

Abstract
Preclinical behavioral pharmacological and neuropharmacological evidence indicates that the NMDA receptor plays an important role in opioid dependence, however, the neural substrates subserving these actions are poorly understood. The central nucleus of the amygdala (CeA) is a critical coordinator of autonomic, behavioral, and emotional systems impacted by opioids, however there is no evidence that the essential NMDA-NR1 (NR1) subunit gene in the amygdala plays a role in opioid dependence. To determine the role of the NR1 subunit gene in the amygdala with respect to physical and psychological opioid withdrawal, a spatial-temporal deletion of this gene was produced by microinjecting a recombinant adeno-associated virus (rAAV) expressing the GFP reporter and Cre recombinase (rAAV-GFP-Cre) into the CeA of adult “floxed” NR1 mice (fNR1). Amygdala microinjection of rAAV-GFP-Cre produced a decrease in NR1 gene expression and protein immunolabeling in postsynaptic sites of neurons without signs of compromised ultrastructural neuronal morphology. Amygdala NR1 gene deletion also did not affect locomotor, somatosensory, or sensory-motor behaviors. In addition, bilateral local NR1 gene deletion did not impact somatic or visceral withdrawal symptoms precipitated by naloxone in morphine-dependent mice. However, there was a significant deficit in the expression of an opioid withdrawal-induced conditioned place aversion in mice with amygdala NR1 deletion. These results indicate that functional amygdala NMDA receptors are involved in aversive psychological processes associated with opioid withdrawal. More generally, spatial-temporal deletion of the NR1 subunit by Cre-loxP technology is an effective means to elucidate the neurogenetic substrates of complex phenotypes associated with drug abuse.
Keywords: Addiction, Cre recombinase, Glutamate, Opioids, Synaptic Plasticity
INTRODUCTION
Chronic exposure to opioids, such as the mu-opioid receptor (μOR) agonist morphine, can lead to dependence, a phenomenon manifested by withdrawal-induced somatic, autonomic, and aversive symptoms (Epstein et al., 2006). Since avoiding withdrawal can contribute to sustained drug use (Kenny et al., 2006), elucidating the neural processes mediating dependence may lead to novel targets for biological treatments of addictive diseases.
Preclinical behavioral pharmacological and neuropharmacological studies indicate that co-administration of NMDA receptor antagonists with morphine attenuate the development or expression of dependence in rodents (Noda and Nabeshima, 2004; Rezayof et al., 2007). A major central mediator of excitatory signaling by the amino acid neurotransmitter glutamate, the NMDA receptor is a heteromer of the essential NMDA-R1 (NR1) subunit and some combination of NMDA-NR2 subunits (Wenthold et al., 2003). Critical features of the NMDA receptor are it’s high permeability to Ca2+ and ability to activate numerous intracellular signaling cascades (Dingledine et al., 1999) involved in cellular, systems, and behavioral plasticity (Tsien, 2000).
Identifying the functional relationships between NMDA receptors and opioid dependence is complicated by the diverse cellular and brain regional localization of these proteins, as well as their complex pharmacology. NMDA receptors are present in extensive areas of the central nervous system, including limbic brain areas that are strongly implicated in opioid plasticity (Gracy et al., 2001), particularly the central nucleus of the amygdala (CeA). Electron microscopic immunohistochemical studies indicate that NMDA receptors have a heterogeneous cellular localization in the extended amygdala and other brain areas implicated in opioid dependence. Although these proteins frequently have a high distribution in somatodendritic (i.e. postsynaptic) sites, NMDA receptors have also been reported to be found in axon terminals (i.e. presynaptic), as well as glia (Gracy and Pickel, 1995; Van Bockstaele et al., 2000). In addition, the CeA may express a population of NMDA receptors that play a role in opioid dependence (Watanabe et al., 2002). However, the behavioral affects of NMDA receptor blockade frequently depend upon the class of NMDA receptor antagonist (i.e. competitive or non-competitive), and other drug-related factors such as time-course of action, drug dose, and receptor selectivity (Svensson et al., 1991; McNally and Westbrook, 1998; Bespalov et al., 1999; Bespalov et al., 2000; Ribeiro Do Couto et al., 2004; Bespalov et al., 2006).
Constitutive gene knockout models offer the possibility of examining the role of NMDA receptors in opioid-dependent behaviors. However, this approach lacks spatial and temporal specificity, and is associated with compensatory developmental effects, and in some cases lethality, as has been shown with NR1 knockout (Forrest et al., 1994). Spatial-temporal NR1 gene deletion in vivo can be produced with a P1 bacteriophage (Cre-loxP) gene splicing system (Schmidt-Supprian and Rajewsky, 2007). This approach relies on the use of transgenic loxP knock-in mice that have strategically placed loxP sites in the NR1 gene (i.e. “floxed NR1” [fNR1] mice). The Cre (cyclization and recombination) gene expresses Cre recombinase, which, when bound to loxP sites, cleaves the intervening genetic sequence, one of the loxP sites, and reattaches the ends to unite the strands. In the absence of Cre, transcription and translation of the regions flanked by loxP sites are unaffected. The Cre-loxP system has been used to produce conditional NR1 gene knockout in various brain regions (Dang et al., 2006; McHugh et al., 2007). However, these studies typically employ a strategy where floxed NR1 mice are crossed with other transgenic mice engineered so that Cre is under the control of neural-site “specific” promoters, which, to our knowledge, are lacking in the amygdala.
An alternative approach involves intracerebral microinjection of a recombinant adeno-associated virus (rAAV) expressing a fusion protein of Cre and a reporter, green fluorescent protein (GFP), termed “rAAV-GFP-Cre” (Kaspar et al., 2002; South et al. 2003). A vector not expressing Cre (rAAV-GFP) is used as a control. This approach has been used to produce a postsynaptic NR1 deletion in spinal cord dorsal horn neurons that selectively blocks NMDA receptor-mediated currents and pain behaviors (South et al., 2003). In the present study, we attempt to delete NR1 in postsynaptic sites of amygdala neurons by directly microinjecting rAAV-GFP-Cre into the CeA of adult fNR1 mice. We also characterize the ultrastructural-neuroanatomical and behavioral consequences of this deletion, particularly with respect to behaviors associated with opioid dependence.
METHODS
Floxed NR1 mice
Experimental protocols involving animals and their care were approved by the Institutional Animal Care and Use Committee at the Weill Medical College of Cornell University and conform to NIH guidelines. Adult (20–30 grams) fNR1 mice were used in these studies. These mice were homozygous for the fNR1 gene, as described previously (South et al 2003). Briefly, these mice had a loxP site placed in the intron that lies between exons 10 and 11 and a second site downstream after exon 22, the last exon. Thus, the two loxP sequences flanked a region of the NR1 gene encoding the 4 membrane domains and the entire C-terminus sequence of the polypeptide chain. The animals used for breeding of the fNR1 line were tested for homozygosity of the loxP sites with the Southern Blot procedure, and the MAX-BAX (Charles River Laboratories, Wilmington, MA) background strain characterization procedure was used to identify breeders that were at least 92–95% C57BL/6 background.
Viral vectors
The rAAV was a single-stranded DNA parvovirus (~4.7 kb) that was engineered without viral coding sequences (South et al. 2003). The inserted transgene included a promoter/enhancer (human cytomegalovirus immediate early gene [CMV]), a multiple cloning site for insertion of the GFP-Cre or GFP coding sequences, and poly A sequences. These were flanked by 145 base pair inverted terminal repeats necessary for rAAV replication and packaging. The Cre enzyme was directed to the nucleus and loxP sites by nuclear localization signals.
Injection of vector
Under deep isoflurane anesthesia, each mouse was placed in a stereotaxic apparatus by securing the head by ear and teeth bars. A 1.2 mm diameter glass pipette (WPI, Sarasota, FL), with a tip pulled to a diameter of ~50 μm, was interfaced to a picospritzer (Picospritzer II, General Valve Corp., Fairfield, NJ) via a pipette holder and plastic tubing. Using bregma as the point of reference, the pipette tip was positioned to the appropriate stereotaxic coordinates, based on the mouse atlas of Hof et al., (2000) as follows: −1.2 mm anterior and 2.5 mm lateral to bregma at a depth of 4.8 mm from the top of the skull. Approximately 200 nl of rAAV-GFP-Cre or rAAV-GFP (5 × 106 viral particles per μl) was slowly injected over a 15-minute interval. To prevent leakage up the pipette tip, the pipette was left in place for 15 minutes. Bone wax was used to cover the hole, and the mice were allowed to recover in their home cages. Since the behavior of untreated and rAAV-GFP fNR1 mice did not differ, it was deemed unnecessary to include a sham-injected group.
Tissue preparation for light microscopy
Mouse brains were prepared for light microscopy according to previously described methods (South et al., 2003). Briefly, mice were deeply anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and their brains were fixed by aortic arch perfusion sequentially with: (a) 15 ml of normal saline (0.9%) containing 1000 units/ml of heparin and (b) 100 ml of 4% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4) all delivered at a flow rate of 100 ml/minute. The brains were removed and post-fixed overnight in 4% paraformaldehyde in PB, and then cryoprotected in 20% sucrose for 24 hours, followed by 15% sucrose for another 1–5 days. Coronal forebrain sections 20 μm thick were cut with a cryostat according to the mouse brain atlas of Hof et al. (2000). For immunoperoxidase labeling, brain sections were incubated for 48 hours in rabbit anti-NR1 antisera (1:100) in 0.1% bovine serum albumin (BSA). After incubation, sections were rinsed in 0.1 M Tris-buffered saline (TBS, pH 7.6) incubated in goat anti-rabbit IgG conjugated to biotin (1:400), rinsed in TBS, and then incubated for 30 minutes in avidin-biotin-peroxidase complex (1:100, Vectastain Elite Kit, Vector Laboratories) in TBS. The bound peroxidase was visualized by reaction for 5–6 minutes in 0.2% solution of 3, 3′-diaminobenzidine and 0.003% hydrogen peroxide in TS. For immunofluorescence labeling, brain section were incubated for 24 hours in a solution containing rabbit anti-GFP along with either mouse anti-neuronal-specific nuclear protein (NeuN: 1:1,000) or mouse anti-glial fibrillary acidic protein (GFAP: 1:1,000) in 0.1% BSA. Sections were rinsed in TBS, and incubated in a solution containing Texas Red-conjugated anti-mouse (1:100) and FITC-conjugated anti-rabbit (1:100) secondary antisera for 2 hours. Following secondary antisera incubations, sections were washed in 0.1% TBS, dehydrated through a series of alcohols and then coverslipped.
