

Abstract
Background
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are vital in treating HIV-1 infection by inhibiting reverse transcriptase (RT). Drug toxicity and resistance drive the need for effective new inhibitors with improved physiochemical properties and potent antiviral activity. Computer-aided and structure-based drug design have guided the addition of solubilizing substituents to the diaryltriazine scaffold. These derivatives have markedly improved solubility and maintain low nanomolar antiviral activity against RT. The molecular and structural basis of inhibition for this series was determined to facilitate future inhibitor development with improved pharmacological profiles.
Methods
The molecular mechanism of inhibition was investigated using transient-state kinetic analysis. Crystal structures of RT in complex with each inhibitor were obtained to investigate the structural basis of inhibition.
Results
The diaryltriazine and its morpholine derivative have RT inhibition constants of 9±2 nM and 14±4 nM, respectively. They adopt differential binding modes within the non-nucleoside inhibitor binding pocket to distort the catalytic site geometry and primer grip regions. The novel morpholinopropoxy substituent extends into the RT/solvent interface of the NNIBP.
Conclusions
Kinetic and structural analyses show that these inhibitors behave as conventional NNRTIs and inhibit the polymerization step. This study confirms appending solubilizing substituents on the azine ring of diaryltriazine class of NNRTIs that extend into the RT/solvent interface effectively maintains low nanomolar potency and improves physiochemical properties.
General Significance
The modification of NNRTI scaffolds with solubilizing substituents, which extend into the RT/solvent interface, yields potent antivirals and is an effective strategy for developing novel inhibitors with improved pharmacological properties.
Keywords: HIV-1, Reverse Transcriptase, NNRTI, Crystallography, Transient Kinetics
1. Introduction1
An estimated 35 million people worldwide are infected with HIV-1. The administration of highly active antiretroviral therapy (HAART), a combination of inhibitors targeting essential viral enzymes of the HIV-1 life cycle, has improved patients’ quality of life. Key among the enzymes inhibited by HAART is HIV-1 reverse transcriptase (RT), a viral polymerase that transforms single-stranded RNA into double-stranded DNA that is subsequently integrated into the host genome [1]. The error prone nature of RT [2] along with inhibitor side effects and dosing regimens stemming from poor physiochemical properties [3,4] necessitate the development of novel antiretroviral inhibitors with improved resistance and pharmacological profiles to combat this disease.
To this end, our previous efforts utilized computer-aided and structure-based rational drug design to develop non-nucleoside reverse transcriptase inhibitors (NNRTIs) with both potent antiviral activity and improved physiochemical properties [5,6]. A key physiochemical property for an effective orally administered drug is aqueous solubility. The second-generation FDA-approved diarylpyrimidine (DAPY) NNRTIs etravirine and rilpivirine, with reported aqueous solubility of ≪1 [7] and 0.02–0.24 μg/ml [8,9], respectively, have poor solubility, limiting the ease of formulation and bioavailability [7]. Given the poor solubility of approved DAPY NNRTIs, our drug design efforts focused on the addition of solubilizing groups to a structurally related class of NNRTIs, the diaryltriazines (DATAs). The impact of structural modifications of DATAs on antiviral activity and physiological properties has been previously examined but a detailed molecular and structural investigation is lacking [10]. Our previous work showed the addition of a solubilizing morpholinopropoxy substituent yielded a novel, structurally diverse DATA compound with 63- to 700-fold greater aqueous solubility than the parent DATA inhibitor and both FDA-approved NNRTIs etravirine and rilpivirine [6] (Fig. 1).
Fig. 1.

Structures of diarylazine NNRTIs. Top row: current DAPY NNRTIs approved for the treatment of HIV-1 infection. Compound 1 and compound 2 are DATA derivatives. Compound 2 contains a morpholinopropoxy substituent at the C-6 position of the triazine ring. Drawings were rendered with Chemdraw 12.0.3 (CambridgeSoft).
