGGATGGGAGCAGGAAATTATAGAGG |
| chr9_125582872 | PDCL | ACGTTGGATGTGACTCTGAAGGAGTTTGCC | ACGTTGGATGATTCGCTGCTTCCGGTACTG |
| chr6_90418252 | MDN1 | ACGTTGGATGTTTGATGGACTTTGACCCAC | ACGTTGGATGTGCAGCTGATTCTAAAAGGG |
| chr20_420894 | TBC1D20 | ACGTTGGATGATGGGTGATGGTGAACCCAG | ACGTTGGATGACCCACTGATGCCGATTTAC |
| chr9_2717768 | KCNV2 | ACGTTGGATGAGCCATGCTCAAACAGAGTG | ACGTTGGATGCCTCATTCTCCGTCGTGTTC |
| chr16_20492206 | ACSM2A | ACGTTGGATGGGTAGAGAATGCACTGATGG | ACGTTGGATGACGGGGTCTGGGCTGCTGAT |
| chr15_41275952 | INO80 | ACGTTGGATGACAACCAAACCAGTGCTGGG | ACGTTGGATGGTCTCAGATACCGTGAATGG |
| chr6_168366533 | MLLT4 | ACGTTGGATGAGACAGCACGACGAGGCGG | ACGTTGGATGTAGTCCCGGGGAAGCGGAG |
| chr3_196921405 | DLG1 | ACGTTGGATGGAACCAATTCTGGACCTATC | ACGTTGGATGGGATGAAGATACACCTCCTC |
| chr1_1470881 | TMEM240 | ACGTTGGATGAGCCGCCTGACCGCCCCTGT | ACGTTGGATGTGCACAGCTTGGGCAGCCAG |
| chr16_16170185 | ABCC1 | ACGTTGGATGTGTCCCTGACATGTCTCTGT | ACGTTGGATGTGAATGTGGCATTCCTCACG |
| chr15_39885853 | THBS1 | ACGTTGGATGTGGCGAGCACCTGCGGAAC | ACGTTGGATGTCCAGGGCTTTGCTTCTTAC |
| chr10_116225553 | ABLIM1 | ACGTTGGATGTGTACACAGGGGAGTTGATG | ACGTTGGATGAGGATGTTCGGGATCGGATG |
| chr7_98602860 | TRRAP | ACGTTGGATGTGCAACACACGCTCCTCTC | ACGTTGGATGTGGCAAGATCTACCCATACC |
| chr6_152536152 | SYNE1 | ACGTTGGATGCTTCCTTCTAGGGACAGATG | ACGTTGGATGGGTAACCTATATCCAAGCTC |
| chr14_21860964 | CHD8 | ACGTTGGATGCACAGCTAGTACTCAGACTC | ACGTTGGATGCGAGGTCAATACGGTTTATC |
| chr7_99160120 | ZNF655 | ACGTTGGATGGATAAACCGAATAATAAGG | ACGTTGGATGACCTCTACAGAGAAGTGATG |
| chr7_87051466 | ABCB4 | ACGTTGGATGGCAGAAGTGCAACATATTCTC | ACGTTGGATGACCTACCTGAAGGAAGAAAG |
Table 5. The PCR primers of all exons of DLG1 and PDCL.
