

Abstract
EAAT2 is a predominantly astroglial glutamate transporter responsible for the majority of synaptic glutamate clearance in the mammalian CNS. Its dysfunction has been linked to many neurological disorders, including amyotrophic lateral sclerosis (ALS). Decreases in EAAT2 expression and function have been implicated in causing motor neuron excitotoxic death in ALS. Nevertheless, increasing EAAT2 expression does not significantly improve ALS phenotype in mouse models or in clinical trials. In the SOD1-G93A mouse model of inherited ALS, the cytosolic carboxy-terminal domain is cleaved from EAAT2, conjugated to SUMO1, and accumulated in astrocytes where it triggers astrocyte-mediated neurotoxic effects as disease progresses. However, it is not known whether this fragment is sumoylated after cleavage or if full-length EAAT2 is already sumoylated prior to cleavage as part of physiological regulation. In this study, we show that a fraction of full-length EAAT2 is constitutively sumoylated in primary cultures of astrocytes in vitro and in the CNS in vivo. Furthermore, the extent of sumoylation of EAAT2 does not change during the course of ALS in the SOD1-G93A mouse and is not affected by the expression of ALS-causative mutant SOD1 proteins in astrocytes in vitro, indicating that EAAT2 sumoylation is not driven by pathogenic mechanisms. Most interestingly, sumoylated EAAT2 localizes to intracellular compartments, while non-sumoylated EAAT2 resides on the plasma membrane. In agreement, promoting desumoylation in primary astrocytes causes increased EAAT2–mediated glutamate uptake. These findings could have implications for optimizing therapeutic approaches aimed at increasing EAAT2 activity in the dysfunctional or diseased CNS.
Keywords: Amyotrophic lateral sclerosis, excitotoxicity, protein trafficking, GLT-1
Introduction
The activity of EAAT2, a Na+-dependent, high affinity glutamate transporter, accounts for the majority of glutamate clearance in the CNS (Tanaka et al. 1997). Primarily expressed by astrocytes, EAAT2 curbs excitatory neurotransmission, maintaining glutamate homeostasis at the synapse (Kim et al. 2011). In organotypic spinal cord cultures, blocking EAAT2 activity or decreasing its expression causes an excitotoxic environment for motor neurons, which can lead to their death (Qian et al. 2011; Wallis et al. 2012; Wisman et al. 2003). Furthermore, decreased expression and function of EAAT2 has been reported in many neurological conditions and neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), a disease primarily of motor neurons (Foran and Trotti 2009). Excitotoxicity due to decreased EAAT2 expression results in some, but not nearly all of the motor neuron toxicity seen in ALS (Boillee et al. 2006). We previously found that a SUMO1-conjugated C-terminal fragment of EAAT2 (termed CTE-SUMO1) accumulates in ALS (Foran et al. 2011; Gibb et al. 2007). Small ubiquitin modifier 1 (SUMO1) is a post-translational modifier protein, which attaches to lysine residues via an isopeptide bond, often at a canonical consensus site (ΨKXE) (Sampson et al. 2001). Proteins modified with SUMO1 have altered behavior; including changes in localization, trafficking, and half-life (Fei et al. 2006; Singh et al. 2013). In physiological conditions, sumoylation is a dynamic process that requires the maturation of unbound, inactive SUMO1 by SUMO1/sentrin specific peptidases (Senps) before conjugation to its target proteins in a multi-step process (Sharma et al. 2013). The SUMO1 E2 conjugating enzyme (UBC9) is necessary and sufficient for SUMO1 conjugation to most target proteins (Su et al. 2012). Sumoylation is a reversible process and SUMO1 can be removed from target proteins, primarily by Senp1 (Hickey et al. 2012). Sumoylation can be mediated by cAMP (Jones et al. 2006), heat shock (Miller et al. 2013), and oxidative stress (de la Vega et al. 2012). Alteration to the sumoylation balance can profoundly alter a protein’s behavior and hence impact cellular functions. Altered balance of post-translational modifications to proteins, including sumoylation, has been found in neurodegenerative diseases including ALS (Foran et al. 2011; Foran et al. 2013; Kaikkonen et al. 2009; Mukherjee et al. 2009).
ALS is a progressive, ultimately fatal paralytic disorder, involving degeneration of both upper and lower motor neurons. Most of the human phenotype is recapitulated by a mouse model using constitutive overexpression of the human disease-causative SOD1-G93A mutation, thus enabling extensive investigation of mechanisms of motor neuron degeneration and the development of new therapeutics (Gurney et al. 1994). In SOD1-G93A mice, CTE-SUMO1 accumulates in the spinal cord with the progression of disease symptoms (Foran et al. 2011; Gibb et al. 2007). Moreover, expression of CTE-SUMO1 in astrocytes induces a toxic phenotype, leading to toxicity of neighboring motor neurons, suggesting that this sumoylated fragment might be pathogenic in ALS (Foran et al. 2011). CTE-SUMO1 could be derived from either a cleavage of already-sumoylated full-length EAAT2 or by the sumoylation of an already caspase-3-cleaved EAAT2 fragment. If the former is the case, questions arise regarding the role and impact of sumoylation on EAAT2: Is sumoylation of EAAT2 normal or part of ALS pathogenesis, and if normal, what is its function? The latter case would imply that sumoylation of the carboxy-terminal fragment of EAAT2 is a disease-driven process and would introduce aspects of disease pathology that have yet to be examined.
Here, we report that a fraction of full-length EAAT2 is conjugated to SUMO1 under physiological conditions both in cultured primary astrocytes and in vivo. Sumoylation of EAAT2 occurs rapidly in primary astrocytes, with or without disease-causative human SOD1 mutations. The extent of full-length EAAT2 sumoylation and the expression levels of primary sumoylation enzymes do not change during the course of ALS, suggesting that the disease mechanisms do not impact this post-translational modification of EAAT2 and possibly the sumoylation of other proteins. Interestingly, sumoylated EAAT2 predominates intracellularly while little to none is seen on the plasma membrane. Additionally, preventing sumoylation of EAAT2 in cultured astrocytes causes a shift from intracellular compartments to the plasma membrane and an increase in EAAT2-mediated glutamate uptake, while enhancing sumoylation increases intracellular localization. The effect of this change in localization on glutamate transport emphasizes the potential for this post-translational mechanism to impact the overall glutamate uptake capacity of astrocytes in the central nervous system. Inhibiting sumoylation of EAAT2 might even prove a viable goal in treating ALS, as it could both increase EAAT2 on the plasma membrane, and thus glutamate clearance from the synapse, and prevent CTE-SUMO1 formation in astrocytes, thus inhibiting related non-cell autonomous neurotoxicity.
Materials and Methods
Animal models
SOD1-G93A transgenic mice model of ALS [B6.Cg-Tg(SOD1 G93A)1Gur/J] were purchased from Jackson Laboratories (stock #004435) and bred in our animal facility. Sprague Dawley rats were purchased from Charles Rivers (strain code 400).
Preparation of homogenates from rat and mouse CNS
Spinal cords of SOD1-G93A or non-transgenic SOD1-WT age-matched littermate mice were homogenized in 20 volumes of modified RIPA buffer containing Protease Inhibitor Cocktail (PIC) tablet (Roche) using a Teflon-glass homogenizer (600 rpm). Similar homogenization procedure was followed for rat forebrains harvested from adult rats. Homogenates were briefly sonicated and heavy nuclei and unbroken cells removed by low-speed centrifugation. Supernatant was snap-frozen in liquid nitrogen for later processing. Protein concentration was determined on a separated aliquot of the supernatant.
Immunoprecipitation and western blotting
250 μl of CNS homogenates in RIPA buffer containing 500 μg of total proteins were pre-cleared (4 hours at 4 °C) by adding 20 μl of Protein A magnetic beads (Qiagen). After beads removal, 10 μg of anti-EAAT2 antibody or control IgG were added to the sample and incubated overnight at 4 °C. Immunoprecipitation was achieved by adding 20 μl of Protein A beads. After 4-hour incubation at 4 °C, beads were washed three times with ice cold RIPA buffer containing PIC tablet (Roche), re-suspended in sample buffer containing 0.05% BME, boiled and the supernatant resolved on SDS-PAGE Tris-Glycine gels (Bio-Rad), transferred to a membrane, probed with antibody or IgG control and developed. For analysis of cytoplasm and membrane enriched fractions, tissue homogenates were prepared in ice-cold PBS buffer containing 1% CHAPS and PIC. Residual intact cells and large tissue debris were removed by centrifugation. Membrane fraction was pelleted at 100,000 × g for 1 hour at 4°C and re-suspended in 1% SDS buffer. Supernatant and pellet were mixed with 1% SDS (enriched cytoplasm fraction) or 1% CHAPS (enriched membrane fraction), pre-cleared, and immunoprecipitated using the Pierce Crosslink IP kit, using 10 μg of anti-EAAT2 antibody (custom Ab against the C-terminal domain). Cytosolic and membrane fractions were analyzed for markers of cross-contamination (e.g. cadherin and GAPDH), while immunoprecipitated fractions were analyzed for EAAT2 (ABR518, 1:500) and SUMO1 (Santa Cruz cat.#5308, 1:100).
Immunofluorescence analysis
Primary astrocytes or HEK-293T cells grown on slides were washed once with warm PBS, fixed with 4% paraformaldehyde for 10 minutes and treated with ice-cold methanol for 2 minutes. After the cells were incubated in blocking buffer (5% donkey serum, 0.5% Triton in PBS) for 1 hour at room temperature, primary antibody was added overnight at 4°C. Cells were washed three times with PBS and incubated in AlexaFluor secondary antibody (Invitrogen; 1h at room temp.). Slides were mounted with Prolong Gold Antifade reagent with DAPI (Invitrogen), imaged with confocal microscope (Olympus FV1000D), and images analyzed with Olympus Fluoview FV1000.
Caspase-3 cleavage assay
Caspase-3 cleavage assay was performed on rat cortex homogenates as previously described (Boston-Howes et al. 2006). Cleavage reaction was run for 5 hours at 37 °C with 200 ng of active Caspase 3 (Sigma). SDS sample buffer with 0.05% BME was added to the samples, which were boiled for 10 minutes to stop the reaction and then immediately resolved by SDS-PAGE.