To understand how the morpholinopropoxy substituent significantly improved solubility while maintaining nanomolar levels of potency against wild-type (WT) HIV-1 [6], we sought to understand the molecular mechanism and structural basis of inhibition of RT by compound 1 and its morpholinopropoxy derivative, compound 2. We evaluated the mechanism of inhibition by transient kinetic analysis using pre-steady state burst experiments to examine single nucleotide incorporation. These studies illustrate the value of this approach for measuring the inhibition constant (Ki) of low nanomolar small molecule inhibitors of RT, a significant challenge arising from limits in assay sensitivity and stoichiometric binding. Transient kinetic analysis indicates that compounds 1 and 2 bind RT with high affinity and behave as conventional NNRTIs by inhibiting the polymerization reaction. Comparative structural analysis reveals that compounds 1 and 2 have different binding modes, with both inhibiting RT by preventing formation of a catalytically competent complex. Extension of the structural analysis provides possible implications on how resistance mutations within the non-nucleoside inhibitor binding pocket (NNIBP) may impact the antiviral activity of these inhibitors.
2. Materials and Methods
2.1 Purification of HIV-1 RT for transient-state analysis
Recombinant HIV-1 RT (p66/p51 heterodimer), a clone kindly provided by Stephen Hughes, Paul Boyer, and Andrea Ferris (Frederick Cancer Research and Development Center, MD), was expressed in E. coli BL21(DE3) pLysS cells and purified as described previously [11]. RT concentration was estimated by UV absorbance at 280 nm using an extinction coefficient of 260,450 M−1cm−1 as previously described [12]. RT purity as judged by SDS-PAGE analysis with Coomassie staining was >90%. RT active site concentration was determined by pre-steady-state burst experiments as previously described [13] and subsequent transient state biochemical experiments were performed using active site concentrations. RT protein samples were stored at −80°C.
2.2 Nucleotides and oligonucleotides
Natural 2′-deoxynucleotides were purchased from GE Healthcare Biosciences (Pittsburgh, PA). DNA oligonucleotides were purchased from Integrated DNA Technologies (Coraville, IA) and further purified using 20% polyacrylamide denaturing gel electrophoresis. The sequences of DNA primers and templates used for single nucleotide incorporation experiments were: D21 primer (5′-TCAGGTCCCTGTTCGGGCGCC-3′) and D36 template (5′-TCTCTAGCAGTGGCGCCCGAACAGGGACCTGAAAGC-3′). D21 primer was 5′-32P-labeled and annealed to the D36 template as previously described [12,14].
2.3 In Vitro radiolabeled-primer extension assay
Single nucleotide incorporation reactions catalyzed by RT in the absence and presence compounds 1 and 2 were performed. RT (10 nM active site) and inhibitor concentrations ranging from 0 to 100 nM were pre-incubated in buffer solution (50 mM Tris pH 7.5, 50 mM NaCl) for 15 minutes at 4°C. Subsequently, (5′-32P)-labeled D21/D36 (30 nM) was added to this mixture and pre-incubated at 4°C for an additional 5 minutes. A RQF-3 rapid chemical quench (KinTek Instruments) was used to rapidly mix the inhibitor•RT•DNA solution with a saturating concentration of dATP (20 μM) in buffer containing 10 mM MgCl2 at 37°C. The reactions were quenched with 0.5 M EDTA pH 8.0. The concentrations are the final concentrations after 1:1 mixing in the instrument and all samples were performed in duplicate. The reaction mixtures were separated on a 20% polyacrylamide denaturing gel (8 M urea), visualized by phosphorimaging (Bio-Rad Molecular Imager FX), and extension of 5′-32P-labeled D21 to D22-mer was quantified with Quantity One 4.6.9 (Bio-Rad). DMSO concentrations were 0.1% in all reactions.
2.4 Data analysis
Data were fit by nonlinear regression using KaleidaGraph (Synergy Software). Single nucleotide incorporation time courses at each inhibitor concentration tested were plotted and fit to a burst equation , where A is the burst phase amplitude, kobs is the observed single exponential rate, kss is the steady-state rate, and t is the time. To generate inhibitor Ki values, the burst phase amplitudes were plotted versus inhibitor concentration and fit to a quadratic equation where A is the burst phase amplitude, E is the enzyme concentration, D is the primer-template concentration, and Ki is the inhibition constant. The data were fit to a quadratic function because the concentration of RT used in the assay was comparable to the Ki values and thus the assumption that the free concentration of inhibitor was equal to the total concentration of inhibitor added was not valid.
2.5 Chemical synthesis
Details of chemical synthesis of 1 and 2 were previously reported [6].