| Primer ID | F-primer sequences (5′-3′) | R-primer sequences (5′-3′) | Annealing temperature (°C) |
| DLG1 | |||
| Exon 1 | CCGACTTCTGTCTGTTCTT | GGACCGTGCTGTCTCAT | 54 |
| Exon 2 | CTCCTCCGTTTTCTAATG | GTTACCGAATGCCTCAG | 51.5 |
| Exon 3 | GTTAAGTAGTTTGCCTGAACTTGTAGC | CAGATGAAGCCTTGTTGAGGTCT | 62 |
| Exon 4 | GGTAAGAAATGAGCAATCAATATTCAG | GGGCGAACCTACATGAAAGAATA | 55.5 |
| Exon 5 | TTTATCTTTATGGCACAGC | AAATGGCAAATCCTGACT | 51.5 |
| Exon 6 | TTCTGTTTGGTGCTGGAG | GGTCTTCGCATTTGTATC | 55.5 |
| Exon 7 | CAGAGAAGGATCGGAGGTTGA | GTAAATGGAAACTCTTGGGACTATC | 58.8 |
| Exon 8 | CCTCCAGAACAAGTCCA | GTATTTATCCCTTATCCAGTC | 51.5 |
| Exon 9 | TGTTCCTTTTGCTGGCCCTT | ATGACTGCACCACTGGACTC | 63 |
| Exon 10 | TTCGTAACTCTAGGAGCAGCTGT | CTGTGCATACAAGCCCTCAAC | 61 |
| Exon 11 | AGACTGGGAGAATAGGAGG | TCACTAATGGCATCACAAC | 55.5 |
| Exon 12 | TTGAGACTAACCTGGGCAACAT | AAGGACAATTTACCAAGCCTCAA C | 61 |
| Exon 13 | CTTCTAAGTAGGGGCAGTG | AATAGGTCCAGTGAAAATAAC | 54 |
| Exon 14 | CAGTAGGCGTGAGAATGTGGC | GCCTGGGCAGTAAGAGTGGA | 63 |
| Exon 15 | GATTACTGCTGTCTGATGC | GCCTCCTTTGCTACTATG | 55.5 |
| Exon 16 | TCAATATAACTTACCATTGGATTACAATC | AGTACTATTACCTGTAGTTGCCATGCT | 57 |
| Exon 17 | TTAAACTCAGAAATGGTGCCTCA | GGTCTGTGAAATGGGTGCTTG | 61 |
| Exon 18 | AGGTATAAATGAACTATGCTGTCTGAA | CCTTGAAGACAATTAGCAACCTG | 58.8 |
| Exon 19 | AGTTTGTCCCCTTTGCC | TCAGAATCCCTCCACCC | 55.5 |
| Exon 20 | AAATAAAGGAGTAGCACATAGC | GAAAGAAGTGGGATAAACAG | 54 |
| Exon 21 | CATCTTTGGTTGATGGTAGAGTGAG | AGAAAGGACAATAATATGGAGGATG | 58.8 |
| Exon 22 | ATCCATCCTCCATATTATTGTCCT | ACCCGGCCCTTATCTCCT | 57 |
| Exon 23 | TTTCATTTCCTATCTAAAGTTTGCTG | ATGGTTCTGCCTCACATTCTGT | 57 |
| Exon 24 | TGTGTCATCTCTCCTTTGCCA | GAGCCGAGTCATACCATTGC | 62 |
| Exon 25 | CTATGGGATTGTACCCAGTTTCC | GGTCAGGCCATTCCATCTTC | 57 |
| PDCL | |||
| Exon 1 | TGTCCTGGAAATTGTAGGATCTCA | GACTAGGTTACCTCTGAAAGTGGGA | 60.5 |
| Exon 2 | ATGTTGGGCATTAGCTTGGC | TTTGACAGGGCTCTATGATTTCTC | 60.5 |
| Exon 3-1 | TCAAGTGATCCGCTCGTCT | AGCTTCAAGGTCCACAGCA | 60.5 |
| Exon 3-2 | GCCAGCAGTCAGTTCACCAG | TTTGACAGGGCTCTATGATTTCTC | 60.5 |
SPSS17.0 statistical software was used for statistical analysis, the measurement data were expressed as means +/− standard deviation (SD). PLINK was performed on analysis of genotype data. P values<0.05 were considered as significant.
Results
Whole Exome Sequencing of the CD Family
Whole exome sequencing was performed on DNA extracted from the peripheral blood of 4 members of Family A using next-generation sequencing technology. As shown in Table 6, we obtained at least 88.5 million reads that mapped to the target region for each exome, more than 98.5% of the target region was covered and the mean depth of the target region was 128.64×, 148.90×, 202.26× and 158.25×. The summary statistics of the total quality-passing SNPs and indels are all listed in Table 6.