Glutamate uptake assay
HEK293T cells (8×105/well) or astrocytes (5×105/well) were plated on 35mm culture plates. Twenty-four hours later, cells were transfected with Lipofectamine LTX (Invitrogen) overnight and washed 3 times with uptake buffer containing 140 mM NaCl, 2.5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 1.2 mM K2HPO4, 15 mM HEPES/Na and 10 mM glucose, pH 7.4. Transport assays were performed at room temperature after the cells reached near confluency. Cells were incubated with uptake buffer containing increasing concentrations of glutamate isotopically diluted with 3H-L-glutamate (S.A. 51.9 Ci/mmol) for 10 min (Trotti et al. 2001). Transport assay was stopped by rinsing three times with ice-cold choline-containing uptake buffer (no Na+ ions). Cells were lysed with NaOH (0.5N) + Triton X-100 (0.1%), and the radioactivity content in solution quantified using a scintillation counter. Uptake values were normalized by total protein content in each well. Na+-dependent transport was calculated to be the difference between the amount of radioactivity measured in cells incubated in Na+-containing uptake buffer and cells incubated in choline+-containing uptake buffer.
Primary astrocyte cultures
Primary cultures of astrocytes were prepared from post-natal mice (P1-P3) (Gibb et al. 2007). Cell monolayers, ≥95% astrocytepure as determined by GFAP immunoreactivity, were trypsinised and plated onto either 6-well plates or glass slides.
Antibodies
EAAT2 antibodies include EAAT2-H85 (SantaCruz, cat# sc-15317), EAAT2 epitope 556-573 (Pierce Scientific, cat# PA3-040A), ABR518-536 (epitope 518-536 of the mouse sequence; custom-made from Affinity BioReagents; this antibody cross-reacts with human, rat and mouse EAAT2). Other primary antibodies used were anti-Myc (Clontech, cat# 631206), SUMO1 (SantaCruz, cat# sc-5308, UBC9 (Cell Signaling, cat# 4786), GFP (Clontech, cat# 632592), Flag (Sigma, cat# F3165), GAPDH (Fitzgerald, cat# 10R-G109A), Senp1 (Abcam, Cat# 58417), HA (Clontech, cat# 631207).
Plasmids and viral constructs
Plasmids encoding CTE or CTE-SUMO1 (in which the N-terminus of SUMO1 is fused in frame with the C-terminus of CTE) fragments were obtained as previously described (Gibb et al. 2007). The reactive C-terminus of the SUMO1 moiety was inactivated by removing one glycine residue of the conserved “GG” active region (Martin et al. 2007), hence preventing unwanted sumoylation reactions. Both CTE and CTE-SUMO1 were inserted into pCMV-Myc (Clontech) at Kpn1 and EcoR1-blunted sites. CTE-Knull was created by single site mutations at each of the 6 lysines of the myc-CTE construct to arginines using QuikChange lightening kit (Agilent). CTE-K570R was created by single site mutation at the single lysine at K570 to arginine. CTE-Knull-R570K was created by mutating the 570 arginine to lysine in the CTE-Knull construct.
Myc-EAAT2 and truncated Myc-EAAT2 (Myc-trEAAT2) were obtained as previously described (Boston-Howes et al. 2006). EAAT2-SUMO1 was created by combining the EAAT2 and CTE-SUMO1 constructs using Kpn1 and blunted EcoR1/Nco1 sites. HA tagged dominant negative UBC9 (DN-UBC9) was obtained from Addgene (#14439) thanks to Peter Howley and Flag-Senp1 was obtained from Addgene (#17357) thanks to Edward Yeh.
To create adenoviral vectors expressing human mutants and wild-type SOD1 fused with GFP, the corresponding cDNAs were inserted into pENTR for further Gateway recombination in adenoviral expression plasmids. Similarly Myc-EAAT2 was inserted into pENTR. Viral vectors were produced and amplified in HEK 293A cells (ViraPower Adenoviral Expression System; Invitrogen). Viral infections were performed on astrocyte monolayer. Adenoviral vectors were added in OPTI-MEM medium containing Primocin (Invivogen), and the medium changed 18 hours later. Multiplicity of infection (m.o.i.) used in these experiments ranged from 3-6.
Statistical analysis
Comparisons were done by two tailed t-test analysis except where noted otherwise in figure legends. Data are expressed as average ± sem. p<0.05 was considered statistically significant.
Results
EAAT2 is sumoylated in astrocytes in primary culture
Caspase-3 cleaves the astroglial glutamate transporter EAAT2 in the SOD1-G93A mouse model of ALS, generating a carboxy-terminal fragment (CTE) that is SUMO1-conjugated and accumulates in spinal cord astrocytes during the progression of the symptomatic phase of ALS (Gibb et al. 2007). These observations prompted us to examine whether full-length EAAT2 is itself a substrate for SUMO1 conjugation, particularly in pertinent cell culture models that endogenously express it, such as primary astrocytes. Evidence of EAAT2 sumoylation in COS7 cells co-transfected with the sumoylating enzyme UBC9, His-tagged SUMO1, and EAAT2 was previously reported by us. The homogenate from these cells was run through nickel columns to purify sumoylated His-tagged proteins, revealing EAAT2 as one protein substrate (see Fig.4B in Gibb et al. 2007). It was also found that UBC9, SUMO1 and Aba2, three proteins involved in the sumoylation reaction, bind to the carboxy-terminal domain of EAAT2 (Gibb et al. 2007). To determine if astrocytes are capable of sumoylate endogenously EAAT2, astrocytes primary culture were transduced with AdV-Myc-EAAT2 (>90% transduction efficiency). An anti-Myc antibody was used to immunoprecipitate EAAT2 at 8-hour intervals post-transfection, in order to isolate the ability, contribution, and time course of SUMO1 conjugation on the de novo expressed Myc-EAAT2 rather than monitoring sumoylation of endogenous EAAT2. SUMO1 conjugation to Myc-EAAT2 was detected within 8 hours post-transfection of Myc-EAAT2 (Fig. 1). We did not see statistically appreciable differences in sumoylation at following 8-hour time increments, suggesting that SUMO1 attachment to EAAT2 can reach steady state levels in less than 8 hours. It is worth noting, however, that the extent of Myc-EAAT2 sumoylation at 8 hours was more variable than at longer time points, suggesting that the kinetics of sumoylation can vary. Though this hypothesis was not explored further, it could be subject to future investigations.
Fig 1. EAAT2 is sumoylated in primary astrocytes.
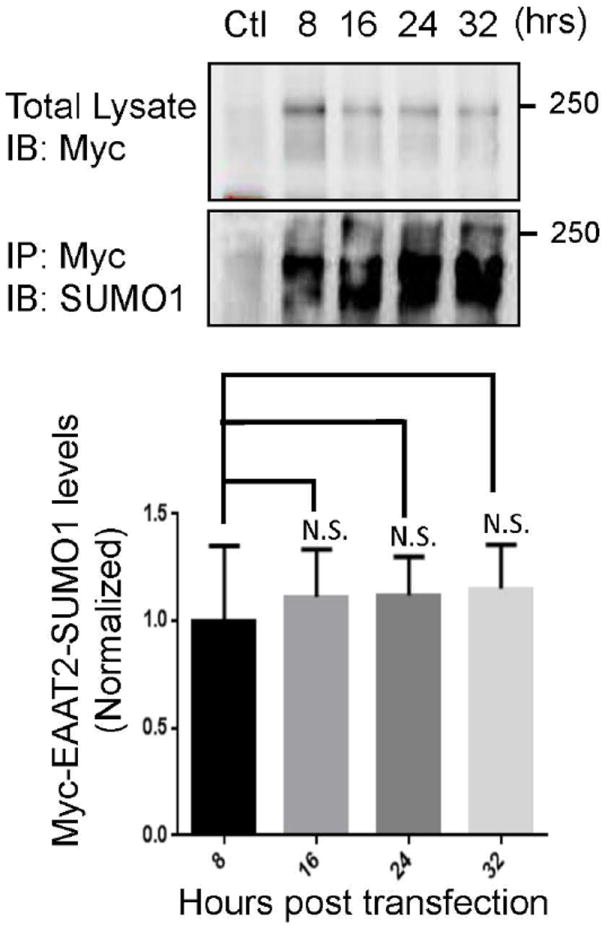
Primary astrocytes were treated with AdV-Myc-EAAT2 (MOI 3) and collected at 8, 16, 24 or 32 hours. Virus treated astrocytes were analyzed for transduction by western blotting for the Myc tag (anti-Myc, Clontech, Cat #631206, 1:1000). Exogenous Myc-EAAT2 was immunoprecipitated using an antibody to the myc tag (anti-Myc, Clontech, Cat #631206, 10 μg) and was immunoblotted for SUMO1 (anti-SUMO1, SantaCruz, cat.# sc-5308, 1:500), showing de novo sumoylation of the exogenous Myc-EAAT2. The immunoprecipitated Myc was also positive for Myc indicating a successful immunoprecipitation (not shown). All immunoprecipitations were normalized to their own immunoprecipitation control. There was no increase in the sumoylation of the exogenous EAAT2 over time, indicating that the SUMO1 conjugation occurs quickly and reaches equilibrium within 8 hours of protein expression (n=3 per group ANOVA ((F3,11)=0.15, p=.92)).