2.6 Crystallization and structure refinement
Recombinant RT52A enzyme was expressed and purified to homogeneity using methods previously described [15]. Crystals of recombinant RT52A in complex with 0.5 mM of compound 1 or compound 2 were prepared using methods similar to those previously described [15]. Briefly, the final optimized condition for crystal growth was 20.0% (w/v) PEG 8000, 100 mM ammonium sulfate, 15 mM magnesium sulfate, 5 mM spermine-HCl, and 50 mM citric acid pH 5.5 or 50 mM HEPES pH 7.0. Crystals grew in approximately 28–30 days at 4°C. Crystals were transferred to a cryo-solution containing 27% (v/v) ethylene glycol and flash cooled with liquid nitrogen.
Several crystals of RT52A:1 and RT52A:2 were screened for diffraction at Brookhaven NSLS on beamline X29A. High-resolution data sets for the best diffracting, single crystals were merged and scaled into the C2 space group using HKL2000 [16]. The best diffracting crystals extended to a resolution of 2.71 Å and 2.89 Å for compound 1 and compound 2, respectively. The impacts of extensive modification of compound 2 relative to compound 1 and other DATAs on the unit cell parameters and orientation of the molecules in the asymmetric unit were unknown and therefore molecular replacement (MR) instead of Difference Fourier Methods was selected to obtain the phases for the RT:1 and RT:2 complexes. MR was performed with Phaser [17,18] using the structure of an RT complex of rilpivirine as the search model (PDB ID: 2ZD1) for compound 1 [19], and of an RT complex with a triazine relative of rilpivirine, Janssen-129385 as a probe model (PDB ID: 1S9E) for compound 2 [20]. Guiding probe model selection were similarity of unit cell dimensions and the observation that RT:1 complexes displayed tighter packing of the crystal lattice and lower solvent content relative to RT:2. Both search models used for MR had the inhibitor and water molecule coordinates removed from the structure, while the atoms for all residues were restored to an occupancy = 1.0. The program COOT [21] was used for model building into the electron density. Maximum-likelihood restrained refinement in Phenix [22] was used to refine the structure after each cycle of model building and modification until acceptable R-factors, geometry statistics (RMSDs for ideal bond lengths and angles), and Ramachandran statistics were achieved for the respective structures. Unbiased omit 2Fo-Fc electron density maps were generated using the Map and Mask Utilities function within the CCP4 program suite [23]. PyMOL [24] was used to visualize and analyze the structures, in addition to Cα backbone structure alignment and RMSD calculations. A summary of data collection parameters and refinement statistics is shown in Table 1. Coordinates and structure factors for the RT:1 and RT:2 complexes are deposited in the Protein Data Bank with PDB IDs 404G and 4O44, respectively.
Table 1.
Data collection parameters and refinement statistics for RT52A in complex with 1 and 2.
| Complex | RT52A:1 | RT52A:2 |
| PDB ID | 4O4G | 4O44 |
| Resolution Limit, Å | 2.71 | 2.90 |
| X-ray Source | NSLS X29A | NSLS X29A |
| Wavelength, Å | 1.075 | 1.075 |
| Space group | C121 | C121 |
| No. Molecules in the asymmetric unit | 1 | 1 |
| Unit cell (a,b,c in Å, β in °) | a=163.037, b=73.400, c=109.728; β=100.06 | a=226.166, b=69.288, c=104.264; β=106.8 |
| Resolution Range, Å | 43.24–2.71 | 36.72–2.89 |
| Last shell, Å | 2.76–2.71 | 2.95–2.90 |
| Rsym, (last shell) | 0.073 (0.413) | 0.114 (0.972) |
| Completeness, % (last shell %) | 98.48 (97.65) | 98.20 (89.44) |
| No. Reflections (Unique Reflections) | 130786 (34315) | 124208 (34385) |
| Redundancy (last shell) | 3.8 (3.9) | 3.6 (3.4) |
| Avg. I/σ (last shell) | 29.5 (3.08) | 19.8 (1.39) |
| Refinement Statistics | ||
| Total Number of Atoms (Protein/Inhibitor/Solvent) | 8104 (8044/25/35) | 8072 (8019/35/18) |
| Rfree, Rfactor | 0.2996, 0.2359 | 0.2902, 0.2328 |
| RMSD bond lengths (Å), angles (°) | 0.005, 0.86 | 0.005, 0.99 |
| Avg. B-factor for All Atoms (Protein/Inhibitor/Solvent) | 63.7 (63.93/51.34/35.5) | 64.0 (64.1/64.7/30.1) |