Nissl staining
Slide mounted brain sections were dehydrated in a series of alcohols (50–100%) and incubated in chloroform/ethanol (1:1) for 30 minutes. Sections were then rehydrated in a series of alcohols (100-50%), washed in distilled water, and stained in thionin for 3 minutes. Sections were then washed in distilled water and dehydrated in a series of alcohols (50–100%), followed by 100% xylene, and then coverslipped.
In situ hybridization measurement of NR1 mRNA
Levels of NR1 gene expression were measured by non-radioactive in situ hybridization using a 2.2 kb antisense riboprobe whose DNA sequence was deleted by Cre-loxP recombination. Slide mounted cryostat-cut brain sections (20 μm) were incubated in 4% paraformaldehyde (PFA), washed in 1x phosphate buffered saline (PBS), incubated in Proteinase K, and washed in 1x PBS. Sections were again incubated in 4% PFA, washed in 4x PBS, incubated in 0.25% acetic anhydride in 0.1 M Triethanolamine, and washed in 2x SSC, DEPC water. Next, sections were incubated in prehybridization solution at 65°C in a chamber containing towels moistened with 4x SSC and 50% formamide. After incubation, the prehybridization solution was drained off the slide onto an RNAse free towel. Then, sections were hybridized with digoxigenin (DIG)-labeled antisense or sense probes for NR1 (1:1000), coverslipped, and placed in a 65° C oven overnight. After incubation, the coverslips were removed and the hybridized brain sections were washed in 5xSSC at 55° C, followed by 2x SSC and 50% formamide at 65° C. Next, sections were incubated in RNAse buffer at 37°C, followed by RNAse A solution, then RNAse buffer. Sections were then washed sequentially in 2x SSC and 50% formamide at 65°C, 2xSSC, washing buffer, and blocking solution. Sections were subsequently incubated in primary anti-DIG primary antisera conjugated to alkaline phosphatase (1:1000), followed by washing buffer. The sections were then incubated in detection buffer, followed by the chromogen NBT/BCIP (nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate). After approximately 16 hours, slides were rinsed in distilled water and dehydrated through a series of alcohols and xylene, then coverslipped in Permount.
Light microscopic cell counting
For light microscopy, the mounted sections were viewed with a Nikon Microphot-FX microscope (Nikon, Garden City, NY) equipped with a digital CoolSNAP camera (Photometrics, Huntington Beach, CA). The light microscopic images were acquired through an interface between the camera and a Macintosh computer. Cell counting was performed using relative optical density measurements via Microcomputer Imaging Device software (MCID, Imaging Research Inc., Ontario, Canada). Pixel intensity thresholding procedures were performed as per manufacturer’s guidelines. Electronic images were imported into MCID, which automatically calculates a relative threshold level for each image, then adjusted using an object enhancement filter that maximizes the contrast between large objects and background. The CeA was captured, and the highlighted cells were tallied automatically. Three rostrocaudal sections from each hemisphere containing the CeA were counted, averaged, and analyzed by ANOVA. Cell counts were also performed manually to verify the consistency of automated tallies.
Tissue preparation for electron microscopy
Mouse brains were prepared for electron microscopy as previously described (Leranth and Pickel, 1989; Chan et al., 1990). Briefly, mice were deeply anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and their brains fixed by aortic arch perfusion sequentially with: (a) 15 ml of normal saline (0.9%) containing 1000 units/ml of heparin, (b) 50 ml of 3.75% acrolein in 2% paraformaldehyde in 0.1 M PB, and (c) 200 ml of 2% paraformaldehyde in PB, all delivered at a flow rate of 100 ml/minute. The brains were removed and post-fixed for 30 minutes in 2% paraformaldehyde in PB. Coronal forebrain sections 40 μm thick were cut with a vibrating microtome. Tissue sections were next treated with 1.0% sodium borohydride in PB followed by a wash in PB. Sections then were immersed in a cryoprotectant solution (25% sucrose and 2.5% glycerol in 0.05 M PB) for 15 minutes. To enhance tissue permeability, sections were then freeze-thawed in liquid freon and liquid nitrogen. Sections were next rinsed in 0.1 M TS (pH 7.6) and incubated for 30 minutes in 0.5% BSA to minimize nonspecific labeling.
Immunocytochemical procedures for electron microscopy
Brainstem sections were incubated for 48 hours in a solution containing antisera for NR1 (1:100) or NR2 (1:100) in 0.1% BSA. After incubation, sections were rinsed in TBS and prepared for peroxidase or immunogold identification. For immunoperoxidase labeling, sections were incubated in anti-rabbit IgG conjugated to biotin, rinsed in TBS, and then incubated for 30 minutes in avidin-biotin-peroxidase complex (1:100, Vectastain Elite Kit, Vector Laboratories) in TBS. The bound peroxidase was visualized by reaction for 5–6 minutes in 0.2% solution of 3, 3′-diaminobenzidine and 0.003% hydrogen peroxide in TBS. For immunogold labeling, sections were rinsed in 0.01 M PBS (pH 7.4), and blocked for 10 minutes in 0.5% BSA and 0.1% gelatin in PBS to reduce non-specific binding of gold particles. Sections then were incubated for 2 hours in anti-rabbit IgG conjugated with 1 nm gold particles (1:50, AuroProbeOne, Amersham, Arlington Heights, IL). The sections were rinsed in PBS and incubated for 10 minutes in 2% glutaraldehyde. The bound gold particles were enlarged by a 6 minute silver intensification using an IntenSE-M kit (Amersham, Piscataway, NJ). Sections were postfixed in 2% osmium tetroxide in PB for one hour, and dehydrated in a series of alcohols, followed by propylene oxide, and embedded in EM BED 812 (EMS, Fort Washington, PA) between 2 sheets of Aclar plastic. Ultrathin sections from the surface of the tissue were cut with a diamond knife using an ultramicrotome (Ultratome, NOVA, LKB, Bromma, Sweden). These sections were collected on grids, and then counterstained with Reynold’s lead citrate and uranyl acetate.
Ultrastructural analysis
From the CeA, three ultrathin sections at the tissue-surface interface were selected for analysis. Electron microscopic images were captured using a digital camera (Advanced Microscopy Techniques, Danvers MA) interfaced with a transmission electron microscope (Technai 12 BioTwin, FEI, Hillsboro, OR). Digital images were captured and analyzed to determine the number of labeled somatodendritic and axonal profiles. The classification of labeled profiles was based upon descriptions by Peters and co-workers (Peters et al., 1991). Dendrites were identified by the presence of postsynaptic densities, as well as ribosomes and endoplasmic reticulum. However, profiles were also considered dendritic whenever postsynaptic densities were observed, independent of endoplasmic reticulum. Dendritic spines constituted the bulbous heads that received mostly asymmetric synapses. Spine necks were <0.1 μm in diameter and were distinguished from small unmyelinated axons by the presence of a spinous apparatus. Synapses were defined as either symmetric or asymmetric, according to the presence of either thin or thick postsynaptic specializations, respectively. Appositions were distinguished by closely spaced plasma membranes lacking recognizable specializations, and without the presence of interposing astrocytic processes. Morphological parameters, including surface area (perimeter) and cross-sectional area were measured using MCID software. The nuclear form factor represented the relationship between nuclear perimeter to area (Chan et al., 2000). A perfectly circular nuclear profile had a form factor of 1. Any deviation from perfect circularity, such as nuclear invagination, resulted in a reduction in the form factor value. A procedure that has been previously used to demonstrate regional differences in the subcellular distribution of surface proteins in rodent brain was employed for the analysis of gold particle distributions in the present study (Glass et al., 2003; Glass et al., 2005). Briefly, digital images of sampled tissue were captured and analyzed by an experimenter blind to the treatment conditions to determine the following: (1) the number of labeled dendritic profiles, and (2) the number of gold-silver particles present in the cytoplasm, or in contact with the plasma membrane of these structures. Gold particles in contact with any portion of the surface membrane were considered as plasmalemmal. Ultrathin sections from the CeA were analyzed at the epon-tissue interface to minimize potential differences due to penetration of reagents.
Antisera used for immunocytochemistry
Previously characterized antisera against the NR1 subunit (anti-rabbit; Chemicon International, Inc. Temecula, CA), the neuronal marker NeuN (anti-mouse; Chemicon), GFP (anti-rabbit; Invitrogen, Carlsbad, CA), the glial-specific marker GFAP (anti-mouse), and anti-NR2 (anti-rabbit; ABR, Golden, CO) were obtained commercially (South et al., 2003; Glass et al., 2004). A commercially available and previously characterized guinea pig anti-μOR (Chemicon) was used to label the opioid receptor (Glass et al., 2005).
Image enhancement
For preparation of figures light and electron microscopic images were adjusted for contrast and brightness using Photoshop 6.0 software, and imported into Powerpoint or Adobe Illustrator, to add lettering and prepare the composite light microscopic figure. For quantification of cross-sectional area, surface area, as well as minor and major axis length digital images were analyzed according to previously characterized methods (Glass et al., 2005) using MCID software.
Statistical analyses
All data were presented as mean ± SEM. Data were analyzed by t-tests, or one-or two-way factorial ANOVA where applicable. For analysis of the data presented as a ratio, the data were converted by arcsine prior to analysis to allow for comparison of proportions. Differences in means were analyzed by Fisher’s PLSD. The criteria for significance was set at p<0.05.
Phenotypic analyses
In order to determine the phenotypic consequences of CeA NR1 deletion, mice were tested in a series of behavioral tests prior to, and following, rAAV injections. The tests were conducted in the order in which they are described below.
General health and neurological assessment
To evaluate the overall health of conditional NR1 knockout mice, animals were visually examined prior to, and two weeks after microinjection of rAAV-GFP-Cre or rAVV-GFP. The presence of established signs of sickness symptoms post-injection were scored. These included labored breathing, presence of encrusted blood in the facial area, signs of grooming, presence of whiskers, and sensitivity to handling (Crawley and Paylor, 1997). In addition, body weight and food intake were measured just prior to, and for two weeks following vector microinjection. Sensory function was assessed by the visual cliff test. Briefly, mice were placed on a platform, and the latency to move to the edge, and the number of times it placed its head or shoulders over the edge were counted. Normal mice place their heads over edge a few times each minute, however mice with sensory or motor impairments will walk over the edge. The eye blink and ear twitch reflexes were evaluated by touching both eyes and tip of each ear with a cotton swab. In normal mice these stimuli should elicit blinking and ear twitching. The whisker-orienting reflex was evaluated by touching whiskers with a cotton swab. Normal mice will continuously move whiskers until touched, after which they stop.