Lysine 570 is a preferential, although not unique, site for sumoylation within the EAAT2 c-terminus
Sumoylation occurs exclusively at lysine residues in target proteins, preferentially within canonical consensus sites identified by the specific amino acid sequence, -ΨKXE-, where Ψ is an aromatic amino acid and X is a generic one (Sampson et al. 2001). EAAT2 has 31 lysine residues throughout its primary structure, six of which are within the c-terminal domain (CTE) that was previously found to be cleaved by caspase-3. Of these lysines, only the residue located near the c-terminal end of EAAT2 (K570) resides within a canonical consensus site for sumoylation. To determine whether K570 was a target site of sumoylation, COS7 cells were co-transfected with constructs encoding a myc-tagged version of the EAAT2 c-terminus fragment (myc-CTE), the sumoylating enzyme UBC9, and SUMO1, to enable sumoylation reactions. COS7 cells were chosen instead of primary astrocytes to maximize the efficiency of transfection and improve chances of detecting sumoylation of CTE by immunoblot analysis. Analysis was restricted to the CTE fragment instead of the full-length transporter for the following reasons: 1) It is the domain in EAAT2 to which UBC9, SUMO1, and Aba2 were found to bind in a yeast two-hybrid screening assay (Gibb et al. 2007); 2) It was found to have neurotoxic effects in a motor neuron-astrocyte cell model system (Foran et al. 2011); 3) It is entirely cytosolic and therefore likely to be a preferred target region for the sumoylation enzyme machinery, which also resides in the cytosol. Twenty-four hours post-transfection of myc-CTE, SUMO1, and UBC9, a myc+ band at approximately 25 kDa was detected by immunoblotting (Fig.2A), indicating that the myc-CTE peptide, seen as a 10 kDa myc+ band (Fig.2A) underwent sumoylation. The 25 kDa myc+ band was also at a molecular mass similar to the artificial, translationally fused myc-CTE-SUMO1 peptide that was expressed in COS7 cells as positive control (Fig.2A) (Gibb et al. 2007). Mutation of all of the lysines in myc-CTE (CTE-Knull) to non-sumoylatable arginines (Steffan et al. 2004) prevented sumoylation as shown by the lack of appearance of the myc+ CTE-SUMO1 band at the 25-kDa mark. Mutating lysine 570 to arginine within the sumoylation consensus site (CTE-K570R), but maintaining the other lysine residues in myc-CTE significantly reduced sumoylation of this fragment (by approx. 50%), indicating that these lysine residues, though not within a canonical consensus site, could be targeted for sumoylation in vitro. Concurrently, restoring the consensus site in CTE-Knull (CTE-Knull-R570K) reinstated sumoylation of myc-CTE to nearly basal levels (Fig.2A,B). This further suggests that while other lysines can be sumoylated, the consensus site is responsible for the majority of sumoylated CTE.
Fig 2. Lysine 570 is the primary target of sumoylation in the c-terminal tail of EAAT2.
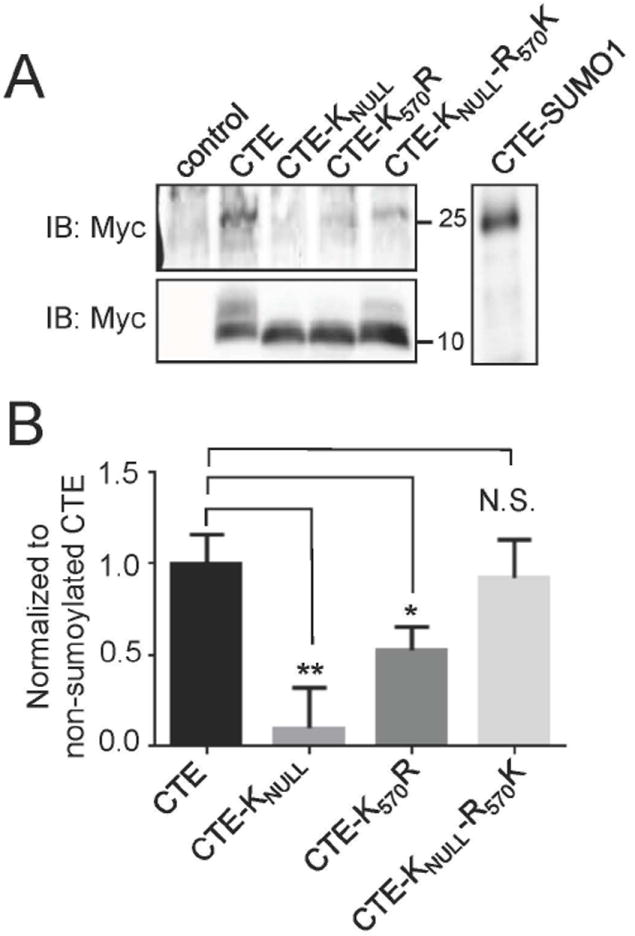
(A) Cos7 cells were transfected with Myc-CTE, Myc-CTEKnull, Myc-CTEK570R or Myc-CTEKnullR570K in conjunction with SUMO1 and UBC9. The immunoblot of Myc (anti-Myc, Clontech, Cat #631206, 10 μg) showed a 25 kDa band in the CTE lane indicating sumoylation of CTE (Gibb et al. 2007). There was no 25 kDa band in the CTEKnull lane indicating that no sumoylation occurs. The band in the CTEK570R lane was reduced indicating that there is less sumoylation of CTE when the consensus site was destroyed. Sumoylation was nearly restored when the only lysine available was the lysine 570. All transfected cells had a 10 kDa band, indicating mostly unsumoylated exogenous CTE. (B) Sumoylation of CTE was quantified with all sumoylated CTE bands normalized to their non-sumoylated CTE bands (n=3, ANOVA ((F3,11)=12.47, p=.002) with post hoc Dunnet test, Myc-CTEKnull p<0.01, Myc-CTEK570R, p<0.05, Myc-CTEKnullR570K, p>0.05).
EAAT2 is SUMO1 conjugated in vivo
We next wanted to determine whether EAAT2 was constitutively sumoylated in vivo. Immunoprecipitations of normal rat cerebral cortex homogenates were performed with anti-EAAT2 and anti-SUMO1 antibodies and showed that EAAT2 was indeed sumoylated in vivo (Fig. 3A, B). The presence of a 25 kDa band immunopositive for both the EAAT2 c-terminus (ABR518-536) and SUMO1 detected following the incubation of rat cortex homogenate with activated caspase-3 (200 ng; 5 hrs. at 37 °C) also confirmed sumoylation of EAAT2 (Fig.3C). Interestingly, following caspase-3 incubation, a band at 10 kDa was immunopositive for EAAT2 but not SUMO1, demonstrating that some but not all EAAT2 was sumoylated at the C-terminal end (arrow, Fig. 3C). Similarly, immunoprecipitation experiments performed in spinal cord homogenates of non-ALS control human post-mortem samples (obtained from Dr. Maragakis, Johns Hopkins University) demonstrated SUMO1 conjugation of EAAT2 (Fig.3D, E). Interestingly, anti-SUMO1 antibody seemed to preferentially probe the multimeric forms of EAAT2, suggesting that sumoylation might drive EAAT2 oligomerization, at least in vitro.
Fig. 3. EAAT2 is sumoylated in human and rat CNS.
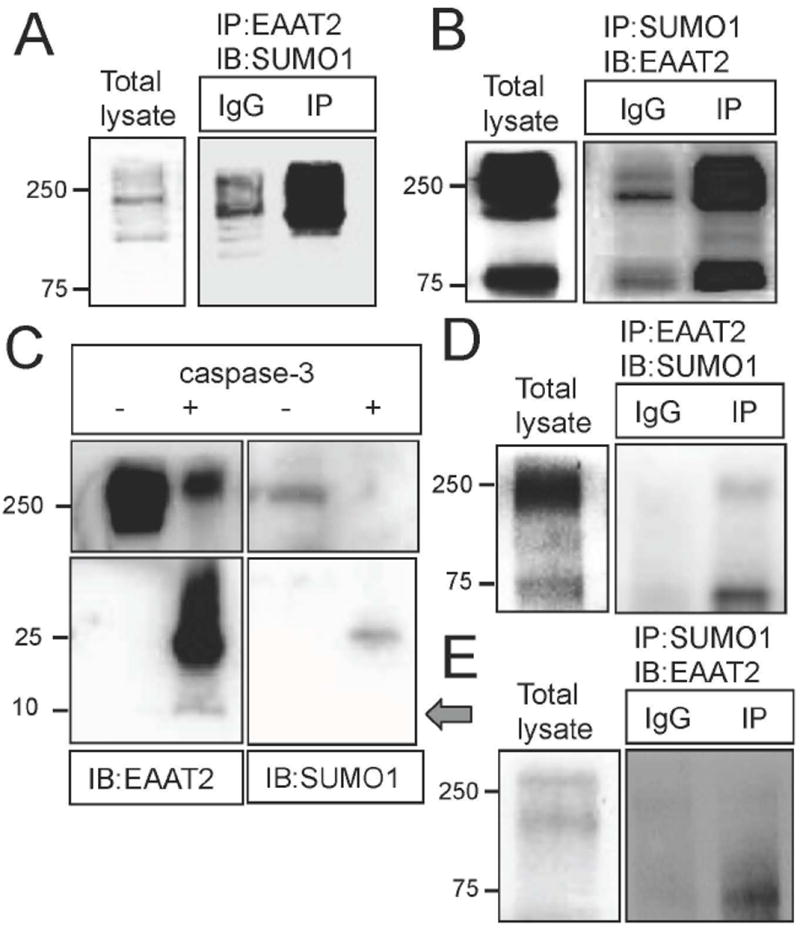
(A) Rat cortices were immunoprecipitated for EAAT2 (using custom antibody against residues 518-536) and analyzed by western blotting with anti-SUMO1 antibody, showing sumoylated EAAT2. IgGs (Santa Cruz, cat.#sc-2027, 10 μg) were used as control for immunoprecipitation. The immunoprecipitated EAAT2 (ABR518-536, 10 μg) was also positive for another epitope of EAAT2 (a.a. 1-85, Santa Cruz, cat.# sc-15317, 1:1000) indicating a successful immunoprecipitation (not shown). (B) Rat cortices were also immunoprecipitated for SUMO1 (anti-SUMO1, Santa Cruz, cat.# sc-5308, 10 μg) and analyzed by western blotting with anti-EAAT2 antibody (ABR518-536, 1:10,000), confirming sumoylated EAAT2. The immunoprecipitated SUMO1 was also positive for SUMO1 indicating a successful immunoprecipitation (not shown). (C) Rat cortex homogenate was treated without (-) or with (+) activate Caspase-3 (Sigma, cat.#C5482, 200 ng, 5hr, 37°C) and the products were analyzed by western blotting with anti-EAAT2 antibody (ABR518-536; 1:10,000). Samples treated with activated caspase-3 showed both a 10 kDa and a 25 kDa band, the latter of which was also positive for SUMO1 (anti-SUMO1, Santa Cruz, cat.# sc-5308, 10 μg), indicating that the EAAT2 had been sumoylated prior to caspase-3 treatment. The lysate incubated without active caspase-3 showed no cleavage of EAAT2, indicating that the caspase-3 cleavage was specific. (D) EAAT2 was immunoprecipitated (ABR518-536, 10 μg) from human post-mortem spinal cord homogenates and stained for SUMO1 (anti-SUMO1, Santa Cruz, cat.# sc-5308, 1:500), indicating some sumoylated EAAT2 in normal human spinal cord. (E) SUMO1 (anti-SUMO1, Santa Cruz, cat.# sc-5308, 10 μg) was also immunoprecipitated from human post-mortem spinal cord homogenates and stained for EAAT2 (ABR518-536, 1:10,000), confirming sumoylated EAAT2 in normal human spinal cord.