| Ramachandran Favored, Allowed, Outliers (%) | 91.82, 7.26, 0.92 | 93.54, 5.33, 1.13 |
3. Results and Discussion
3.1 Affinity and molecular mechanism of inhibition of 1 and 2 for HIV-1 RT
The mechanism by which compound 1 and compound 2 inhibit HIV-1 RT was investigated using pre-steady-state kinetic analysis and the inhibition constant (Ki) of each inhibitor was determined. Under burst conditions we examined the effect of each inhibitor on the burst amplitude observed from single nucleotide incorporation reactions catalyzed by RT. RT and the 5′-32P-labeled-D21/D36 were pre-incubated with increasing concentrations of inhibitor and then rapidly mixed with MgCl2 and the next correct nucleotide, dATP, to start the reaction. After varying lengths of time, the reaction was quenched and the products were separated with gel electrophoresis. Autoradiographic analysis enabled quantification of incorporation of the single nucleotide to form the D22 product. The concentration of product formed was plotted against reaction time for each inhibitor concentration tested and fit to a burst equation to generate the observed burst amplitude (Fig. 2A & 2C). Increasing concentrations of 1 or 2 conferred dose-dependent reductions in burst phase amplitudes but did not affect the burst rates. This is consistent with slow equilibration of inhibitor binding observed in Spence et al [25]; therefore the pre-steady-state burst phase reflects the uninhibited, active sites of RT remaining upon treatment with inhibitor. The observed burst phase amplitudes were plotted as a function of inhibitor concentration and fit to a quadratic function to generate Ki values (Fig. 2B & 2D). We obtained Ki values of 9 ± 2 nM and 14 ± 4 nM for compounds 1 and 2 respectively, correlating well with observed EC50 values in the HIV-1 antiviral activity assay of 2.3 nM and 8.1 nM [6,26]. The reduction of burst phase amplitude with increasing concentrations of inhibitors suggests compounds 1 and 2 behave as conventional NNRTIs [25] by inhibiting the polymerization pathway.
Fig. 2.

Pre-steady-state kinetic analysis of single nucleotide incorporation by WT HIV-1 RT in absence and presence of inhibitor and determination of the inhibition constant, Ki. (A) and (C) Kinetic traces in the absence and presence of varying concentrations of compound 1 and 2, respectively, were fit to burst equations. Each point represents a single observation; experiments were performed in duplicate. (B) and (D) Burst phase amplitudes are plotted as a function of inhibitor concentration to obtain inhibition constants. (B) Kinetic traces at varying concentrations of 1 were fit to a quadratic equation to define a Ki value of 9 ± 2 nM. (D) Kinetic traces at varying concentrations of 2 were fit to a quadratic equation to define a Ki value of 14 ± 4 nM. Each point represents the average of two burst phase amplitudes at the concentration tested.
Moreover, we were interested in understanding the utility of the radiolabeled assay for quantifying low- nanomolar inhibition. An accurate determination of Ki requires that the enzyme concentration used in the assay should be near the inhibitor Ki to ensure that concentration of free inhibitor is similar to bound inhibitor. If the concentration of enzyme used is much greater than the inhibitor affinity for the enzyme, (the concentration of free inhibitor is very low at low concentrations of inhibitor tested), this is primarily measuring binding stoichiometry. Previously reported cell potencies for compound 1 and compound 2 [6,26] suggested using low nanomolar RT concentrations. However, the limits of assay sensitivity require enzyme concentrations suitable for detecting primer extension by a single nucleotide. This was particularly challenging as the number of enzyme active sites decreases with increasing concentrations of inhibitor tested. Therefore, we optimized the amount of radiolabeled primer to increase signal:noise threshold and enable detection of 5′-32P-labeled-D21 extension by a single nucleotide. This report demonstrates the utility of a radiolabeled primer-template and single nucleotide incorporation experiment for determining the Ki of low nanomolar inhibitors of RT.