Table 6. Summary of original exome sequencing data of four familial individuals.
| Exome Capture Statistics | Daughter | Father | Grandmother | Mother |
| Target region (bp) | 48959543 | 49062223 | 48959543 | 48959543 |
| Raw reads | 243896508 | 204592452 | 253503938 | 192147514 |
| Raw data yield (Mb) | 21951 | 18413 | 22815 | 17293 |
| Reads mapped to genome | 204193470 | 145810600 | 202993882 | 156292035 |
| Reads mapped to target region(2) | 88581652 | 101995817 | 138982919 | 108872112 |
| Data mapped to target region (Mb) | 6298.25 | 7305.56 | 9902.46 | 7748.06 |
| Mean depth of target region (X) | 128.64 | 148.90 | 202.26 | 158.25 |
| Coverage of target region (%) | 98.77 | 98.56 | 98.81 | 98.75 |
| Average read length (bp) | 89.87 | 89.84 | 89.78 | 89.85 |
| Total quality-passing SNPs | 116950 | 114204 | 119780 | 117371 |
| Total quality-passing indels | 7442 | 7361 | 7773 | 7500 |
Bioinformatic Analysis Identifies 22 Candidate Genes
In total, 82 variants shared by the 2 cases remained through the exclusion of 4 public genetic databases (the procedures are shown in Table 7), and no reported IBD single nucleotide variant was found. After performing filtering steps for gene function and mutation prediction, we obtained 22 candidate genes (Table 8). Using 4 internet tools, we acquired the top 6 genes from the 22 candidates: THBS1, KLF4, SYNE1, CHD8, PDCL and DLG1. These genes were the most likely to be the genetic cause of the 2 affected patients.
Table 7. Filtration of SNPs/Indels.
| Individual ID | Grandmother | Mother | Daughter | Father |
| Total SNPs and indels | 173991+7773 | 172848+7500 | 172005+7442 | 173055+7361 |
| Quality-passing SNPs and indels | 119780+7773 | 117371+7500 | 116950+7442 | 114204+7361 |
| Protein-disrupting SNPs and indels (PDSI) | 14553+1369 | 14345+1314 | 14581+1280 | 14547+1294 |
| PDSI after filtering against dbSNP | 2144+382 | 2138+366 | 2108+348 | 2129+359 |
| PDSI after filtering against dbSNP+1000 Genomes | 1459+220 | 1498+221 | 1469+189 | 1448+208 |
| PDSI after filtering against dbSNP+1000 Genomes+HapMap | 1457+220 | 1497+221 | 1467+189 | 1446+208 |
| PDSI after filtering against dbSNP+1000 Genomes+HapMap+YH | 1420+220 | 1460+221 | 1438+189 | 1413+208 |
| PDSI after filtering against dbSNP+1000 Genomes+HapMap+YH+inhouse dataand fitting a dominant model (shared by two cases) | 0 | 0 | 82+0 | |
| Filtered candidate genes | 22 | |||
| Sanger sequence for validation | 22 | |||
Table 8. List of 22 candidate genes and mutations prediction.
| NO | Chromosome | Position | Reference | Gene name | Codons | SIFT Prediction | MutationTaster Prediction | ConsScore GERP |
| 1 | chr6 | 90418252 | C | MDN1 | GAC7861CAC | DAMAGING | polymorphism | 2.23 |
| 2 | chr14 | 21860964 | C | CHD8 | CGT5636CAT | DAMAGING | disease causing | 5.34 |