Our previous work showed that a SUMO1-conjugated fragment of EAAT2 (CTE-SUMO1) accumulates in the spinal cord of symptomatic SOD1-G93A mice over progression of ALS (Gibb et al. 2007). This accumulation is not paralleled by an increase in EAAT2 conjugated to SUMO1. Analysis of spinal cord lysates from pre-symptomatic (75 days old) and symptomatic (140 days old) SOD1-G93A mice showed comparable levels of sumoylation for the immunoprecipitated EAAT2 (Fig. 4A, B), suggesting that perhaps more EAAT2-SUMO1 may be produced during disease progression, but a portion of modified protein is then cleaved by caspase-3, generating the CTE-SUMO1 fragment.
Fig 4. Sumoylation of EAAT2 is not altered by disease progression in the SOD1-G93A mouse.
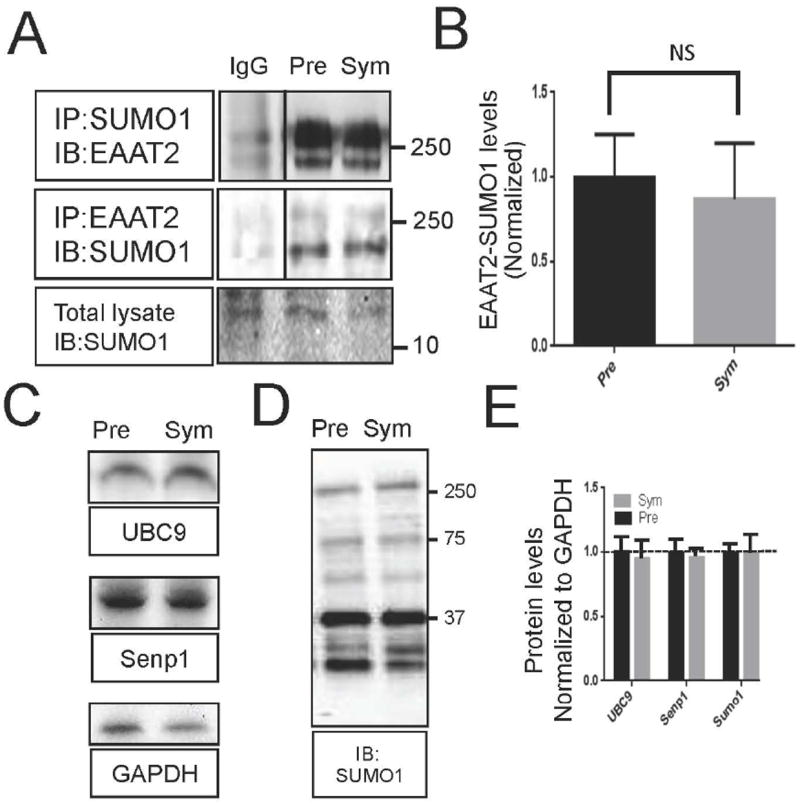
Spinal cord homogenate from both presymptomatic (65 days old) and symptomatic (120 days old) G93A-SOD1 mice were analyzed by western blotting for key proteins involved in sumoylation. (A) Homogenates were immunoprecipitated with anti-SUMO1 antibody (anti-SUMO1, Santa Cruz, cat.# sc-5308, 10 μg) and immunoblotted for EAAT2 (ABR518-536, dil. 1:10,000) or immunoprecipitated with anti-EAAT2 antibody (ABR518-536, 10 μg) and immunoblotted for SUMO1 (anti-SUMO1, Santa Cruz, cat.# sc-5308, dil. 1:500), demonstrating similar levels of sumoylation of EAAT2. (B) Three spinal cords from each age group were analyzed for quantification of EAAT2-SUMO1. (n=3, Two-tailed T-test (P>0.05)). (C) Homogenates were stained for proteins involved in sumoylation; UBC9 (Cell Signaling, cat.# 4786, dil.1:1000) and Senp1 (Abcam, cat.# 58417, dil. 1:1000). (D.) Homogenates were stained with anti-SUMO1 antibody (Santa Cruz, cat. # sc-5308, dil. 1:500). (E) Three spinal cords from each age group were used for analysis. All blots were normalized to the expression levels of GAPDH (Fitzgerald, cat.# 10R-G109A, dil. 1:25,000) and quantified. No change was seen between the two groups (n=3, two tailed T-test, p>0.05).
To further test whether EAAT2 sumoylation could be affected by the presence of ALS-causative mutant SOD1 proteins, primary mouse spinal cord astrocytes were transduced with human SOD1-WT as control, human SOD1-G93A, or human SOD1-A4V (another ALS-causative SOD1 mutation that is most severe in humans) and formation of EAAT2-SUMO1 was then assessed by immunoprecipitation. Quantification of the sumoylated EAAT2 in the immunoprecipitates normalized by the total amount of EAAT2 in astrocytes showed similar levels of EAAT2 sumoylation across the different groups (Suppl.Fig. 1A). It is worth noting that the total expression levels of EAAT2 decreased upon expression of the ALS-linked SOD1 mutations, in accordance with previous reports (Bristol and Rothstein 1996; Sasaki et al. 2000) (Suppl.Fig. 1B). The expression levels of the human SOD1 transgenes were verified to be comparable in all groups (Suppl.Fig. 1B), ruling out any unintended variation in EAAT2 sumoylation due to differential expression of the SOD1 transgenes.
Enzymes of the sumoylation machinery are not altered in the SOD1-G93A mouse spinal cord over the course of disease
As sumoylation of EAAT2 did not change with the occurrence of symptoms in the SOD1-G93A mice, we next wanted to determine if expression of enzymes involved in sumoylation of substrates did change. Expression levels of key proteins of sumoylation were determined by immunoblot analysis in spinal cords of SOD1-G93A mice at presymptomatic (75 days old) and symptomatic stage (140 days old). There were no differences in the expression levels of the SUMO1 E2 conjugating enzyme, UBC9, nor of the SUMO1 maturation and de-sumoylation protein, Senp1, suggesting steady sumoylation activity in the mouse model of ALS (Fig. 4C-E).
Intracellular localization of EAAT2 is regulated by SUMO1 conjugation
Previous studies have established that sumoylation of proteins impacts their cellular distribution and acts to redirect these substrates to different cellular compartments (Huang et al. 2012). We wanted to examine whether SUMO1 conjugation to EAAT2 could affect its cellular partition. HEK 293T cells were used as a heterologous cell model system in which to express EAAT2 and study the impact of sumoylation on localization and function. The EAAT2 proteolytic profile in these cells suggested that they were incapable of endogenously conjugating SUMO1 to EAAT2. Addition of active caspase-3 to a homogenate of EAAT2-expressing HEK 293T cells produced a single EAAT2 fragment (≤10 kDa) detected by the carboxy-terminal domain antibody, ABR518-536 (Suppl.Fig.2). This fragment likely represented the non-sumoylated CTE fragment since a band comparable in size was produced when a cell-free reaction mixture consisting of in vitro translated 35S-EAAT2 was treated with active caspase-3 (Boston-Howes et al. 2006). Conversely, mouse spinal cord homogenates treated with active caspase-3 produced a ~25 kDa fragment immunopositive for ABR518-536, which we characterized as the sumoylated CTE fragment (Gibb et al. 2007) (Suppl.Fig.2, Fig. 3C), indicating that EAAT2 was sumoylated in vivo (Suppl.Fig. 2). Therefore, HEK 293T cells offered an ideal system in which sumoylation of EAAT2 could be ectopically manipulated.
To study whether sumoylation is involved in governing the localization of EAAT2 within the cell, we first transfected HEK293T cells with a plasmid encoding Myc-EAAT2 and analyzed its cellular distribution by confocal microscopy. The non-sumoylated transporter displayed exclusive localization to the plasma membrane (Fig. 5A). However, when HEK cells were transfected with Myc-EAAT2, along with the sumoylating enzyme UBC9 and SUMO1, there was an increased amount of EAAT2 trapped in the cytoplasm (Fig. 5B). Confocal analysis revealed a preponderance of intracellular SUMO1 despite the fact that a fraction of EAAT2 is expressed at the plasma membrane. Similarly, HEK293T cells transfected with Myc-EAAT2-SUMO1, in which SUMO1 is artificially fused to EAAT2 via a peptide bond on lysine 573 and therefore is not prone to desumoylation, displayed extensive clustering of EAAT2 immunoreactivity inside the cells (Fig. 5C). It appeared, however, that some EAAT2-SUMO1 escaped intracellular retention and was directed to the plasma membrane, likely due to overexpression of the construct (Fig. 5C).
Fig 5. Sumoylation of EAAT2 alters EAAT2 localization in a heterologous system.