Motor coordination
Each mouse was placed on a Rota Rod (Life Sciences IITC, Woodland Hills, CA) set at 40 rpm (speed level 10). The amount of time (sec) that the mouse remains on the rod was recorded automatically. A cutoff of 60 seconds was observed (South et al., 2003).
Open field activity
Locomotor activity was conducted in an automated chamber (38.1 cm × 53.3 cm) [MED Associates, Albans VT]. Total ambulatory time and distance was determined over a 30-minute period.
Mechanical stimulus threshold
Sensitivity to a non-noxious mechanical stimulus was determined by use of von Frey hairs. Each mouse was placed on a Plexiglas cage with mesh flooring suspended above the experimenter. The von Frey hairs were applied perpendicularly against the mid-plantar surface of the paw using the up-down method of Dixon (Chaplan et al., 1994).
Thermal paw withdrawal threshold
Thermal paw withdrawal was assessed using the hot plate test. Mice were placed on a pre-heated glass plate maintained at 52° C, and the latency (sec) for the withdrawal of the paw was determined.
Thermal tail withdrawal threshold
A hot water bath was heated to a constant temperature of 52.5° C. Mice were held loosely in a towel, and the lower half of the mouse’s tail was dipped into the water bath. A timer was stopped immediately when the mouse flicked its tail. A tail withdrawal latency of 10 seconds was used as a cutoff (Bilsky et al., 1996).
Induction of morphine dependence
The protocol used to induce morphine dependence was derived from our previous data (unpublished) showing that 8–10 days of morphine pellet exposure resulted in opioid withdrawal behaviors and conditioned place aversion (CPA) in mice. Briefly, under deep isoflurane anesthesia, mice were implanted with one 25 mg morphine pellet subcutaneously. Every fourth day the pellet was removed, and animals were implanted with a fresh pellet. Dependence was determined by observations of diarrhea, wet dog shakes, and jumping after naloxone (1mg/kg, i.p.) during place aversion training, and subsequent quantification of these symptoms following the place aversion test.
Conditioned place aversion (CPA)
The CPA apparatus consisted of a two-chamber box inserted into an activity monitor unit (MED Associates). Each box had distinct visual and tactile cues. During preconditioning, mice were placed in the apparatus, and the time spent in each chamber was recorded to serve as the baseline chamber selection. During training, one compartment was paired with naloxone and the other with saline, such that there were no significant differences between the amounts of time spent in the saline-paired or naloxone-paired compartments across groups. On four alternate training days morphine dependent mice were injected with saline (0.9 %, i.p.) or naloxone (1 mg/kg, i.p.) and then placed in the respective chamber for 30 minutes (Schulteis et al., 1998). On day 5, subjects were allowed to freely explore both chambers for 30 minutes. The difference in time spent in the naloxone-paired chamber during the preconditioning phase and testing phase served as the measure of place aversion.
RESULTS
Expression and distribution of NR1 in the CeA of untreated fNR1 mice
In situ hybridization for NR1 mRNA yielded a robust signal in brain areas surrounding the amygdala (Figure 1A), including the CeA (Figure 1B). In tissue processed for electron microscopic immunogold labeling of NR1, gold-silver deposits were observed in somata and dendrites of CeA neurons (Figures 1C–D). In somata, immunogold particles for NR1 were present in association with Golgi complexes, and endoplasmic reticula, known sites of protein synthesis, packaging and transport (Figure 1C). Immunogold particles for NR1 were also present in proximal dendritic processes directly branching from the cell body (Figure 1C). Moreover, in dendritic profiles NR1 immunogold particles were present in association with tubulovesicular organelles (Figure 1D), as well as near the plasma membrane adjacent to asymmetric postsynaptic specializations indicative of excitatory synaptic transmission (Figure 1D). The majority of labeled profiles were dendrites (226/247; 6,516 μm2; N=3;), although there was also a small number of NR1 labeled axon terminals (21/247). Since dendritic processes were most numerous, the subcellular distribution of NR1 was counted in these structures. In dendritic profiles, immunogold labeling of NR1 was mainly found in intracellular sites relative to the surface membrane (Figure 1E). The percentage of immunogold particles associated with the plasma membrane and intracellular sites were comparable to that seen in non-transgenic C57BL/6 mice (15±2% and 85±2%, respectively; n=262; 7,392 μm2, N=3;). Dual immunogold labeling of NR1 and immunoperoxidase labeling of μOR demonstrated that NMDA and the opioid receptor were co-expressed in common CeA neurons (Figure 1F), indicating that some NR1 expressing neurons in the CeA are responsive to opioids.
Figure 1. The NR1 gene is expressed in CeA neurons and trafficked to appropriate subcellular locations in somatodendritic processes that can be shown to express μOR in the CeA of fNR1 mice.
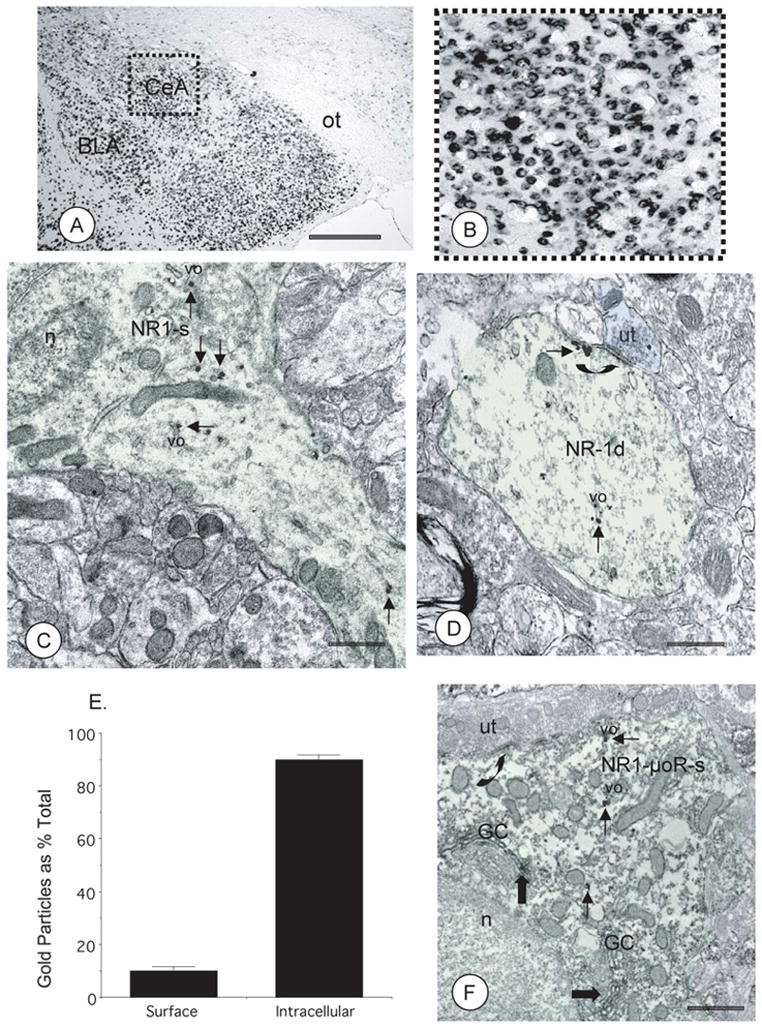
(A). In the absence of local rAAV-GFP-Cre microinjection, NR1 mRNA is present throughout the forebrain, including the CeA, as seen in a coronal brain section. Scale Bar = 1 mm. (B). A magnified view of the area bounded by the box in Fig 1A showing NR1 mRNA in neurons in the CeA. (C). Immunogold particles for NR1 (thin arrows) are clustered near a Golgi complex (GC), as well as tubulovesicular organelles (vo), within a soma (NR1-s, light green shading) in the CeA. (D). In a dendritic profile from the CeA (NR1-d, light green shading) a cluster of NR1 immunogold particles (thin arrows) is associated with a tubulovesicular organelle (vo). An aggregate of gold-silver deposits is also associated with the perisynaptic plasmalemma apposed by an unlabeled axon terminal (ut; blue shading) forming an asymmetric excitatory type synapse (large arrow). (E). Quantification of the subcellular distribution of NR1 immunogold particles shows that these receptors are present on the plasma membrane, but are more abundant in intracellular sites of CeA neurons from untreated fNR1 mice. (F). A soma (NR1-μOR-s) from the CeA is contacted by an unlabeled axon terminal (ut) forming an unidentified synapse (curved arrow). This soma shows immunogold labeling for NR1 (thin arrows) and immunoperoxidase labeling for μOR (thick arrows). While present in the same neuron, labeling is segregated in distinct subcellular compartments. Immunoperoxidase reaction product for μOR is present with the lumen of Golgi Complexes (GC), while NR1 labeling is found near vesicular organelles (vo). BLA: basolateral nucleus of the amygdala, n: nucleus, ot: optic tract. EM scale bars = 0.5 μm.
Unilateral NR1 gene deletion produced by microinjection of rAAV-GFP-Cre in the CeA of fNR1 mice
Unilateral microinjection of rAAV-GFP-Cre into the CeA of adult fNR1 mice produced ipsilateral gene deletion covering approximately 0.5 – 0.6 mm after a minimum of 14 days post-injection. Light microscopic visual analysis showed that there was an overlapping expression of the GFP reporter, reduced NR1 gene expression (Figures 2A–D), and immunolabeling for the NR1 protein (Figures 2E–F).
Figure 2. Unilateral central amygdala microinjection of rAAV-GFP-Cre results in an apparent ipsilateral NR1 gene deletion in fNR1 mice.