| 3 | chr9 | 125582872 | T | PDCL | GAT398GGT | DAMAGING | disease causing | 5.47 |
| 4 | chr17 | 55183450 | G | AKAP1 | GTG625ATG | DAMAGING | polymorphism | 4.22 |
| 5 | chr15 | 39885853 | G | THBS1 | GGA3251GAA | DAMAGING | disease causing | 5.78 |
| 6 | chr16 | 16170185 | G | ABCC1 | GGG1915TGG | DAMAGING | disease causing | 4.11 |
| 7 | chr1 | 1470881 | G | TMEM240 | TCG380TTG | DAMAGING | disease causing | 3.37 |
| 8 | chr7 | 73279329 | C | WBSCR28 | CAG79AAG | DAMAGING | polymorphism | 4.43 |
| 9 | chr9 | 2717768 | C | KCNV2 | TCC29TGC | DAMAGING | disease causing | 4.45 |
| 10 | chr7 | 99160120 | A | ZNF655 | – | – | – | 3.92 |
| 11 | chr15 | 41275952 | G | INO80 | – | – | – | 2.69 |
| 12 | chr10 | 116225553 | G | ABLIM1 | CGG1345TGG | DAMAGING | disease causing | 3.37 |
| 13 | chr1 | 26368197 | T | SLC30A2 | ATG685GTG | DAMAGING | disease causing | 5.6 |
| 14 | chr20 | 30232655 | T | COX4I2 | GTG464GCG | DAMAGING | disease causing | 4.38 |
| 15 | chr6 | 167570520 | G | GPR31 | ACG800ATG | DAMAGING | polymorphism | 2.65 |
| 16 | chr6 | 168366533 | G | MLLT4 | GGA4993AGA | DAMAGING | disease causing | 5.03 |
| 27 | chr16 | 20492206 | C | ACSM2A | ACG1472ATG | DAMAGING | polymorphism | 3.26 |
| 18 | chr20 | 420894 | C | TBC1D20 | GTG766ATG | DAMAGING | disease causing | 5.65 |
| 19 | chr9 | 110249480 | G | KLF4 | – | – | – | 3.45 |
| 20 | chr3 | 196921405 | A | DLG1 | ATC374ACC | TOLERATED | disease causing | 5.17 |
| 21 | chr6 | 152536152 | C | SYNE1 | CGT22022CAT | TOLERATED | disease causing | 5.07 |
| 22 | chr7 | 87051466 | T | ABCB | ATT2287GTT | TOLERATED | polymorphism | 4.85 |
Sanger Sequencing and Genotyping Combined with Bioinformatic Analyses Identifies DLG1 as a Potential Susceptibility Gene
Sanger sequencing confirmed the presence of the 22 mutations in the affected father and daughter. 10 healthy members of family A were sequenced to test for these variants. We found that one family member carried the variant in the KLF4 gene. The other 21 mutations absent in healthy family members showed co-segregation.
The genotyping of the 22 SNVs indicated that 8 variants in THBS1, SYNE1, CHD8, TMEM240, AKAP1, COX4I2, ZNF655 and KCNV2 were positive in 401 HC, whereas the other 14 variants were negative. We again focused on the 6 top candidate genes (THBS1, KLF4, SYNE1, CHD8, PDCL and DLG1) identified through the prioritization analysis. In contrast to THBS1, KLF4, SYNE1 and CHD8, none of the 401 HC was found to carry PDCL or DLG1 mutations. Subsequent genotyping of 22 SNVs in 401 sporadic IBD cases indicated that one female CD patient aged 21 years carried a mutation in DLG1 (Table 9), and no patients had variation in PDCL. A PLINK analysis showed the variant in DLG1was of significance (P<0.05).
Table 9. Distributions of rare variants in the DLG1 gene.