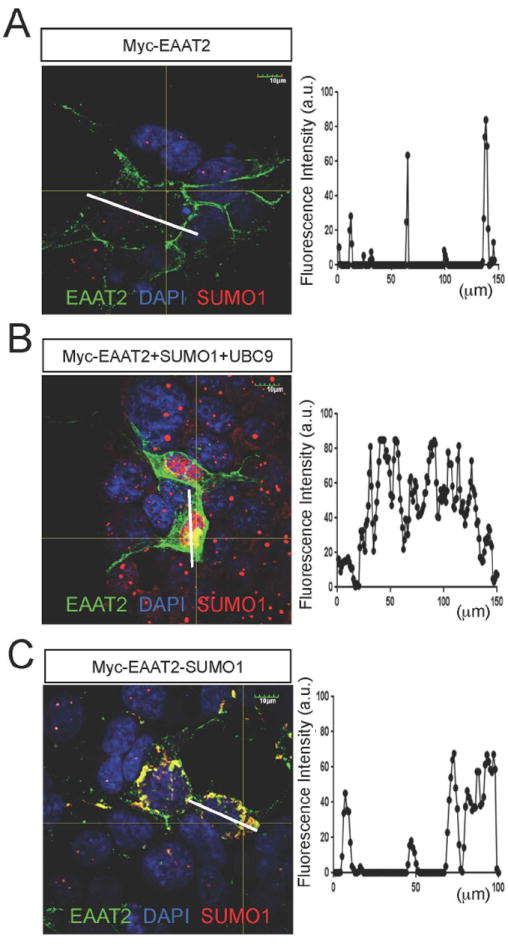
HEK293T cells were transfected with myc-EAAT2 alone (A), myc-EAAT2 with UBC9 and SUMO1 (B), or myc-EAAT2-SUMO1 (C). (A) Confocal analysis of HEK293T cells expressing Myc-EAAT2 and stained with anti-EAAT2 (ABR518-536, dil. 1:100) and anti-SUMO1 (Santa Cruz, cat.# sc-5308, dil. 1:50) showed that Myc-EAAT2 was primarily localized to the plasma membrane. The intensity of the fluorescence along the central axis of a representative astrocyte was graphed to show the subcellular localization of EAAT2 (ImageJ). (B) HEK293T cells transfected with myc-EAAT2 as well as SUMO1 and UBC9 showed increased cytoplasmic localization of EAAT2 (anti-EAAT2, ABR518-536, dil. 1:100, anti-SUMO1, Santa Cruz, cat# sc-5308, 1:50). (C.) Myc-EAAT2-SUMO1 had a propensity to form cytoplasmic aggregates (anti-EAAT2, ABR518-536, dil. 1:100, anti-SUMO1, Santa Cruz, cat# sc-5308, 1:50).
In concordance, overexpression of wild type UBC9 in astrocytes increased distribution of EAAT2 into intracellular pools (Fig. 6A), leading to the clearing of EAAT2 from the plasma membrane. Conversely, the expression of a dominant negative form of UBC9, with the active cysteine mutated to serine, (DN-UBC9) allowed EAAT2 to traffic more to the plasma membrane (Fig. 6B). Moreover, preferential plasma membrane localization was observed with a truncated form of Myc-EAAT2, in which the last 63 amino acids of the C terminus were removed in order to avoid endogenous sumoylation of the transporter (Fig. 6C). In sharp contrast, the endogenous EAAT2, which was immunostained by an antibody reactive to the c-terminal tail of EAAT2, retained its cytoplasmic localization (Fig. 6C).
Fig 6. Sumolyation alters EAAT2 localization in primary astrocytes.
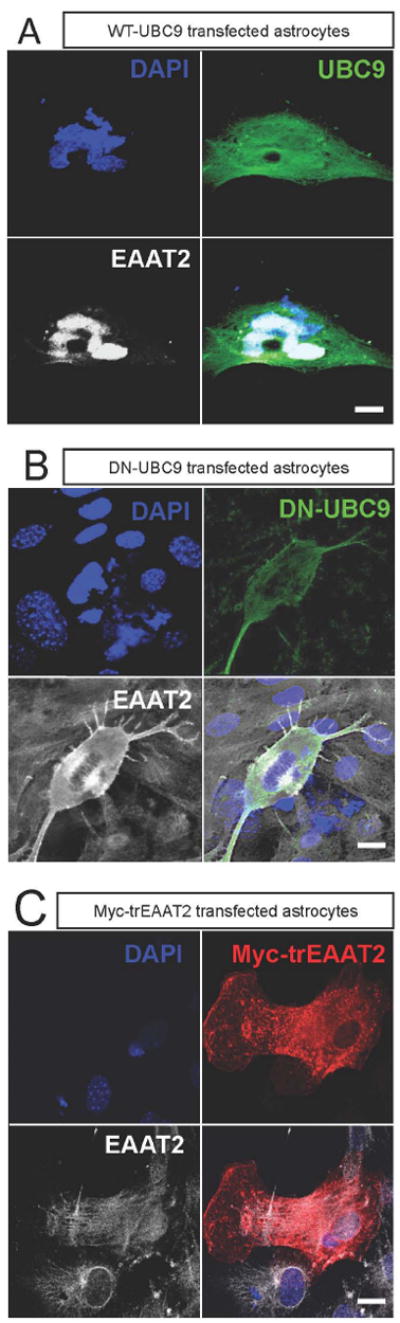
Astrocytes in culture were transfected (0.8 μg plasmid DNA/condition) with: (A) WT-UBC9, (B) dominant negative-UBC (HA-dn-UBC9), or (C) Myc-trEAAT2 (EAAT2 missing the last 67 amino acids of the c-terminus). (A) Intracellular localization of EAAT2 is increased by overexpression of wild type UBC9. Astrocytes were stained with anti-EAAT2 antibody (ABR518-536, dil. 1:100) and anti-UBC9 (Cell Signaling, cat.# 4786,1:100). (B) Expression of dominant negative UBC9 (HA-dnUBC9) causes dispersal of EAAT2 relative to wild type UBC9 overexpression. Astrocytes were stained with anti-EAAT2 antibody (ABR518-536, 1:100) and anti-HA (Clontech, cat #631207, dil.1:100). (C) The truncated form of EAAT2 (myc-trEAAT2), which lacks the c-terminus domain, is robustly expressed at the plasma membrane, away from the intracellular pools of the endogenous EAAT2. Staining for trEAAT2 was performed with anti-Myc (Clontech, cat.# 631206, dil. 1:1000), while staining for endogenous EAAT2 was completed with the anti-EAAT2 antibody (ABR518-536, dil. 1:100).
Primary astrocytes, under our culturing conditions, endogenously express EAAT2. Plasma membrane localization of EAAT2 was also detectable in confocal micrographs (arrow, Fig. 7A); however, a large portion of EAAT2 was retained in the cytoplasm (Fig. 7A). Transfection of the desumoylating enzyme, Senp1, promoted a redistribution of the EAAT2 pool, with much less intracellular EAAT2 aggregates and more EAAT2 localized at the plasma membrane (Fig. 7B), suggesting that desumoylation of the intracellular pool of EAAT2 was required to allow partition of the transporter to the cell surface. In agreement with the redistribution towards the plasma membrane induced by the desumoylating enzyme, Senp1, overall glutamate uptake in Senp1-transfected astrocytes increased by ~30% (Fig. 7C), despite modest levels of transfection efficiency (~30%), while the EAAT2-mediated uptake component, parsed out by DHK treatment, increased by ~60% (Fig. 7D). Interestingly, the DHK-insensitive, non-EAAT2 mediated, glutamate uptake of the astrocytes was not affected by Senp1 transfection as uptake levels in control and Senp1-transfected astrocytes were not significantly different (Fig. 7C), suggesting that SUMO1 conjugation/deconjugation impacts only the glutamate transport activity credited to EAAT2.
Fig 7. Increasing desumoylation disperses intracellular pools of EAAT2.
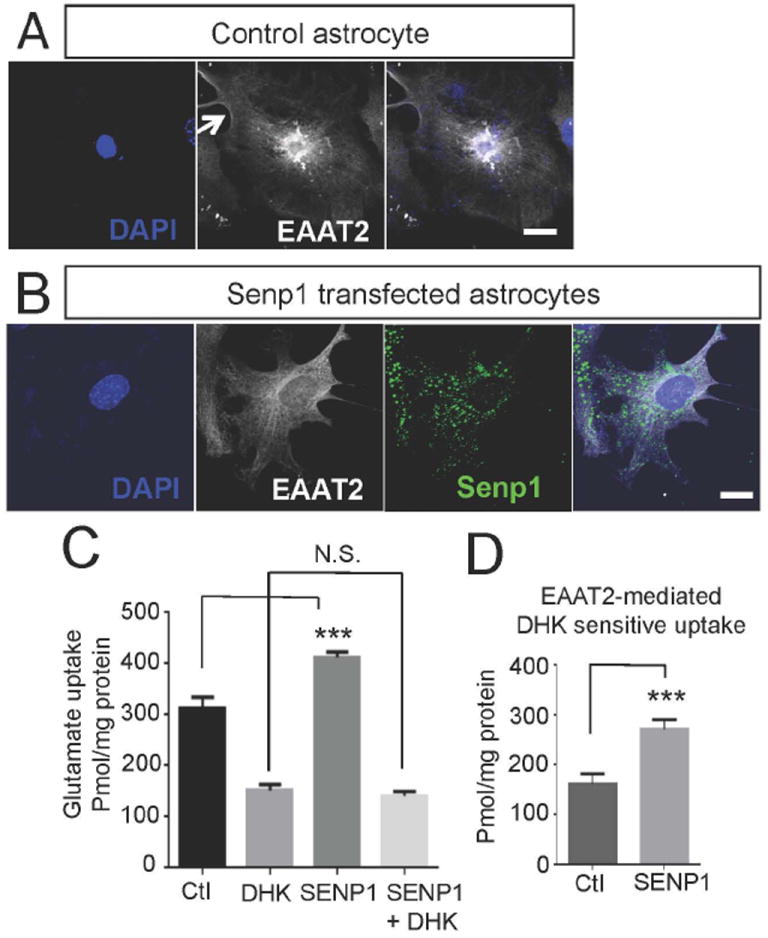
Primary astrocytes were transfected with control plasmid (pcDNA3.1+) or Flag-Senp1 (0.8 μg of plasmid DNA/condition). (A) EAAT2 formed intracellular pools in control transfected astrocytes transfected. Similar pattern of staining is observed in non-transfected astrocytes (not shown), suggesting that transfection of pcDNA3.1 per se does not affect the physiological localization pattern of EAAT2 (anti-EAAT2, ABR518-536, dil. 1:100). (B) These intracellular pools were not present when astrocytes were transfected with Flag-SENP1 (anti-EAAT2, ABR518-536, 1:100, anti-Flag, Sigma, cat.# F3165, dil. 1:200). (C, D) Uptake of 3H-L-glutamate was measured at room temperature for 10 minutes without (-) or with (+) DHK (1mM, Tocris, cat.# 0111). Overexpression of Senp1 increased the total uptake in astrocytes, but did not alter the DHK-insensitive uptake. (D) The DHK-sensitive, or EAAT2-dependent uptake, was increased in the astrocytes overexpressing SENP1 (n=3, two tailed T-test, ***p<0.05).