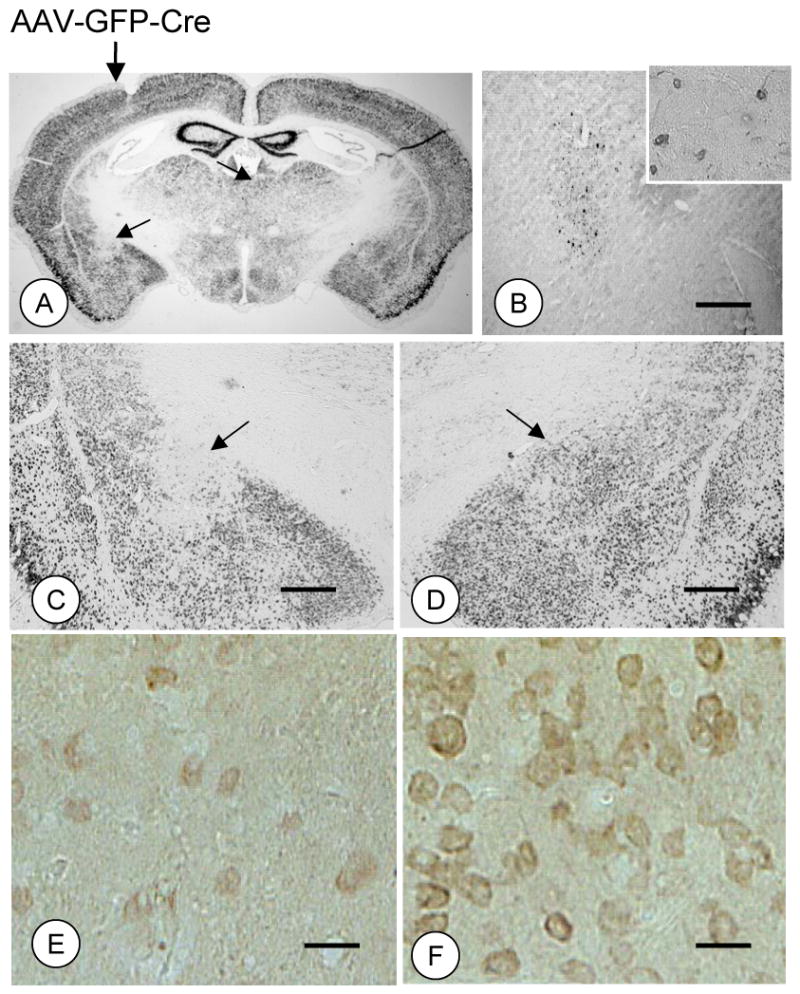
(A–D). Unilateral microinjection of rAAV-GFP-Cre in the CeA produces an ipsilateral NR1 gene knockout centered on the target site that correlates with immunolabeling of the reporter protein GFP (B) in a serial section. (E–F). There is also a concomitant decrease in NR1 immunolabeling in the hemisphere microinjected with rAAV-GFP-Cre. Scale Bars = 1mm (B–D), 40 μm (E–F).
In order to identify the cell types where recombination occurred, forebrain sections containing the central amygdala previously microinjected with rAAV-GFP-Cre were processed for dual immunofluorescence labeling. Labeling was performed for the reporter protein GFP and either the neuronal marker NeuN or the glial marker GFAP. Dual labeled cells were seen in the injected amygdala of brain sections processed for dual labeling with GFP and NeuN (Figures 3A–C–E), but not GFP and GFAP (Figures 3B–D–F), demonstrating that recombination occurred in neurons and not glial cells.
Figure 3. Unilateral microinjection of rAAV-GFP-Cre in the CeA results in recombination within neurons.
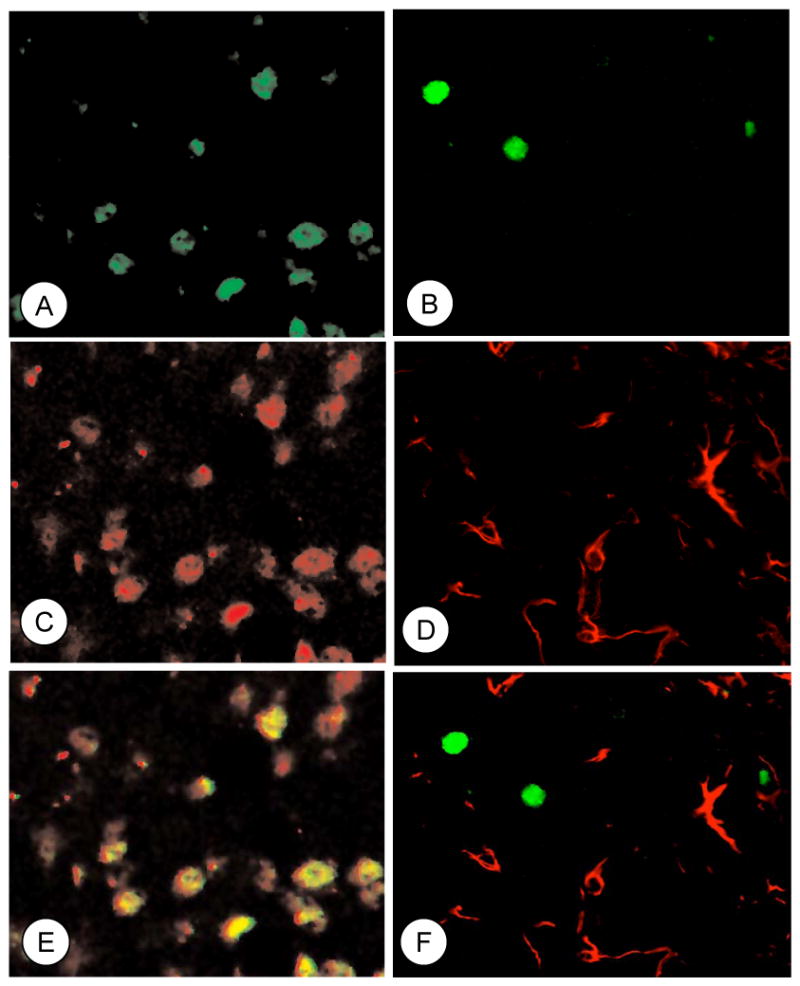
Immunofluorescence labeling for GFP (green; A), NeuN (red; C), and their co-localization (merge; E) in a subpopulation of neurons. Immunofluorescence labeling for GFP (green; B) and GFAP (red; D) do not show overlapping distributions (merge; F).
Quantitatively, there was a significant reduction in NR1 mRNA in the CeA of the injected hemisphere of mice receiving rAAV-GFP-Cre relative to rAAV-GFP (p<0.05; N=8; Figure 4A). In order to determine if expression of this protein was reduced in dendrites, the major functional site of NMDA receptors, single labeling electron microscopic immunocytochemistry with a peroxidase marker was used to quantify the number of NR1 labeled dendritic profiles in the CeA of mice receiving vector microinjection. Significant reductions in immunolabeling for NR1 occurred in dendritic profiles (p<.05; 23,490 μm2; n=1,556; N=3; Figure 4A) in mice injected with rAAV-GFP-Cre. There was no significant difference in the number of NR1 labeled axon terminals (0.28 ± .05 versus 0.31 ± .05; p=.7) in the CeA injected with rAAV-GFP-Cre relative to the contralateral hemisphere. In order to determine if rAAV-GFP-Cre produced compensatory changes in the NR2 protein, electron microscopic analysis was used to quantify the number of NR2 labeled somatodendritic profiles. There was no significant difference in NR2 immunoperoxidase labeled dendritic processes in the hemisphere microinjected with rAAV-GFP-Cre compared to the contralateral hemisphere (p=.4; 17,000 μm2; N=3; Figure 4B). In order to determine if rAAV-GFP-Cre affected cellular survival, thionin staining was used to identify central amygdala cells for quantification. There were no differences in the number of stained cells in the CeA microinjected with rAAV-GFP-Cre relative to the contralateral hemisphere (p=.9, N=4; Figure 4C–E).
Figure 4. Unilateral central amygdala rAAV-GFP-Cre microinjection produces quantitative ipsilateral reductions in NR1 gene expression and ultrastructural labeling of NR1 but not NR2, or cell number.
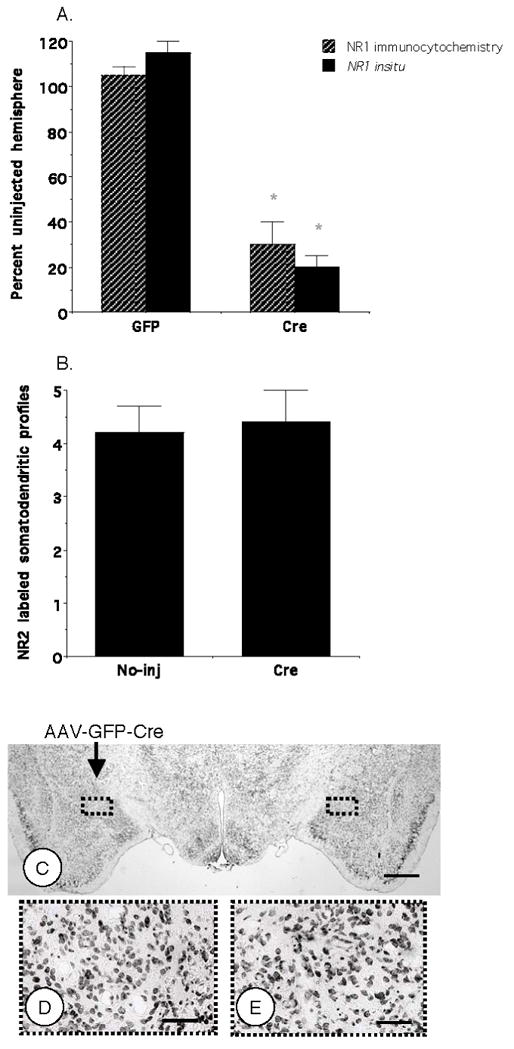
(A). Expression of the NR1 gene, as well as protein immunolabeling in somatodendritic processes is reduced in the hemisphere microinjected with rAAV-GFP-Cre, but not rAAV-GFP, relative to the contralateral hemisphere. (B). There is no difference in the mean number of NR2 labeled somatodendritic profiles (per 55 μm2) in the hemisphere microinjected with rAAV-GFP-Cre compared to the contralateral hemisphere. (C–E). There were no differences in the number of thionin stained central amygdala cells in the injected versus contralateral hemisphere. Values represent means±SEM. * p<0.05. Scale Bars = 1 mm (A), 100 μm (B).
To establish whether rAAV-GFP-Cre was impacting neuronal morphology, an indicator of cell viability, neuronal cell bodies were examined qualitatively and by quantitative morphometry. In the CeA microinjected with rAAV-GFP-Cre and the contralateral hemisphere, neuronal somata were medium in size and contained round to oval shaped nuclei (Figure 5A–B). These cell bodies had numerous intracellular organelles, including Golgi Complexes, endoplasmic reticula, and mitochondria, as described previously (Figure 1C). Quantitative ultrastructural morphometric analysis of the hemisphere receiving rAAV-GFP-Cre and the contralateral hemisphere showed that there were no distinctions in neuronal cross-sectional area (p= .46; n=100; N=3; 10,979 μm2; Figure 5C), surface area (p=.23; Figure 5D), nuclear form factor (p=.12; Figure 5E), or ratio of major-to-minor axes (p= .35; Figure 5F).