| Patient ID | Gender | Age (years) | Nucleotidechange | Amino acidchange | ChromosomePosition | Exon | Sequencing method |
| CD Family A | |||||||
| Father(diagnosed) | Male | 44 | c.374T>C | p.I125T | chr3_196921405 | 4 | Exome |
| Daughter(diagnosed) | Female | 16 | c.374T>C | p.I125T | chr3_196921405 | 4 | Exome |
| Mother(unaffected) | Female | 42 | – | – | – | – | Exome |
| Grandma(unaffected) | Female | 81 | – | – | – | – | Exome |
| CD Family B | |||||||
| Case 1(diagnosed) | Male | 39 | c.833G>A | p.R278Q | chr3_196865242 | 9 | Direct PCR sequencing |
| Case 2(diagnosed) | Male | 42 | c.833G>A | p.R278Q | chr3_196865242 | 9 | Direct PCR sequencing |
| Case 3(diagnosed) | Female | 32 | c.833G>A | p.R278Q | chr3_196865242 | 9 | Direct PCR sequencing |
| Case 4(diagnosed) | Female | 24 | c.833G>A | p.R278Q | chr3_196865242 | 9 | Direct PCR sequencing |
| CJ 5(undiagnosed) | Female | 56 | c.833G>A | p.R278Q | chr3_196865242 | 9 | Direct PCR sequencing |
| CJ 6(undiagnosed) | Female | 6 | c.833G>A | p.R278Q | chr3_196865242 | 9 | Direct PCR sequencing |
| CJ 7(undiagnosed) | Female | 6 | c.833G>A | p.R278Q | chr3_196865242 | 9 | Direct PCR sequencing |
| CJ 8(undiagnosed)) | Male | 7 | c.833G>A | p.R278Q | chr3_196865242 | 9 | Direct PCR sequencing |
By examining all of the exons of PDCL and DLG1 in 25 young and intractable CD patients, we found two cases (Table 2, Patient ID are 3 and 4) who carried another variant in DLG1 (Figure 2, exon 9, c.833G>A, p.R278Q). We traced Patient 3, 4 and their families, and found that two cousin sisters (Cases CJ2 and CJ3) and one brother (Case CJ4) of Patient 4 who were unexpectedly found to have ulcers in the terminal ileum by endoscopy, and a biopsy showed non-specific chronic inflammation. After being treated with 5-ASA and azathioprine, four affected cases in this family have almost achieved their colonic mucosal healing. Cases CJ2, CJ3 and CJ4 were all found to be carriers of mutation R278Q (c.833G>A) by Sanger sequencing, and the family was called family B. We found 4 unaffected carriers (CJ5, CJ6, CJ7 and CJ8) of this variant after sequencing the other 15 members of family B, and these individuals will be followed up. CJ5 received a diagnosis of rheumatic heart disease with arthritis. The variants and carriers of DLG1 are listed in Table 9. Neither of the monozygotic CD twins carried any mutation in all 3 exons of PDCL or in all 25 exons of DLG1.
Figure 2. Chromatogram of DLG1 gene mutations.

The Sanger sequence traces from normal human controls are shown in panel A and B; the mutations were heterozygous at the corresponding locus (orange arrows indicating) in panel C and D.
Bioinformatics analyses were used to dissect the two non-synonymous mutations of DLG1 found in the study described above. MutationTaster showed that the variant in DLG1 (Figure 2, c.374T>C, p.I125T) was likely to be disease-causing. We compared the SNV sequence of species at different evolutionary distances by GERP and found that the amino acid substitution of DLG1 was highly conserved. Regarding another variant of DLG1 (Figure 2, exon 9, c.833G>A, p.R278Q), the PMut analysis of the mutation indicated that it is pathological (http://mmb.pcb.ub.es/PMut/), and the prediction from PolyPhen-2 was that the mutation was most likely damaging; however, the MutationTaster analysis indicated polymorphism, and SIFT predicted the mutation to be tolerated.
Discussion
Rare and low-frequency variants might have substantial effect sizes in complex disorders such as IBD [18]. A main goal of human genetic studies is to identify uncommon variants that play important roles in pathogenesis and reveal the familial transmission of diseases [6], [8]. Furthermore, uncommon alleles shared by affected individuals in a family are more prone to familial clustering of disease than common alleles carried in a population.
In this study, we applied whole exome sequencing to anatomize the genetic background of a Chinese family with CD and successfully identified genetic variants in the coding regions of the DLG1 gene that may be associated with increased risk of CD. We first identified a novel SNV c.374T>C (p.I125T) in exon 4 of DLG1 through whole exome sequencing and bioinformatic analysis. In subsequent validation studies, we also identified 4 CD patients of another Han Chinese family harbored the variant c.833G>A. Altogether these data suggest that DLG1 is a susceptible gene for CD.