Previous analysis of rat brain sections by post-embedding electron microscopic immunocytochemistry revealed that localization of EAAT2 along glial plasma membranes was approximately one order of magnitude higher than in other types of intracellular membranes (i.e. endoplasmic reticulum, mitochondria, nuclear) (Chaudhry et al. 1995). To investigate whether intracellular localized EAAT2 was sumoylated in vivo, plasma membrane- and cytosol-enriched fractions were purified by differential centrifugation of normal rat forebrain homogenate and analyzed by immunoprecipitation. The degree of cross-contamination between these two cellular compartments was minimal as confirmed by specific proteic markers including cytosolic GAPDH and plasma membrane-localized cadherin (Suppl.Fig. 3). A much larger portion of EAAT2 was immunoprecipitated from the plasma membrane compared to the cytosolic fraction, confirming that the prevalent localization of EAAT2 in vivo is at the plasma membrane (Fig. 8). However, SUMO1 immunoreactivity was much higher in EAAT2 present in the cytosolic fraction, showing that most sumoylated EAAT2 is in the cytosolic compartment while more EAAT2 overall is on the plasma membrane (Fig. 8). This suggests that sumoylation could also impact EAAT2 distribution and, hence, EAAT2-mediated glutamate uptake activity in vivo.
Fig 8. Sumoylated EAAT2 partitions prevalently to intracellular compartments in vivo.
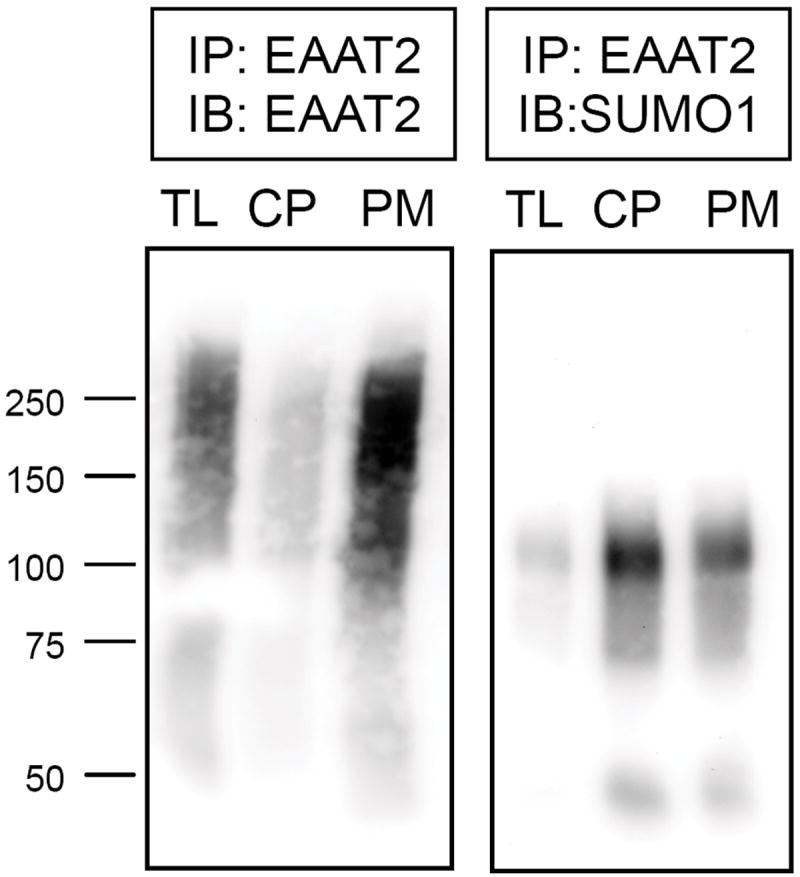
EAAT2 was immunoprecipitated from 1 mg each of normal mouse brain homogenate (total lysate = TL), cytoplasm-enriched fraction (CP), or plasma membrane-enriched fraction (PM). (A) Half of the immunoprecipated protein from each fraction was resolved on 7.5% SDS-PAGE gel and stained for EAAT2 (ABR518-536, dil. 1:500), or (B) stained for SUMO1 (Santa Cruz cat# 5308, dil. 1:100) demonstrating unsumoylated EAAT2 is highly enriched in the plasma membrane fraction and SUMO1-bound EAAT2 is enriched in the cytoplasm fraction of astrocytes in vivo.
Sumoylation affects glutamate transport ability of EAAT2
Having demonstrated that sumoylation of EAAT2 affects its localization; we sought to determine if it also affected its function. HEK293T cells transfected with Myc-EAAT2 (Fig. 9A) compared to HEK293T cells transfected with Myc-EAAT2-SUMO1 (Fig. 9B) showed approximately twice the glutamate uptake (Myc-EAAT2, Vmax=1660±153 pmol/min/mg prot.; Myc-EAAT2-SUMO1, Vmax=882±18 pmol/min/mg prot.) without significant changes in affinity (Myc-EAAT2, Km=143.8±37.3 μM; Myc-EAAT2-SUMO1, Km=120.1±17.6 μM; not significant) (Fig. 9C). This Vmax decrease in HEK293T cells expressing Myc-EAAT2-SUMO1 suggested a drastic reduction in functional transporters at the plasma membrane, likely due to the preponderant partition of EAAT2 to intracellular compartments, since the overall expression levels were similar between Myc-EAAT2-SUMO1 and Myc-EAAT2 expressing HEK 293T cells (Fig. 9D). The HEK293T cells are an artificial cell system in which overexpression of Myc-EAAT2-SUMO1 could explain the residual uptake activity measured in these cells. In support, confocal analysis of HEK293T cells expressing Myc-EAAT2-SUMO1 showed plasma membrane localization of the transporter, even though there was also considerable cytosolic localization.
Fig 9. SUMO1 attachment to EAAT2 dramatically reduces its plasma membrane and glutamate uptake in a heterologous system.
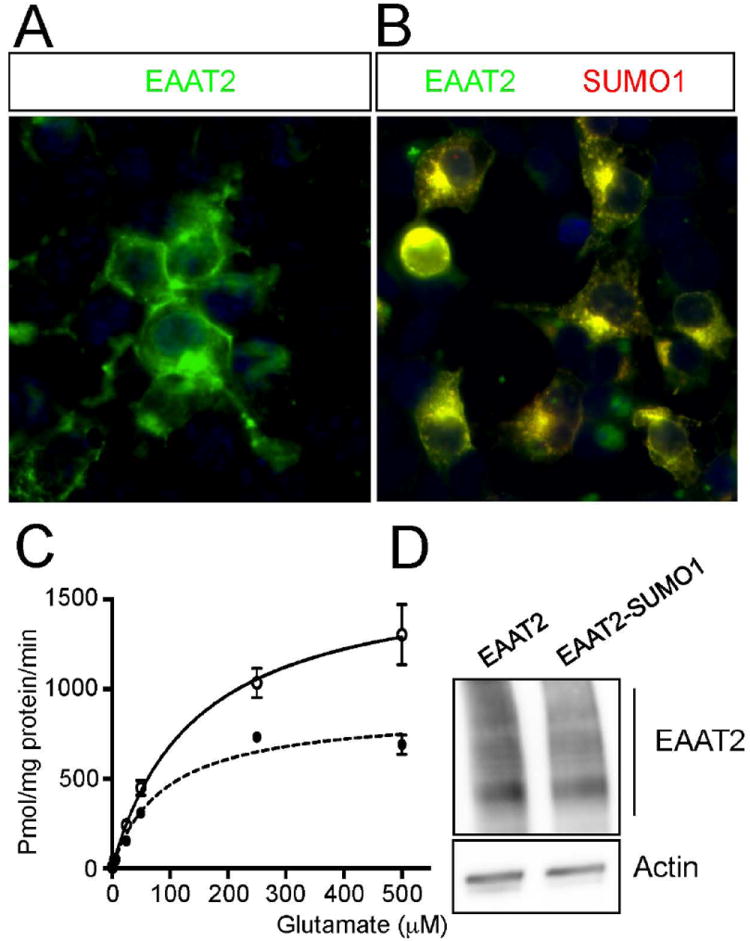
HEK293T cells were plated on 3mm plates and transfected with either myc-EAAT2, (A) or myc-EAAT2-SUMO1, (B) (1.5 μg of plasmid DNA/condition). (C) Uptake of 3H-L-glutamate was measured for 10 minutes at room temperature and compared between the groups. The HEK293T transfected with myc-EAAT2 had nearly double the maximum uptake compared to HEK293T transfected with myc-EAAT2-SUMO1. The uptake rate, however, was similar between the two groups. (D) Transfections showed similar levels of EAAT2 expression by western blot analysis (anti-Myc, Clontech, cat.# 631206, dil. 1:1000), eliminating the possibility that different translation levels accounted for the difference in uptake capacity.