Figure 5. Microinjection of rAAV-GFP-Cre in the CeA does not affect neuronal morphology.
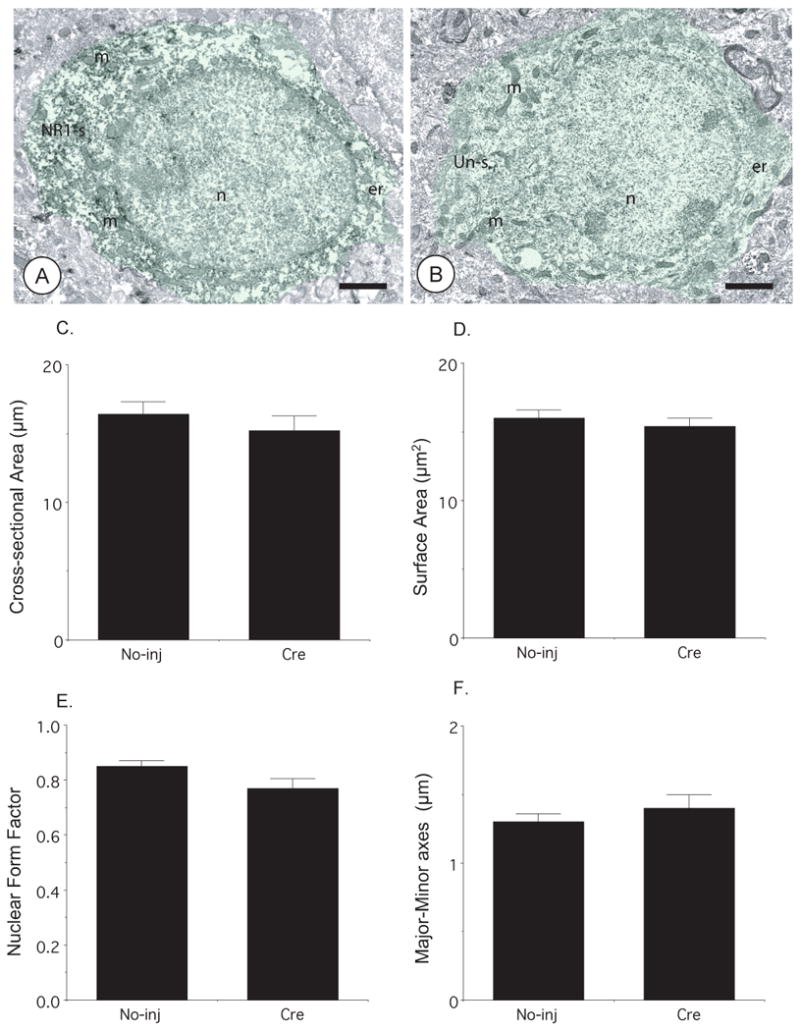
(A–B). Neuronal somata in the contralateral (A) and rAAV-GFP-Cre (B) injected CeA show round nuclei and contain numerous intracellular organelles including mitochondria. The neuron from the amygdala not injected with rAAV-GFP-Cre (NR1-s, green shading) shows diffuse intracellular immunoperoxidase reaction product for NR1, whereas the cell body from the injected hemisphere (Un-s, green shading) is devoid of NR1 immunoreactivity. Ultrastructural morphometric analysis in the CeA showed that there were no significant differences in (C) cross-sectional area, (D) surface area, (E) nuclear form factor, or (F) major-to-minor axis length in neuronal somata of rAAV-GFP-Cre injected and contralateral hemispheres. Scale Bars = 1 μm. Values represent means±SEM.
To examine whether rAAV-GFP-Cre was impacting dendritic spine morphology, an indicator of cytotoxicity, spiny appendages were examined qualitatively and by quantitative morphometry. By visual inspection, dendritic spines in both hemispheres had bulbous heads, lacked intracellular organelles, and received asymmetric excitatory-type synapses from unlabeled axon terminals (Figure 6A–B). However, many of the postsynaptic densities in the rAAV-GFP-Cre hemisphere did not show immunoperoxidase labeling for NR1 (Figure 6B). Using quantitative ultrastructural morphometry, there were no differences in dendritic spine morphology, including cross-sectional area (p=.1; 8,104 μm2; n=148; N=3; Figure 6C), surface area (p=.14: Figure 6D), or major-to-minor axis length (2.2 ± .01 versus 2.0 ± .07, p=.09). There were also no differences in dendritic spine number in single thin sections (165 ± 19 versus 174 ± 20; p=.16) in rAAV-GFP-Cre injected and contralateral hemispheres, respectively.
Figure 6. Microinjection of rAAV-GFP-Cre in the CeA does not affect dendritic spine morphology.
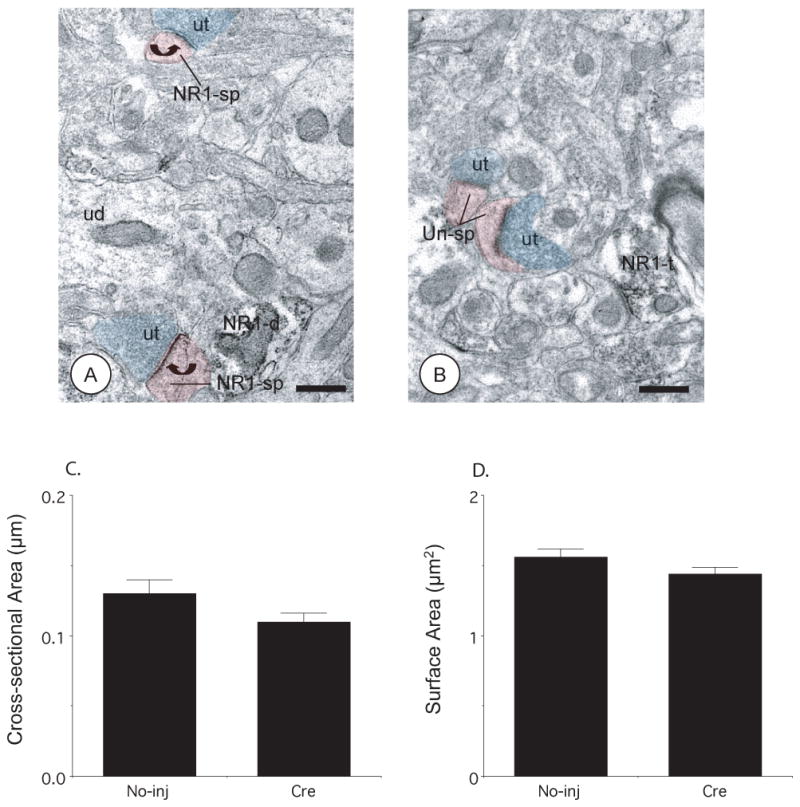
(A–B). Dendritic spines in the uninjected and rAAV-GFP-Cre injected hemispheres show bulbous spine heads, lack intracellular organelles, and receive asymmetric excitatory-type synapse from unlabeled axon terminals (ut, blue shading). The postsynaptic densities of dendritic spines (NR1-sp, red shading) from the CeA not receiving vector microinjection show immunoperoxidase labeling for NR1 (curved arrows), whereas the dendritic spines (Un-sp, red shading) from the contralateral hemisphere receiving rAAV-GFP-Cre do not. Ultrastructural morphometric analysis of dendritic spines showed that there were no significant differences in (C) cross-sectional and (D) surface areas in rAAV-GFP-Cre injected and contralateral hemispheres. NR1-d: NR1 labeled dendritic profile, NR1-t: NR1 labeled axon terminal. Scale Bars= 0.5 μm. Values represent means±SEM
Basal phenotypic effects of bilateral NR1 gene deletion in the CeA by rAAV-GFP-Cre injection
In order to determine the phenotypic consequences of NR1 subunit deletion in the amygdala, mice were tested on a variety of behaviors before, and a minimum of two weeks following bilateral rAAV-GFP-Cre or rAAV-GFP microinjection. Observations of general health and neurological function showed that there were no differences in any of these measures pre-and post-injection of either vector (not shown). Similarly, when locomotor activity was measured in an automated open field, there were no differences in ambulatory time in mice before or after microinjection of rAAV-GFP-Cre or rAAV-GFP (p=.4; N=13: Figure 7A). Additionally, there were no differences in sensory-motor function as measured by distance traveled on the rotarod (p=.59; N=13; Figure 7B). Further, mice did not differ in tests of nociceptive function, including thermal tail withdrawal thresholds in a water bath (p=.76, N=13; Figure 7C), and thermal paw withdrawal using a hot plate (p=.93; N=13; Figure 7D), as well as hindpaw mechanical thresholds measured by the von Frey response (right: p=.16; left: p=.2; N=13; Figure 7E). In addition, there was no difference in body weight in either rAAV-GFP-Cre or rAAV-GFP injected mice before of after vector microinjection (p=.34; N=13; Figure 7F).
Figure 7. Bilateral central amygdala NR1 deletion does not affect basal behaviors in fNR1 mice.