DLG1 encodes a multi-domain scaffolding protein, which may have a role in septate junction formation, signal transduction, cell proliferation, synaptogenesis and lymphocyte activation (http://www.ncbi.nlm.nih.gov/gene/). The DLG1 protein is composed of an N-terminal L27b oligomerization domain, a proline-rich domain (PRD), three PDZ (PSD-95, Dlg and ZO-1) domains, an SH3 (Src Homology 3) domain and a catalytically inactive GUK (GUanylate Kinase) domain. During antigen recognition, these modular domains allow DLG1 to co-localize with synaptic actin, translocate into sphingolipid-rich microdomains within the IS and associate with Lck, ZAP-70, Vav, WASp Ezrin and p38 [19]. DLG1 has been shown to play roles in T cell polarity and T cell receptor signal specificity [20], [21], and be involved in the generation of memory T cells [22]. The loss of DLG1 leads to increased invasion in response to pro-tumorigenic cytokines, such as IL-6 and TNF-α [23], [24].
In accord with the suggested autoimmune nature of CD, strong evidence has implicated T cells and T-cell migration to the gut in initiating and perpetuating the intestinal inflammatory process and tissue destruction [25], [26]. Anti-cytokine agents are therefore likely to be useful in the treatment of IBD [27], [28]. After intravenous injection with six cycles of infliximab, the affected daughter in Family A has almost achieved mucosal healing of her colonic disease and was likely to have a better prognosis than those DLG1 mutation carriers who did not accept infliximab treatments in our study. It was corroborative evidence that DLG1 was causative for the CD patients of the two Chinese families.
Complex human disease is a large collection of individually rare, even private variants [29]. A single locus can harbor both common variants of weak effect and rare variants of strong effect [30]. The results of our study of two CD families indicated genetic heterogeneity and susceptibility. We analyzed family A using an autosomal dominant model, and several factors were important to the success of this study.
First, according to the database at our center [31], although the incidence of CD and UC is still low, the number of cases and severity of disease are increasing in China [32], [33], which provides the appropriate conditions to recruit patients for the subsequent validations.
Second, a stepwise approach was taken to help narrow down the list of genetic variants responsible for this disease. For the genetic susceptibility of CD, despite the success of GWAS in identifying significantly associated loci [34], the currently identified variants are estimated to account for less than a quarter of the predicted heritability [35]. Uncommon alleles may be maintained at a lower frequency in the population through negative selection, and it is not possible to create a complete catalog in the general population [36]. Therefore, rare causal variants are not likely to be found in public SNP databases and control exomes [37]. We did not find mutations in any reported susceptibility genes that were shared by the affected father and daughter, which suggested that other variants may be associated with CD in these 2 individuals. To predict the impact of nonsynonymous variants, we applied 4 popular methods (PolyPhen2, SIFT, MutationTaster and PMut) [38], [39]. However, none of these methods was perfectly sensitive or specific. Regarding the mutation c.374T>C, SIFT and MutationTaster predicted it to be tolerated and disease causing, respectively. Different prediction algorithms used different information, and each had its own relative merits. It is thought to be better to use predictions from multiple algorithms rather than relying on a single one [40], [41]. We also used several different bioinformatic methods to filter and prioritize the SNVs and genes to increase the robustness of the analysis results.
Finally, to confirm the results and identify the susceptibility gene, we used genotyping and Sanger sequencing methods for validation. Traditional Sanger sequencing is the gold standard for mutation detection [9]. We were able to narrow the scope to only a few genes through these steps. By scanning all exons of DLG1 and PDCL, a nonsynonymous variant c.833G>A of DLG1 was found in family B, thus confirming that DLG1 is a gene whose mutation is associated with high risk.
Some limitations must be addressed. First, IBD patients with family history are rare among Han Chinese. In this family study, there were only two affected members, so the size of the pedigree was small. Second, the patients studied did not have an onset as early as those were previous reported in Caucasian population [42], [43]. Third, because of genetic heterogeneity, the variants appear to be present only in a subset of CD patients, and were not carried by the pair of monozygotic twins studied. Furthermore, in complex diseases, a central problem is that each variant only makes a small contribution to the disorder [44]. Other candidate genes discovered by us, such as THBS1, KLF4, SYNE1, CHD8 and PDCL, may also contribute to CD. However, variation in these genes must be identified in more cases and controls. Additionally, considering that the variant was also present in the unaffected individuals of family B, other disease-causing factors lying outside of our set of candidate genes may also exist [45]. Finally, functional analyses are needed to elucidate the biological role of this gene in CD susceptibility.