Discussion
The astroglial glutamate transporter EAAT2 (GLT1) is responsible for the reuptake of the majority of glutamate released in the synapses of the adult CNS (Furness et al. 2008; Rothstein et al. 1996; Tanaka et al. 1997). Several lines of evidence linked the loss of EAAT2 expression and activity to mechanisms of pathogenesis in ALS and other neurodegenerative or neurological conditions (Foran and Trotti 2009; Ikematsu et al. 2002; Kim et al. 2011). However, therapies aimed at increasing levels of EAAT2 expression in animal models and patients have been mostly ineffective; for example, ceftriaxone, which considerably increased EAAT2 by approximately 10-fold (Lee et al. 2008; Rothstein et al. 2005), failed a phase III clinical trial for ALS (Berry et al. 2013). Similarly, a 2-fold increase in expression levels of EAAT2 in the SOD1-G93A mouse model of ALS (Guo et al. 2003) also failed to convincingly improve the course of ALS, casting skepticism on the role for this transporter’s dysfunction in the pathogenesis of this disease. However, elevation of the overall expression levels of EAAT2 is often not matched by a corresponding increase in EAAT2-mediated glutamate uptake activity (Rothstein et al. 2005; Schlag et al. 1998), implying that EAAT2 function has effective regulatory cellular checkpoints. Therefore, simply increasing EAAT2 expression levels with the intent to boost glutamate uptake may not necessarily lead to the expected outcome. Strategies involving augmentation of EAAT2 trafficking to the plasma membrane and/or overcoming regulatory cellular checkpoints might have improved chances of increasing EAAT2-mediated glutamate uptake. We showed here that a fraction of EAAT2 undergoes SUMO1 conjugation under physiological conditions. SUMO1 attachment to EAAT2 appears to impact the localization pattern of this transporter as sumoylated EAAT2 preferentially localizes intracellularly. In the future, blocking sumoylation of EAAT2 might enable increased EAAT2-mediated glutamate transport.
In vivo, EAAT2 is primarily localized to lipid rafts (Butchbach et al. 2004) of astrocyte processes (Melone et al. 2011), unlike in primary astrocytes in vitro. EAAT2 function and localization is highly dependent on neuronal input in vivo (Benediktsson et al. 2012). Other factors that impact EAAT2 localization and function include hypoxia (Imasawa et al. 2005) and oxidative stress (Zagami et al. 2005). Additionally, it has been shown that EAAT2 requires a region of the c-terminal tail to properly localize to the plasma membrane (Leinenweber et al. 2011). However, how these factors work together to determine the localization of EAAT2 has not been elucidated. As sumoylation is often linked to alterations in subcellular localization and trafficking of protein substrates (Watts 2013), it is certainly conceivable that sumoylation of EAAT2 could affect its distribution within the cell. In this report, it was demonstrated that astrocytes are capable of sumoylating EAAT2 in vitro, predominantly at lysine 570, which is part of a consensus sequence. In HEK293T cells, which lack the ability to endogenously sumoylate EAAT2, this transporter was essentially entirely localized to the plasma membrane, whereas EAAT2 translationally fused to SUMO1 (in frame at the c-terminus domain) was largely retained intracellularly. Interestingly, a fraction of EAAT2-SUMO1 did make it to the plasma membrane, implying that sumoylation of EAAT2 is not a restricted signal for cytoplasmic retention, although it is likely that this fraction could have escaped SUMO1-mediated regulation because of the overexpression of the protein. In further support for a role of sumoylation as a cytoplasmic retention signal for EAAT2, when increased EAAT2 sumoylation was forced in cultured astrocytes by overexpression of active UBC9 and SUMO1, large intracellular clusters of EAAT2 and an almost complete clearing of EAAT2 from the plasma membrane were observed. Concurrently, intracellular EAAT2 retention was counteracted by the overexpression of the SUMO1 desumoylating enzyme, Senp1, which drastically reduced the intracellular pooling of EAAT2 and increased its redistribution to the plasma membrane. Additionally, while more EAAT2 was immunoprecipitated from membrane-enriched fractions of rat brain homogenate, more sumoylated EAAT2 was detected in cytoplasm-enriched fractions. Ectopic expression of Senp1 in astrocytes not only decreased retention of EAAT2 in cytosolic compartments, but also consistently increased EAAT2-mediated glutamate uptake by the astrocytes. Interestingly, the non-EAAT2 mediated glutamate uptake by these cells, likely due to the contribution of the other major glial transporter, EAAT1 (or GLAST), is not affected by the Senp1 treatment, suggesting that EAAT1 may not be subject to regulation by sumoylation.
Primary astrocytes in culture have been accepted as a cell model system to examine the physiological actions of astrocytes in vivo (Skytt et al. 2010). Indeed, primary astrocytes have exposed the complicated regulation of EAAT2. However, differences have been seen between primary astrocytes and astrocytes in vivo. Many groups have seen that EAAT1 is consistently and highly expressed by primary astrocytes, but EAAT2 levels in primary astrocytes are suppressed (Kondo et al. 1995; Skytt et al. 2010). Additionally, while EAAT2 expression can be triggered by cAMP (Eng et al. 1997) or neuronal co-culture, EAAT2 dependent transport does not increase (Swanson et al. 1997). The localization of EAAT2 in primary astrocytes also differs from that seen in vivo, which may be determined by the sumoylation status of EAAT2. While we have shown that a fraction of EAAT2 is sumoylated in vivo, the stimulus triggering sumoylation or desumoylation in vivo and in primary astrocytes is not yet known. The isolation from other cell types may have removed the vital stimulus leading to desumoylation of EAAT2 in the primary cell culture.
We propose that the function of sumoylation of EAAT2 is to retain the transporter in cytosolic compartments, preventing its degradation, and making it available for fast deployment to the plasma membrane. We cannot distinguish at this point whether sumoylation can occur on EAAT2 when it is at the plasma membrane or if the sumoylation event occurs rapidly after translation, preventing EAAT2 from moving to the plasma membrane. Interestingly, an internalization signal triggered by a phosphorylation event on the transporter and involving ubiquinylation of critical lysine residues located in the c-terminus of EAAT2 has been recently described by Zafra and colleagues (Martinez-Villarreal et al. 2012). It is worth noting that ubiquitinylation and sumoylation target and potentially compete for the same lysine residues in the c-terminal domain of EAAT2 and that ubiquitinylation often acts as post-translational modification with biological effects that are antagonistic to sumoylation. It is, therefore, tempting to hypothesize that sumoylation acts as a cytosolic retention signal for EAAT2, which also prevents its degradation, whereas ubiquitinylation acts as a retrieval signal for the non-sumoylated EAAT2 at the plasma membrane, which also directs it to degradation pathways. We have shown that sumoylated EAAT2 is missing from membrane-enriched brain homogenates, but robustly expressed in the cytoplasm-enriched fraction. Since sumoylation is a dynamic process, sumoylation of EAAT2 could be an immediate and simple protein switch to redirect EAAT2 localization. The presence of consistent EAAT2-SUMO1 immunoprecipitation shows that in normal healthy rodents and humans, a portion of EAAT2 is conjugated with SUMO1. Although we have not quantified the extent of EAAT2 sumoylation, in most proteins that can be sumoylated, only ~5% of substrate is sumoylated. The sumoylation and desumoylation process is dynamic and regulated by cell cycle, cellular stress, and neuronal activity (Loriol et al. 2013; Watts 2013). The speed and consistency at which exogenous EAAT2 is sumoylated in primary astrocytes also indicates that sumoylation of EAAT2 is a vital physiological event. This dynamic regulation could be responsible for the lack of improvement seen with overexpression of EAAT2 in the mouse models of ALS and in clinical trials: While more EAAT2 might be translated, it might be trapped intracellularly, thus preventing increased EAAT2-mediated glutamate uptake. Additionally, it suggests that the disease-related accumulation of CTE-SUMO1 is due to cleavage of physiological EAAT2-SUMO1 rather than sumoylation of cleaved EAAT2. Rather than aiming to increase EAAT2 expression, it might be worthwhile to decrease sumoylation of EAAT2, thus increasing EAAT2-mediated glutamate uptake with an accompanying decrease in excitotoxicity and decreasing CTE-SUMO1 accumulation with an accompanying decrease in astrocyte-mediated neurotoxicity.
EAAT2-SUMO1 levels remain consistent in the spinal cord of transgenic SOD1-G93A mice and the expression level of enzymes of the sumoylation and de-sumoylation pathway are unchanged over disease. It is interesting that we do not see changes to EAAT2-SUMO1 levels considering the facts that 1) EAAT2 levels overall decrease in the SOD1-G93A rat model of ALS (Howland et al. 2002) and 2) that the levels of the sumoylated, cleaved, c-terminal tail of EAAT2 accumulation over disease (Gibb et al, Foran et al). This indicates that levels of sumoylated EAAT2 are under the control of a pathway that is not impacted by ALS dysfunction, despite the overall vulnerability of EAAT2 to ALS and linked stressors (Miralles et al 2001). It is possible that in ALS, more EAAT2-SUMO1 is cleaved by caspase-3 causing the accumulation of CTE-SUMO1 and a decrease in overall EAAT2 levels in order to maintain the sumoylation equilibrium. While the levels of proteins of the sumoylation machinery and overall levels of sumoylation do not significantly change with symptom presence, predominant targets of sumoylation might be altered. Indeed it has been seen that cellular stress can alter the sumoylation status of vital proteins.
Our study introduces an additional avenue to EAAT2 therapeutics in ALS. Targeting and blocking the sumoylation of EAAT2 increases EAAT2 localization to the plasma membrane, in vitro, and therefore elevates glutamate transport. In situations, like ALS, where overexpressing EAAT2 alone does not prove efficacious in isolation; additionally altering the localization may prove to be a more successful paradigm.
Supplementary Material
Main Points.
EAAT2 is constitutively sumoylated in astrocytes;
Sumoylation governs EAAT2 compartmentalization favoring intracellular vs. plasma membrane localization;
Sumoylation of EAAT2 is a regulatory checkpoint, which impacts the overall EAAT2-mediated glutamate uptake in vitro and in vivo.