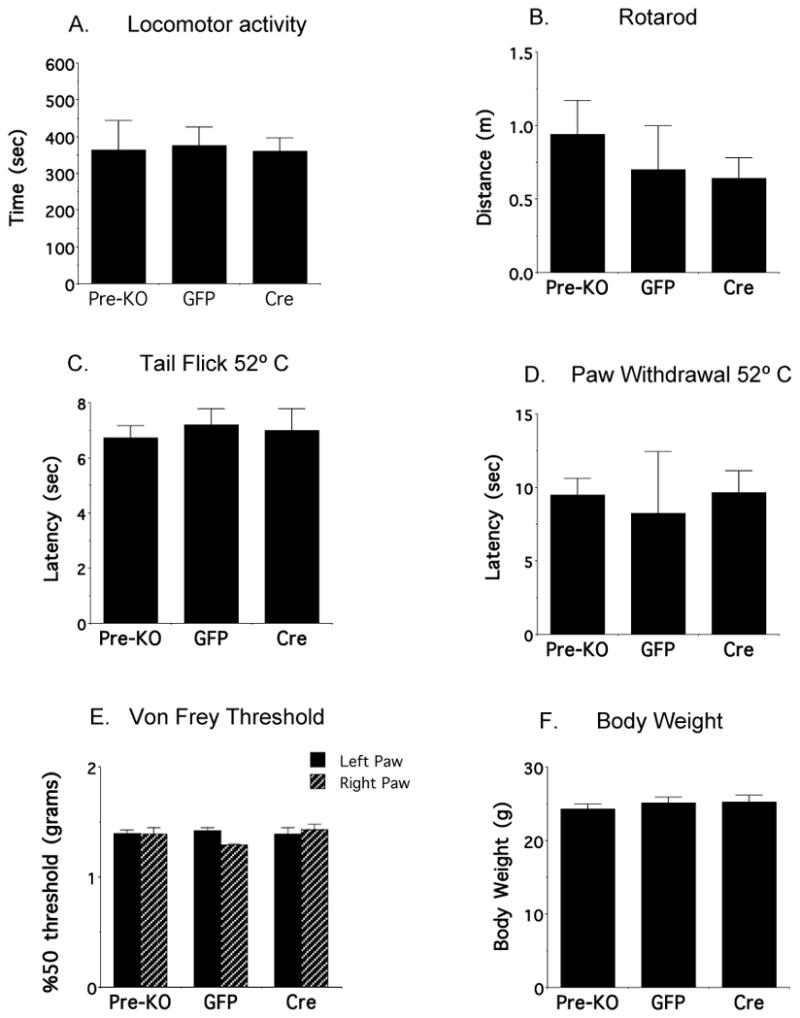
There are no differences in (A) activity time, (B) distance traveled on the rotarod, (C) tail flick latency in the water bath, (D) hindpaw withdrawal latency in the hot plate (E) midplantar forepaw 50% gram thresholds using von Frey hairs, or (F) body weight in adult floxed NR1 mice prior to injection (Pre-KO), or in response to rAAV-GFP or rAAV-GFP-Cre microinjection in the amygdala two weeks later.
Effect of bilateral CeA NR1 gene deletion on opioid withdrawal-induced physical and psychological symptoms
Separate groups of fNR1 mice, including those given no injection, or bilaterally microinjected with either rAAV-GFP or rAAV-GFP-Cre, received subcutaneous morphine pellets. On the eighth day after initial morphine exposure, each group of mice received an injection of naloxone to elicit withdrawal. Shortly after injection with the opioid antagonist, mice from each group were observed to express stereotypical signs of withdrawal including burrowing, forepaw tremor and penis grooming (not shown). Mice also exhibited wet dog shakes, jumping, and diarrhea, all of which were quantified. Mice receiving rAAV-GFP-Cre injection in the CeA did not significantly differ from uninjected or rAAV-GFP injected animals in somatic (wet dog shakes + jumping) [p=.89, N=17; Figure 8A] or autonomic (diarrhea) symptoms (p=.35, N=17; Figure 8B). Mice were also trained to develop a CPA to naloxone-precipitated withdrawal. Unlike the case of physical withdrawal symptoms, there were significant differences with respect to naloxone-induced place aversion in the different groups of morphine dependent fNR1 mice (p=.03, N=18; Figure 8C). Mice injected with rAAV-GFP-Cre in the CeA did not spend less time in the withdrawal-paired chamber, indicating an inability to express a CPA in response to opioid withdrawal (Figure 8C). Reductions in NR1 mRNA were seen selectively in the animals injected with rAAV-GFP-Cre (Figure 9).
Figure 8. Bilateral central amygdala NR1 deletion selectively attenuates psychological properties of opioid withdrawal in morphine-dependent fNR1 mice.
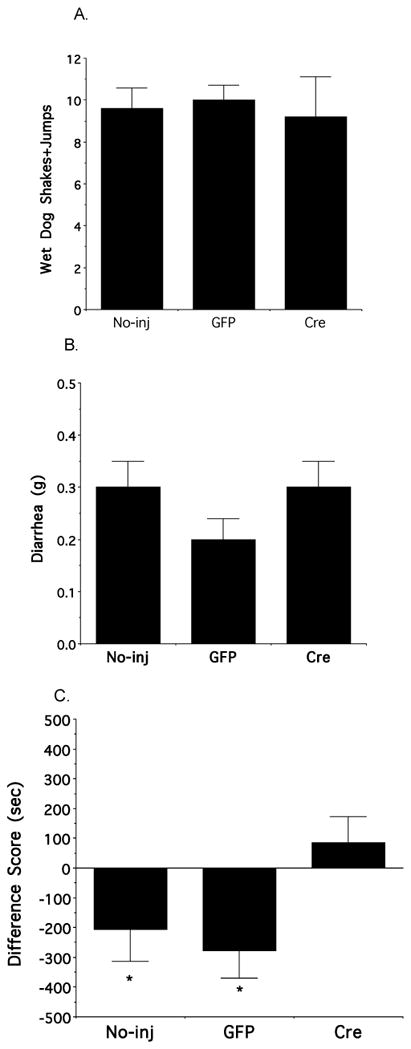
(A–B) There are no differences in the number of naloxone-induced somatic (wet dog shakes+jumps) or autonomic (diarrhea) withdrawal symptoms in morphine pelleted uninjected, rAAV-GFP injected and rAAV-GFP-Cre injected mice. (C). In distinction to uninjected and rAAV-GFP injected fNR1 mice, rAAV-GFP-Cre injected animals do not spend significantly less time in a chamber where they have experienced opioid withdrawal. Values represent means±SEM. * p<0.05
Figure 9. Bilateral central amygdala rAAV-GFP-Cre microinjections reduce NR1 gene expression in morphine-dependent fNR1 mice.
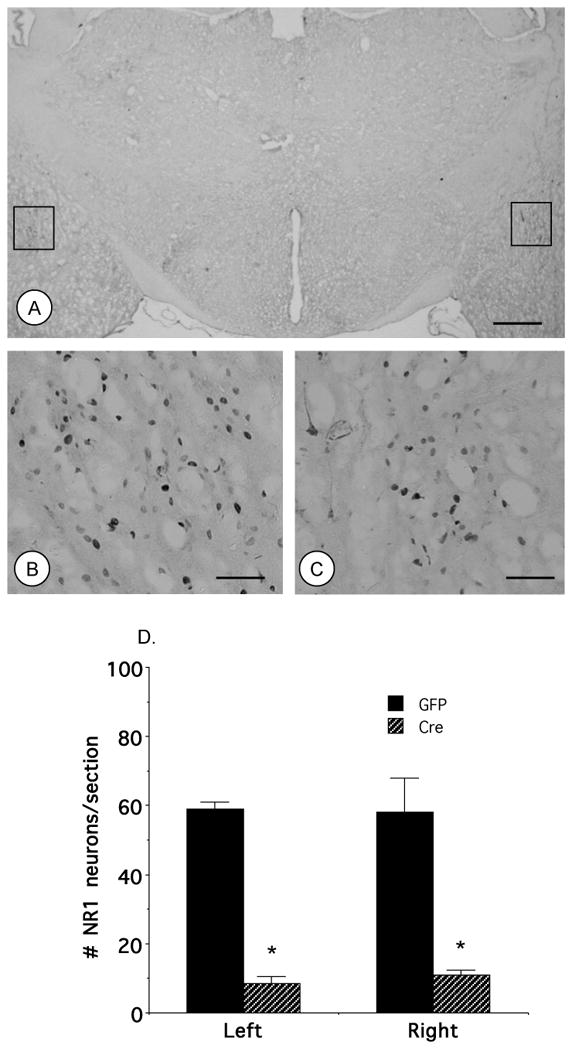
(A–C). Mice receiving bilateral rAAV-GFP-Cre in the CeA express the GFP reporter in the target region. Areas in the boxes in A are shown at a higher magnification in B and C. (D). There are reductions in NR1 mRNA in the CeA receiving rAAV-GFP-Cre microinjection. Scale Bars= 1 mm (A), 100 μm (B–C). Values represent means±SEM. * p<0.05
DISCUSSION
Characterization of CeA NR1 knockout by rAAV-GFP-Cre in fNR1 mice
Given the nature of conditionally manipulating gene expression in vivo, there was concern that these methods may produce significant off-target effects that can compromise cellular structure and function (Alvarez et al., 2006; Schmidt-Supprian and Rajewsky, 2007). However, unlike other adenovirus vectors, the rAAV has not been shown to produce pathogenic or immune reactions, as indicated by the lack of vascular cuffing, as well as the absence of glial-, CD4-, or CD8-cell infiltration in the target site following intracranial microinjection (Kaspar et al., 2002). Moreover, it has also been shown that rAAV injection in the spinal cord of fNR1 mice does not produce neuronal loss, as measured by NeuN labeling (South et al., 2003).
In our study, we used electron microscopic immunocytochemistry and morphometry to examine the ultrastructural receptor distribution of NMDA receptor subunits, as well as the morphological status of central amygdala neurons in response to local rAAV-GFP-Cre microinjection. By electron microscopic immunocytochemistry, there was a decrease in the number of NR1 labeled somatodendritic profiles, but not axon terminals, or NR2 labeled somatodendritic processes in response to rAAV-GFP-Cre. These results indicated that NR1 deletion appropriately reduced NR1 labeling in their major sites of expression and function, but did not result in compensatory responses in presynaptic NR1, or postsynaptic NR2 distribution. Ultrastructural morphological analysis demonstrated that there were no signs of swelling or condensation in neuronal somata or nuclei, as shown by a number of parameters including the nuclear form factor, a widely used measure of nuclear pathology (Chan et al., 2000). Dendritic spine morphology has also been shown to be associated with neural toxicity, which may involve NMDA receptor activation (Ackermann and Matus, 2003; Gisselsson et al., 2005; Zeng et al., 2007). Ultrastructural morphometric analysis showed no differences in spine cross-sectional or surface areas in rAAV-GFP-Cre administered hemispheres, indicating that NR1 deletion was not associated with changes in basal dendritic spine integrity. This ultrastructural evidence, when coupled with a previous report that spinal cord parenchymal rAAV-GFP-Cre administration selectively inhibited NMDA receptor-mediated currents (South et al., 2003), indicated that NR1 knockout was producing highly specific effects involving NMDA receptor function.
Phenotypic effects of CeA NR1 deletion: Basal behavior
Knockout of the NR1 gene in the CeA did not produce obvious behavioral deficits. There were no significant performance differences in a battery of gross neurological tests of sensory or motor function (Crawley and Paylor, 1997) before and after microinjection of either rAAV-GFP or rAAV-GFP-Cre in fNR1 mice. The viability of the phenotype was further supported by the use of more refined behavioral analytical procedures. Mice with CeA NR1 knockout exhibited no significant differences in tests of locomotor activity, body weight, sensory-motor coordination, thermal nociception, or somatosensation before and after they received microinjection of the vectors. We thus provided evidence that NR1 receptor subunit gene deletion elicited by microinjection of rAAV-GFP-Cre into the CeA of NR1 floxed mice produced a stable phenotype.