In conclusion, we report the discovery of coding region variants in DLG1 in human CD through whole exome sequencing and bioinformatic analysis and identify DLG1 as a potential susceptibility gene for CD in the Chinese population. Our study also demonstrates that whole exome sequencing is an efficient and cost-effective genetic strategy. Bioinformatic approaches are likely to become useful tools for the discovery of genes and to provide important guidance for finding rare variants in a complex disorder. Finding different, rare and pathogenic mutations in the same gene in unrelated individuals with the same phenotype provides important support for our study. However, confirmation of DLG1’s involvement in CD pathogenesis still requires validation in further functional experiments and clinical trials. Personalized medicine is also anticipated to be developed based on definite biological processes and molecular causes.
Acknowledgments
The authors would like to thank the patients with IBD and the healthy controls who volunteered to participate in this study. The works of whole exome sequencing and MassARRAY genotyping were performed in BGI-Shenzhen. We thank editors in American Journal Experts for the valuable contribution to edit and revise the manuscript.
Funding Statement
The study was supported by the Hubei Clinical Center and Key Laboratory of Intestinal and Colorectal Diseases, the Natural Science Foundation of China (No. 81070280) and a grant from the Ministry of Public Health of China (201002020). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Van Assche G, Dignass A, Panes J, Beaugerie L, Karagiannis J, et al. (2010) The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: Definitions and diagnosis. J Crohns Colitis 4: 7–27. [DOI] [PubMed] [Google Scholar]
- 2. Mowat C, Cole A, Windsor A, Ahmad T, Arnott I, et al. (2011) Guidelines for the management of inflammatory bowel disease in adults. Gut 60: 571–607. [DOI] [PubMed] [Google Scholar]
- 3. Freeman HJ (2002) Familial Crohn’s disease in single or multiple first-degree relatives. J Clin Gastroenterol 35: 9–13. [DOI] [PubMed] [Google Scholar]
- 4. Brant SR (2011) Update on the heritability of inflammatory bowel disease: the importance of twin studies. Inflamm Bowel Dis 17: 1–5. [DOI] [PubMed] [Google Scholar]
- 5. Majewski J, Schwartzentruber J, Lalonde E, Montpetit A, Jabado N (2011) What can exome sequencing do for you? J Med Genet 48: 580–589. [DOI] [PubMed] [Google Scholar]
- 6. Teer JK, Mullikin JC (2010) Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet 19: R145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, et al. (2009) Finding the missing heritability of complex diseases. Nature 461: 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nazarian A, Sichtig H, Riva A (2012) Knowledge-Based Method for Association Studies on Complex Diseases. PLoS One 7: e44162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilissen C, Hoischen A, Brunner HG, Veltman JA (2012) Disease gene identification strategies for exome sequencing. Eur J Hum Genet 20: 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, et al. (2011) Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med 13: 255–262. [DOI] [PubMed] [Google Scholar]
- 11. Mao H, Yang W, Lee PP, Ho MH, Yang J, et al. (2012) Exome sequencing identifies novel compound heterozygous mutations of IL-10 receptor 1 in neonatal-onset Crohn’s disease. Genes Immun 13: 437–442. [DOI] [PubMed] [Google Scholar]
- 12. Christodoulou K, Wiskin AE, Gibson J, Tapper W, Willis C, et al. (2013) Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut 62: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- 14. Li R, Li Y, Fang X, Yang H, Wang J, et al. (2009) SNP detection for massively parallel whole-genome resequencing. Genome Re Li R, Li Y, Fang X, Yang H, Wang J, search 19: 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. (2010) The genome analysis toolkit: a MapReduc framework for analyzing next-generation DNA sequencing data. Genome Research 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J, Wang W, Li R, Li Y, Tian G, et al. (2008) The diploid genome sequence of an Asian individual. Nature 456: 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho JH, Brant SR (2011) Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 140: 1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O’Neill AK, Gallegos LL, Justilien V, Garcia EL, Leitges M, et al. (2011) Protein kinase Cα promotes cell migration through a PDZ-dependent interaction with its