Acknowledgments
This work was supported by the National Institute of Health grant RO1-NS44292 (to DT), F31-NS073351 (to EF), and the Muscular Dystrophy Association (to DT). The Weinberg Unit for ALS research is also supported by the Farber Family Foundation.
Footnotes
CONFLICT OF INTEREST
The Authors declare that they have no conflict of interest
References
- Benediktsson AM, Marrs GS, Tu JC, Worley PF, Rothstein JD, Bergles DE, Dailey ME. Neuronal activity regulates glutamate transporter dynamics in developing astrocytes. Glia. 2012;60:175–88. doi: 10.1002/glia.21249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry JD, Shefner JM, Conwit R, Schoenfeld D, Keroack M, Felsenstein D, Krivickas L, David WS, Vriesendorp F, Pestronk A, et al. Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS One. 2013;8:e61177. doi: 10.1371/journal.pone.0061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boston-Howes W, Gibb SL, Williams EO, Pasinelli P, Brown RH, Jr, Trotti D. Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J Biol Chem. 2006;281:14076–84. doi: 10.1074/jbc.M600653200. [DOI] [PubMed] [Google Scholar]
- Bristol LA, Rothstein JD. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol. 1996;39:676–9. doi: 10.1002/ana.410390519. [DOI] [PubMed] [Google Scholar]
- Butchbach ME, Tian G, Guo H, Lin CL. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem. 2004;279:34388–96. doi: 10.1074/jbc.M403938200. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–20. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- de la Vega L, Grishina I, Moreno R, Kruger M, Braun T, Schmitz ML. A redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol Cell. 2012;46:472–83. doi: 10.1016/j.molcel.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Eng DL, Lee YL, Lal PG. Expression of glutamate uptake transporters after dibutyryl cyclic AMP differentiation and traumatic injury in cultured astrocytes. Brain Res. 1997;778:215–21. doi: 10.1016/s0006-8993(97)01093-7. [DOI] [PubMed] [Google Scholar]
- Fei E, Jia N, Yan M, Ying Z, Sun Q, Wang H, Zhang T, Ma X, Ding H, Yao X, et al. SUMO-1 modification increases human SOD1 stability and aggregation. Biochem Biophys Res Commun. 2006;347:406–12. doi: 10.1016/j.bbrc.2006.06.092. [DOI] [PubMed] [Google Scholar]
- Foran E, Bogush A, Goffredo M, Roncaglia P, Gustincich S, Pasinelli P, Trotti D. Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia. 2011;59:1719–31. doi: 10.1002/glia.21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran E, Rosenblum L, Bogush AI, Trotti D. Sumoylation of critical proteins in amyotrophic lateral sclerosis: emerging pathways of pathogenesis. Neuromolecular Med. 2013;15:760–70. doi: 10.1007/s12017-013-8262-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran E, Trotti D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11:1587–602. doi: 10.1089/ars.2009.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, et al. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2) Neuroscience. 2008;157:80–94. doi: 10.1016/j.neuroscience.2008.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb SL, Boston-Howes W, Lavina ZS, Gustincich S, Brown RH, Jr, Pasinelli P, Trotti D. A caspase-3-cleaved fragment of the glial glutamate transporter EAAT2 is sumoylated and targeted to promyelocytic leukemia nuclear bodies in mutant SOD1-linked amyotrophic lateral sclerosis. J Biol Chem. 2007;282:32480–90. doi: 10.1074/jbc.M704314200. [DOI] [PubMed] [Google Scholar]
- Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA, Lin CL. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet. 2003;12:2519–32. doi: 10.1093/hmg/ddg267. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–5. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol. 2012;13:755–66. doi: 10.1038/nrm3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–9. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, Zhu C, Chen C, Liu X, Cheng J, et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun. 2012;3:911. doi: 10.1038/ncomms1919. [DOI] [PubMed] [Google Scholar]
- Ikematsu K, Tsuda R, Kondo T, Nakasono I. The expression of excitatory amino acid transporter 2 in traumatic brain injury. Forensic Sci Int. 2002;130:83–9. doi: 10.1016/s0379-0738(02)00344-4. [DOI] [PubMed] [Google Scholar]
- Imasawa M, Kashiwagi K, Iizuka Y, Tanaka M, Tsukahara S. Different expression role among glutamate transporters in rat retinal glial cells under various culture conditions. Brain Res Mol Brain Res. 2005;142:1–8. doi: 10.1016/j.molbrainres.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Jones MC, Fusi L, Higham JH, Abdel-Hafiz H, Horwitz KB, Lam EW, Brosens JJ. Regulation of the SUMO pathway sensitizes differentiating human endometrial stromal cells to progesterone. Proc Natl Acad Sci U S A. 2006;103:16272–7. doi: 10.1073/pnas.0603002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaikkonen S, Jaaskelainen T, Karvonen U, Rytinki MM, Makkonen H, Gioeli D, Paschal BM, Palvimo JJ. SUMO-specific protease 1 (SENP1) reverses the hormone-augmented SUMOylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol Endocrinol. 2009;23:292–307. doi: 10.1210/me.2008-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, Dasgupta S, Barral PM, Hedvat M, Diaz P, et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol. 2011;226:2484–93. doi: 10.1002/jcp.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283:13116–23. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinenweber A, Machtens JP, Begemann B, Fahlke C. Regulation of glial glutamate transporters by C-terminal domains. J Biol Chem. 2011;286:1927–37. doi: 10.1074/jbc.M110.153486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loriol C, Khayachi A, Poupon G, Gwizdek C, Martin S. Activity-dependent regulation of the sumoylation machinery in rat hippocampal neurons. Biol Cell. 2013;105:30–45. doi: 10.1111/boc.201200016. [DOI] [PubMed] [Google Scholar]
- Martinez-Villarreal J, Garcia Tardon N, Ibanez I, Gimenez C, Zafra F. Cell surface turnover of the glutamate transporter GLT-1 is mediated by ubiquitination/deubiquitination. Glia. 2012;60:1356–65. doi: 10.1002/glia.22354. [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Ducati A, Iacoangeli M, Conti F. Cellular and Synaptic Localization of EAAT2a in Human Cerebral Cortex. Front Neuroanat. 2011;4:151. doi: 10.3389/fnana.2010.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Scalf M, Rytz TC, Hubler SL, Smith LM, Vierstra RD. Quantitative proteomics reveals factors regulating RNA biology as dynamic targets of stress-induced SUMOylation in Arabidopsis. Mol Cell Proteomics. 2013;12:449–63. doi: 10.1074/mcp.M112.025056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Thomas M, Dadgar N, Lieberman AP, Iniguez-Lluhi JA. Small ubiquitin-like modifier (SUMO) modification of the androgen receptor attenuates polyglutamine-mediated aggregation. J Biol Chem. 2009;284:21296–306. doi: 10.1074/jbc.M109.011494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Guan T, Tang X, Huang L, Huang M, Li Y, Sun H, Yu R, Zhang F. Astrocytic glutamate transporter-dependent neuroprotection against glutamate toxicity: an in vitro study of maslinic acid. Eur J Pharmacol. 2011;651:59–65. doi: 10.1016/j.ejphar.2010.10.095. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–86. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes-Hoberg M, Vidensky S, Chung DS, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–7. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sampson DA, Wang M, Matunis MJ. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J Biol Chem. 2001;276:21664–9. doi: 10.1074/jbc.M100006200. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Komori T, Iwata M. Excitatory amino acid transporter 1 and 2 immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol. 2000;100:138–44. doi: 10.1007/s004019900159. [DOI] [PubMed] [Google Scholar]
- Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol. 1998;53:355–69. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]
- Sharma P, Yamada S, Lualdi M, Dasso M, Kuehn MR. Senp1 is essential for desumoylating sumo1-modified proteins but dispensable for sumo2 and sumo3 deconjugation in the mouse embryo. Cell Rep. 2013;3:1640–50. doi: 10.1016/j.celrep.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Pradhan AK, Chakraborty S. SUMO1 negatively regulates the transcriptional activity of EVI1 and significantly increases its co-localization with EVI1 after treatment with arsenic trioxide. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbamcr.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Skytt DM, Madsen KK, Pajecka K, Schousboe A, Waagepetersen HS. Characterization of primary and secondary cultures of astrocytes prepared from mouse cerebral cortex. Neurochem Res. 2010;35:2043–52. doi: 10.1007/s11064-010-0329-6. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–4. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Su YF, Yang T, Huang H, Liu LF, Hwang J. Phosphorylation of Ubc9 by Cdk1 enhances SUMOylation activity. PLoS One. 2012;7:e34250. doi: 10.1371/journal.pone.0034250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17:932–40. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Trotti D, Peng JB, Dunlop J, Hediger MA. Inhibition of the glutamate transporter EAAC1 expressed in Xenopus oocytes by phorbol esters. Brain Res. 2001;914:196–203. doi: 10.1016/s0006-8993(01)02802-5. [DOI] [PubMed] [Google Scholar]
- Wallis N, Zagami CJ, Beart PM, O’Shea RD. Combined excitotoxic-oxidative stress and the concept of non-cell autonomous pathology of ALS: insights into motoneuron axonopathy and astrogliosis. Neurochem Int. 2012;61:523–30. doi: 10.1016/j.neuint.2012.02.026. [DOI] [PubMed] [Google Scholar]
- Watts FZ. Starting and stopping SUMOylation : What regulates the regulator? Chromosoma. 2013 doi: 10.1007/s00412-013-0422-0. [DOI] [PubMed] [Google Scholar]
- Wisman LA, van Muiswinkel FL, de Graan PN, Hol EM, Bar PR. Cells over-expressing EAAT2 protect motoneurons from excitotoxic death in vitro. Neuroreport. 2003;14:1967–70. doi: 10.1097/00001756-200310270-00017. [DOI] [PubMed] [Google Scholar]
- Zagami CJ, O’Shea RD, Lau CL, Cheema SS, Beart PM. Regulation of glutamate transporters in astrocytes: evidence for a relationship between transporter expression and astrocytic phenotype. Neurotox Res. 2005;7:143–9. doi: 10.1007/BF03033783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.