Phenotypic effects of CeA NR1 deletion: Opioid dependence
Animals given continuous morphine develop dependence, as shown by withdrawal symptoms in response to termination of opioid action (Martin et al., 1963). The neural adaptations mediating opioid dependence may critically depend upon functional NMDA receptors (Trujillo and Akil, 1991; Elliott et al., 1994; Rasmussen et al., 1995). In order to assess the importance of amygdala NMDA receptors in opioid dependence, CeA NR1 knockout mice were exposed chronically to morphine by subcutaneously implanted morphine pellets and then administered naloxone. The CeA NR1 knockouts did not differ from control animals with respect to somatic signs, such as jumping and wet dog shakes, or autonomic symptoms, notably diarrhea and weight loss. These results indicated that CeA NMDA receptors were not necessary to produce some of the major opioid withdrawal symptoms.
In addition to somatic and autonomic symptoms, opioid withdrawal has also been shown to result in aversion (Maldonado et al., 1992; Schulteis et al., 2000), a phenomenon most commonly measured by the CPA paradigm. Moreover, there is also evidence that physical and psychological symptoms can be dissociated by opioid antagonist dose in dependent animals. Very low doses of naloxone elicited place aversion, yet failed to produce physical withdrawal signs (Schulteis et al., 1994). Previous findings have also shown that the CeA plays an important role in the aversive properties of opioid withdrawal, but has a more ambiguous involvement in physical withdrawal symptoms. Direct microinjection of methylnaloxonium in the CeA has been shown to produce place aversion in morphine dependent mice without producing physical withdrawal symptoms such as wet dog shakes (Stinus et al., 1990), although other signs such as mastication and ptosis, have been reported (Maldonado et al., 1992). In our study, we showed that, contrary to physical withdrawal, amygdala NR1 gene deletion impaired naloxone withdrawal-induced CPA, which indicated that functional NMDA receptors in the CeA were necessary for producing a significant negative affective state associated with withdrawal. Deletion of NR1 may have attenuated opioid withdrawal-induced place aversion by interfering with two distinct processes, namely the development or expression of aversion. While a definitive judgment on these two possibilities would have required conditional deletion of the NR1 gene after the CPA had been established, available evidence has shown that NMDA receptors are involved in the development of conditioned aversion. For example, when NMDA receptor antagonists were injected prior to each training session, this prevented withdrawal-induced place aversion (Higgins et al., 1992; Popik and Danysz, 1997; Watanabe et al., 2002). The latter findings were also consistent with other evidence that NMDA receptor antagonists interfered with the induction (Goosens and Maren, 2003), but not necessarily expression of other kinds of emotional learning, including associative fear conditioning (Miserendino et al., 1990; Kim et al., 1991).
In contrast to our finding that suppression of NMDA receptor expression in the amygdala only interfered with opioid withdrawal-induced aversion, it is known that systemic administration of NMDA receptor antagonists can attenuate broad-spectrum withdrawal symptoms, including autonomic and somatic responses (Trujillo and Akil, 1991; Popik and Danysz, 1997). Within the context of the prior literature, our results indicate that the diverse types of withdrawal symptoms, and the neural systems that mediate them, are distinct and dissociable.
With respect to the specificity of the ability of NR1 deletion in the central amygdala to attenuate place aversion, it should be noted that the CeA is a component of a larger structural-functional ventral forebrain system involved in emotional behaviors (Heimer and Van Hoesen, 2006). Other critical components of this system include the bed nucleus of the stria terminalis (BNST) and the shell of the nucleus accumbens (Acb). Both of these areas have been shown to be involved in the aversive properties of withdrawal (Stinus et al., 1990; Delfs et al., 2000; Nakagawa et al., 2005). Whether NMDA receptor deletion in these regions would produce a pattern similar to that of CeA NMDA receptor knockout in regards to withdrawal place aversion, at present, remains an open question.
Potential mechanisms mediating NMDA-μOR interactions and dependence aversion
The ability of amygdala NR1 knockout to impair CPA may reflect interference with cellular processes involving opioid and NMDA receptor interactions that mediate affective behaviors. These interactions may occur at distinct synaptic levels, namely “post”- or “pre”-synaptic. Firstly, NMDA and mu-opioid receptors may co-modulate shared intracellular signaling pathways in common neurons (i.e. a “postsynaptic” effect). This possibility is supported by our observations that these receptors are co-expressed in CeA neurons, and other reports that μOR activation has direct inhibitory actions on excitatory postsynaptic signaling in CeA neurons (Zhu and Pan, 2004). Morphine initiates an array of intracellular transduction pathways involving intracellular calcium, cAMP, protein kinases (Bernstein and Welch, 1998; Fan et al., 1999; Lim et al., 2005) and transcription factors (Rasmussen et al., 1995; Shaw-Lutchman et al., 2002). These pathways may impact intermediate- and long-term cellular processes including gene expression (Ammon-Treiber and Hollt, 2005), glutamate receptor trafficking (Glass et al., 2003; Glass et al., 2005), or neuronal morphology (Robinson and Kolb, 2004). Many of these processes are also co-modulated by NMDA receptor activation, and are critical for the development of experience-dependent neural and behavioral plasticity (Wang et al., 2006). Secondly, opioid-NMDA receptor interactions may be mediated by opioid receptor modulation of a glutamate pathway upstream from the NMDA receptor (i.e. a “presynaptic” effect). For example, it has been shown that activation of amygdala μOR inhibits presynaptic glutamate release by amygdala neurons (Zhu and Pan, 2005). Anatomical evidence indicates that there is a large and diverse group of glutamate afferents that terminate in the CeA. These include projections from brain areas known to express μOR (Mansour et al., 1995), including the basolateral nucleus of the amygdala (LeDoux, 2000), a pathway associated with emotional learning, as well as glutamate afferents from the subiculum (LeDoux, 2000), an area that plays an important role in context learning. In addition, indirect multisynaptic effects involving modulation of GABAergic interneurons (Nugent et al., 2007) presynaptic to glutamatergic projection neurons may be another synaptic alternative.
In summary, we have shown that spatial-temporal deletion of the NR1 subunit in the amygdala can be made by local microinjection of an rAAV expressing Cre recombinase in adult fNR1 mice. Using this approach we achieved significant postsynaptic deletion in amygdala neurons with highly specific phenotypic consequences. These results indicate that brain delivery of rAAV-GFP-Cre in floxed mice is a viable tool for identifying relationships between the NR1 gene, the amygdala, and opioid dependence.
Acknowledgments
Supported by: DA-016735 (MJG), DA001457, DA000198, DA007174 (CEI), and DA-05130 (VMP)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackermann M, Matus A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nature Neurosci. 2003;6:1194–1200. doi: 10.1038/nn1135. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Ridenour DA, Sabatini BL. Retraction of synapses and dendritic spines induced by off-target effects of RNA interference. J Neurosci. 2006;26:7820–7825. doi: 10.1523/JNEUROSCI.1957-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammon-Treiber S, Hollt V. Morphine-induced changes of gene expression in the brain. Addiction Biology. 2005;10:81–89. doi: 10.1080/13556210412331308994. [DOI] [PubMed] [Google Scholar]
- Bernstein MA, Welch SP. mu-Opioid receptor down-regulation and cAMP-dependent protein kinase phosphorylation in a mouse model of chronic morphine tolerance. Mol Brain Res. 1998;55:237–242. doi: 10.1016/s0169-328x(98)00005-9. [DOI] [PubMed] [Google Scholar]
- Bespalov A, Dravolina O, Belozertseva I, Adamcio B, Zvartau E. Lowered brain stimulation reward thresholds in rats treated with a combination of caffeine and N-methyl-D-aspartate but not alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate or metabotropic glutamate receptor-5 receptor antagonists. Behav Pharmacol. 2006;17:295–302. doi: 10.1097/01.fbp.0000205014.67079.be. [DOI] [PubMed] [Google Scholar]
- Bespalov AY, Balster RL, Beardsley PM. N-Methyl-D-aspartate receptor antagonists and the development of tolerance to the discriminative stimulus effects of morphine in rats. J Pharmacol Exp Therap. 1999;290:20–27. [PubMed] [Google Scholar]
- Bespalov AY, Dravolina OA, Zvartau EE, Beardsley PM, Balster RL. Effects of NMDA receptor antagonists on cocaine-conditioned motor activity in rats. Eur J Pharmacol. 2000;390:303–311. doi: 10.1016/s0014-2999(99)00927-9. [DOI] [PubMed] [Google Scholar]
- Bilsky EJ, Inturrisi CE, Sadee W, Hruby VJ, Porreca F. Competitive and non-competitive NMDA antagonists block the development of antinociceptive tolerance to morphine, but not to selective mu or delta opioid agonists in mice. Pain. 1996;68:229–237. doi: 10.1016/s0304-3959(96)03185-5. [DOI] [PubMed] [Google Scholar]
- Chan J, Aoki C, Pickel VM. Optimization of differential immunogold-silver and peroxidase labeling with maintenance of ultrastructure in brain sections before plastic embedding. J Neurosci Methods. 1990;33:113–127. doi: 10.1016/0165-0270(90)90015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RKW, Peto CA, Sawchenko PE. Fine structure and plasticity of barosensitive neurons in the nucleus of solitary tract. J Comp Neurol. 2000;422:338–351. [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Meth. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Hormones and Behavior. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. PNAS. 2006;103:15254–15259. doi: 10.1073/pnas.0601758103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Elliott K, Minami N, Kolesnikov YA, Pasternak GW, Inturissi CE. The NMDA receptor antagonists, LY274614 and MK-801, and the nitric oxide synthase inhibitor, N-nitro-L-arginine, attenuate analgesic tolernace to the mu-opioid morphine but not to k opioids. Pain. 1994;56:69–75. doi: 10.1016/0304-3959(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Epstein DH, Preston KL, Jasinski DR. Abuse liability, behavioral pharmacology, and physical-dependence potential of opioids in humans and laboratory animals: lessons from tramadol. Biol Psychol. 2006;73:90–99. doi: 10.1016/j.biopsycho.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan GH, Wang LZ, Qiu HC, Ma L, Pei G. Inhibition of calcium/calmodulin-dependent protein kinase II in rat hippocampus attenuates morphine tolerance and dependence. Mol Pharmacol. 1999;56:39–45. doi: 10.1124/mol.56.1.39. [DOI] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 1994;13:325–338. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Gisselsson LL, Matus A, Wieloch T. Actin redistribution underlies the sparing effect of mild hypothermia on dendritic spine morphology after in vitro ischemia. J Cer Blood Flow Metab. 2005;25:1346–1355. doi: 10.1038/sj.jcbfm.9600131. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Frys KA, Iadecola C, Pickel VM. Changes in dendritic distribution of NMDA-R1 receptor subunit in the dorsomedial nucleus tractus solitarius of rats with angiotensin II-induced hypertension. Soc for Neurosci 2003 [Google Scholar]