novel substrate discs large homolog 1 (DLG1). J Biol Chem 286: 43559–43568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Humphries LA, Shaffer MH, Sacirbegovic F, Tomassian T, McMahon KA, et al. (2012) Characterization of in vivo Dlg1 deletion on T cell development and function. PLoS One 7: e45276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xavier R, Rabizadeh S, Ishiguro K, Andre N, Ortiz JB, et al. (2004) Discs large (Dlg1) complexes in lymphocyte activation. J Cell Biol 166: 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gmyrek GB, Graham DB, Sandoval GJ, Blaufuss GS, Akilesh HM, et al. (2013) Polarity gene discs large homolog 1 regulates the generation of memory T cells. Eur J Immunol 43: 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chatterjee S, Seifried L, Feigin ME, Gibbons DL, Scuoppo C, et al. (2012) Dysregulation of cell polarity proteins synergize with oncogenes or the microenvironment to induce invasive behavior in epithelial cells. PLoS One 7: e34343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Surena AL, de Faria GP, Studler JM, Peiretti F, Pidoux M, et al. (2009) DLG1/SAP97 modulates transforming growth factor alpha bioavailability. Biochim Biophys Acta 1793: 264–272. [DOI] [PubMed] [Google Scholar]
- 25. Strober W, Fuss IJ (2011) Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140: 1756–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacDonald TT, Monteleone I, Fantini MC, Monteleone G (2011) Regulation of homeostasis and inflammation in the intestine. Gastroenterology 140: 1768–1775. [DOI] [PubMed] [Google Scholar]
- 27. Plevy SE, Targan SR (2011) Future therapeutic approaches for inflammatory bowel diseases. Gastroenterology 140: 1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Danese S (2012) New therapies for inflammatory bowel disease: from the bench to the bedside. Gut 61: 918–932. [DOI] [PubMed] [Google Scholar]
- 29. McClellan J, King MC (2010) Genetic heterogeneity in human disease. Cell 141: 210–217. [DOI] [PubMed] [Google Scholar]
- 30. Altshuler D, Daly MJ, Lander ES (2008) Genetic mapping in human disease. Science 322: 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao J, Ng SC, Lei Y, Yi F, Li J, et al. (2013) First Prospective, Population-Based Inflammatory Bowel Disease Incidence Study in Mainland of China: The Emergence of “Western” Disease. Inflamm Bowel Dis 19: 1839–1845. [DOI] [PubMed] [Google Scholar]
- 32. Ng SC, Tang W, Ching JY, Wong M, Chow CM, et al. (2013) Incidence and Phenotype of Inflammatory Bowel Disease Based on Results From the Asia-Pacific Crohn’s and Colitis Epidemiology Study. Gastroenterology 145: 158–165. [DOI] [PubMed] [Google Scholar]
- 33. Yi F, Chen M, Huang M, Li J, Zhao J, et al. (2012) The trend in newly diagnosed Crohn’s disease and extraintestinal manifestations of Crohn’s disease in central China: a retrospective study of a single center. Eur J Gastroenterol Hepatol 24: 1424–1429. [DOI] [PubMed] [Google Scholar]
- 34. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, et al. (2010) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet 42: 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clarke AJ, Cooper DN (2010) GWAS: heritability missing in action? Eur J Hum Genet 18: 859–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibson G (2012) Rare and common variants: twenty arguments. Nat Rev Genet 13: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, et al. (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31: 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7: 575–576. [DOI] [PubMed] [Google Scholar]
- 40. Liu X, Jian X, Boerwinkle E (2011) dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 32: 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brunham LR, Hayden MR (2013) Hunting human disease genes: lessons from the past, challenges for the future. Hum Genet 132: 603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Glocker EO, Frede N, Perro M, Sebire N, Elawad M, et al. (2010) Infant colitis–it’s in the genes. Lancet 376: 1272. [DOI] [PubMed] [Google Scholar]
- 43. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, et al. (2009) Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 361: 2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kevin J (2012) Mitchell (2012) What is complex about complex disorders? Genome Biol 13: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gibson G (2009) Decanalization and the origin of complex disease. Nat Rev Genet 10: 134–140. [DOI] [PubMed] [Google Scholar]