- Glass MJ, Kruzich PJ, Kreek M, Pickel VM. Decreased plasma membrane targeting of NMDA-NR1 receptor subunit in dendrites of medial nucleus tractus solitarius neurons in rats self-administering morphine. Synapse. 2004;53:191–201. doi: 10.1002/syn.20049. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Kruzich PJ, Colago EE, Kreek MJ, Pickel VM. Increased AMPA GluR1 receptor subunit labeling on the plasma membrane of dendrites in the basolateral amygdala of rats self-administering morphine. Synapse. 2005;58:1–12. doi: 10.1002/syn.20176. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S. Pretraining NMDA receptor blockade in the basolateral complex, but not the central nucleus, of the amygdala prevents savings of conditional fear. Behav Neurosci. 2003;117:738–750. doi: 10.1037/0735-7044.117.4.738. [DOI] [PubMed] [Google Scholar]
- Gracy KN, Pickel VM. Comparative ultrastructural localization of the NMDAR1 glutamate receptor in the rat basolateral amygdala and bed nucleus of the stria terminalis. J Comp Neurol. 1995;362:71–85. doi: 10.1002/cne.903620105. [DOI] [PubMed] [Google Scholar]
- Gracy KN, Dankiewicz LA, Koob GF. Opiate withdrawal-induced FOS immunoreactivity in the rat extended amygdala parallels the development of conditioned place aversion. Neuropsychopharmacol. 2001;24:152–160. doi: 10.1016/S0893-133X(00)00186-X. [DOI] [PubMed] [Google Scholar]
- Heimer L, Van Hoesen GW. The limbic lobe and its output channels: implications for emotional functions and adaptive behavior. Neurosci Biobehav Rev. 2006;30:126–147. doi: 10.1016/j.neubiorev.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Nguyen P, Sellers EM. The NMDA antagonist dizocilpine (MK801) attenuates motivational as well as somatic aspects of naloxone precipitated opioid withdrawal. Life Sci. 1992;50:PL167–172. doi: 10.1016/0024-3205(92)90452-u. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Vissel B, Bengoechea T, Crone S, Randolph-Moore L, Muller R, Brandon EP, Schaffer D, Verma IM, Lee K-F, Heinemann SF, Gage FH. Adeno-associated virus effectively mediates conditional gene modification in the brain. Proc Nat Acad Sci. 2002;99:2320–2325. doi: 10.1073/pnas.042678699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Chen SA, Kitamura O, Markou A, Koob GF. Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J Neurosci. 2006;26:5894–5900. doi: 10.1523/JNEUROSCI.0740-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, DeCola JP, Landeira-Fernandez J, Fanselow MS. N-methyl-D-aspartate receptor antagonist APV blocks acquisition but not expression of fear conditioning. Behav Neurosci. 1991;105:126–133. doi: 10.1037//0735-7044.105.1.126. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Ann Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Leranth C, Pickel VM. Electron microscopic pre-embedding double immunostaining methods. In: Heimer L, Zaborszky L, editors. Tract tracing methods 2, recent progress. New York: Plenum; 1989. pp. 129–172. [Google Scholar]
- Lim G, Wang S, Zeng Q, Sung B, Yang L, Mao J. Expression of spinal NMDA receptor and PKCgamma after chronic morphine is regulated by spinal glucocorticoid receptor. J of Neurosci. 2005 doi: 10.1523/JNEUROSCI.3768-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R, Stinus L, Gold LH, Koob GF. Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Ther. 1992;261:669–677. [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- Martin WR, Wikler A, Eades CG, Pescor FT. Tolerance to and physical dependence on morphine in rats. Psychopharmacologia. 1963;4:247–260. doi: 10.1007/BF00408180. [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, Lowell BB, Fanselow MS, Wilson MA, Tonegawa S. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007;317:94–99. doi: 10.1126/science.1140263. [DOI] [PubMed] [Google Scholar]
- McNally GP, Westbrook RF. Effects of systemic, intracerebral, or intrathecal administration of an N-methyl-D-aspartate receptor antagonist on associative morphine analgesic tolerance and hyperalgesia in rats. Behav Neurosci. 1998;112:966–978. doi: 10.1037//0735-7044.112.4.966. [DOI] [PubMed] [Google Scholar]
- Miserendino MJ, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yamamoto R, Fujio M, Suzuki Y, Minami M, Satoh M, Kaneko S. Involvement of the bed nucleus of the stria terminalis activated by the central nucleus of the amygdala in the negative affective component of morphine withdrawal in rats. Neurosci. 2005;134:9–19. doi: 10.1016/j.neuroscience.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Noda Y, Nabeshima T. Opiate physical dependence and N-methyl-D-aspartate receptors. Eur J Pharmacol. 2004;500:121–128. doi: 10.1016/j.ejphar.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster H. The fine structure of the nervous system. New York: Oxford University Press; 1991. [Google Scholar]
- Popik P, Danysz W. Inhibition of reinforcing effects of morphine and motivational aspects of naloxone-precipitated opioid withdrawal by N-methyl-D-aspartate receptor antagonist, memantine. J Pharmacol Exp Therap. 1997;280:854–865. [PubMed] [Google Scholar]
- Rasmussen K, Brodsky M, Inturrisi CE. NMDA antagonists and clonidine block c-fos expression during morphine withdrawal. Synapse. 1995;20:68–74. doi: 10.1002/syn.890200110. [DOI] [PubMed] [Google Scholar]
- Rezayof A, Golhasani-Keshtan F, Haeri-Rohani A, Zarrindast MR. Morphine-induced place preference: involvement of the central amygdala NMDA receptors. Brain Res. 2007;1133:34–41. doi: 10.1016/j.brainres.2006.11.049. [DOI] [PubMed] [Google Scholar]
- Ribeiro Do Couto B, Aguilar MA, Manzanedo C, Rodriguez-Arias M, Minarro J. Effects of NMDA receptor antagonists (MK-801 and memantine) on the acquisition of morphine-induced conditioned place preference in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:1035–1043. doi: 10.1016/j.pnpbp.2004.05.038. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacol. 2004;47:33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nature Immunol. 2007;8:665–668. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Stinus L, Risbrough VB, Koob GF. Clonidine blocks aquisition but not expression of conditioned opiate withdrawal in rats. Neuropsychpharmacol. 1998;19:406–416. doi: 10.1016/S0893-133X(98)00036-0. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Markou A, Gold LH, Stinus L, Koob GF. Relative sensitivity to naloxone of multiple indices of opiate withdrawal: a quantitative dose-response analysis. J Pharmacol Exper Ther. 1994;271:1391–1398. [PubMed] [Google Scholar]
- Schulteis G, Ahmed SH, Morse AC, Koob GF, Everitt BJ. Conditioning and opiate withdrawal. Nature. 2000;405:1013–1014. doi: 10.1038/35016630. [DOI] [PubMed] [Google Scholar]
- Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S, Duman RS, Storm D, Nestler EJ. Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci. 2002;22:3663–3672. doi: 10.1523/JNEUROSCI.22-09-03663.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South SM, Kohno T, Kaspar BK, Hegarty D, Vissel B, Drake CT, Ohata M, Jenab S, Sailer AW, Malkmus S, Masuyama T, Horner P, Bogulavsky J, Gage FH, Yaksh TL, Woolf CJ, Heinemann SF, Inturrisi CE. A conditional deletion of the NR1 subunit of the NMDA receptor in adult spinal cord dorsal horn reduces NMDA currents and injury-induced pain. J of Neurosci. 2003;23:5031–5040. doi: 10.1523/JNEUROSCI.23-12-05031.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinus L, Le Moal M, Koob GF. Nucleus accumbens and amygdala are possible substrates for the aversive stimulus effects of opiate withdrawal. Neurosci. 1990;37:767–773. doi: 10.1016/0306-4522(90)90106-e. [DOI] [PubMed] [Google Scholar]
- Svensson A, Pileblad E, Carlsson M. A comparison between the non-competitive NMDA antagonist dizocilpine (MK-801) and the competitive NMDA antagonist D-CPPene with regard to dopamine turnover and locomotor-stimulatory properties in mice. J Neural Transm. 1991;85:117–129. doi: 10.1007/BF01244704. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- Tsien JZ. Linking Hebb’s coincidence-detection to memory formation. Curr Opin Neurobiol. 2000;10:266–273. doi: 10.1016/s0959-4388(00)00070-2. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Saunders A, Commons KG, Liu XB, Peoples J. Evidence for coexistence of enkephalin and glutamate in axon terminals and cellular sites for functional interactions of their receptors in the rat locus coeruleus. J Comp Neurol. 2000;417:103–114. doi: 10.1002/(sici)1096-9861(20000131)417:1<103::aid-cne8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Wang H, Hu Y, Tsien JZ. Molecular and systems mechanisms of memory consolidation and storage. Prog Neurobiol. 2006;79:123–135. doi: 10.1016/j.pneurobio.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Nakagawa T, Yamamoto R, Maeda A, Minami M, Satoh M. Involvement of glutamate receptors within the central nucleus of the amygdala in naloxone-precipitated withdrawal-induced conditioned place aversion in rats. Jpn J Pharmacol. 2002;88:399–406. doi: 10.1254/jjp.88.399. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Ann Rev Pharmacol and Toxicol. 2003;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Rensing NR, Sinatra PM, Rothman SM, Wong M. Kainate seizures cause acute dendritic injury and actin depolymerization in vivo. J of Neurosci. 2007;27:11604–11613. doi: 10.1523/JNEUROSCI.0983-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Pan ZZ. Synaptic properties and postsynaptic opioid effects in rat central amygdala neurons. Neurosci. 2004;127:871–879. doi: 10.1016/j.neuroscience.2004.05.043. [DOI] [PubMed] [Google Scholar]
- Zhu W, Pan ZZ. μ-opioid-mediated inhibition of glutamate synaptic transmission in rat central amygdala neurons. Neurosci. 2005;133:97–103. doi: 10.1016/j.neuroscience.2005.02.004. [DOI] [PubMed] [Google Scholar]