While antiviral activity in cell culture suggested that low nanomolar potencies were maintained for compounds 1 and 2, a mechanistic understanding of modifications of the DATA scaffold was unexplored. It was unclear whether the flexible morpholinopropoxy substituent would alter the ability of the DATA scaffold to bind to and induce formation of the hydrophobic NNIBP of RT in vitro. The biochemical inhibition data suggests that the DATA can be elaborated with carefully placed substituents to improve physiochemical properties without adversely impacting the inhibitory power. Importantly, a potent inhibitor with increased solubility enables ease in formulation and improvements in bioavailability enabling lower doses to be administered to achieve an efficacious concentration [7]. Furthermore, the observed reduction in burst phase amplitude indicates 1 and 2 behave as conventional NNRTIs and inhibit the polymerization pathway. Some speculation can be made about the binding mode of compound 1 due to its similarity to etravirine and rilpivirine, and the available structures of these complexes with wild-type RT (PDB IDs: 3MEC, 2ZD1). However, a structural evaluation of compound 2 was essential to understand the binding mode of the novel morpholinopropoxy substituent. Thus, we pursued crystal structures of RT in complex with 1 and 2.
3.2 Elucidation of molecular interactions for RT:1 and RT:2 complexes
The DATA scaffold of the two compounds adopts a horseshoe conformation commonly observed for the diaryl NNRTIs [19,20,27] (Fig. 3 & Fig. S1). Like DAPY inhibitors, the DATA scaffold of both compounds participates in numerous van der Waals interactions with residues Leu100, Lys101, Lys103, Val179, Tyr181, Tyr188, Phe227, Trp229, Leu234, His235, and Y318 in the NNIBP of the p66 subunit. The Cα superposition of RT:1 and RT:2 yield root mean square deviations (RMSD) of 1.496 Å, indicating overall conformational similarity and accounting for differences in unit cell dimensions, % solvent content, and inhibitor binding. With respect to compound 1, compound 2 binds deeper and is rotated in the NNIBP (Fig. 4). Figure 4 depicts the differences in spatial location for compounds 1 and 2 with respect to the residues in the NNIBP. The distance as measured between the N3 atoms of the triazine rings indicates compound 2 translates approximately 3.4 Å deeper in the NNIBP and rotates ~3°, as measured by the dihedral angle between the planes of the two triazine rings, towards the tunnel region of the NNIBP. The reorientation of the triazine ring scaffold of modified DATAs within the NNIBP is not unusual. In their crystallographic investigation of modified DATAs, Das et al. observe that substitution of a chloroindole ring resulted in the triazine scaffold of R120393 binding ~3 Å deeper in the NNIBP relative to its unmodified analog R106168 [28]. Additionally, the cyanophenyl ring is tilted by approximately 23° and directed towards the His235 residue in the RT:2 structure relative to the RT:1 structure.
Fig. 3.
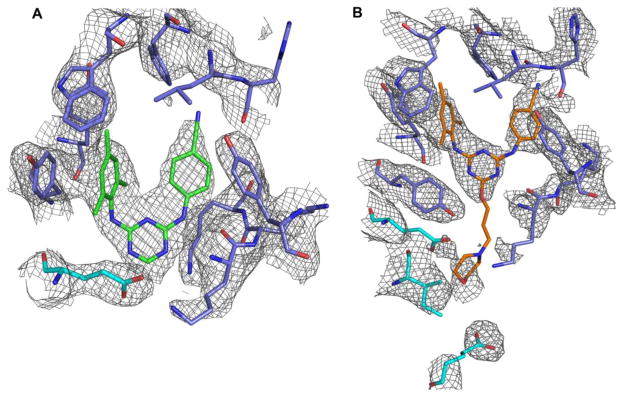
Structure of RT in complex with compounds 1 and 2. 2Fo-Fc of electron density maps of (A) Compound 1 and (B) Compound 2 in the NNIBP of HIV-1 RT. The map contour level is set to 1σ. The RT p66 subunit is shown in blue and the p51 subunit is shown in cyan.
Fig. 4.

Molecular interactions and altered binding modes of compounds 1 and 2. Superposition of RT(blue):1(green) crystal structure and RT(cyan):2(orange) crystal structure. Compound 2 sits approximately 3.4 Å deeper and is shifted approximately 3° towards the tunnel region of the NNIBP. The cyanophenyl ring of 2 is tilted by approximately 23° relative to 1, directed towards His235 of the primer grip region. Note the repositioning of the side chains of residues Glu138 and Lys101 in the RT:2 complex to accommodate extension of the morpholinopropoxy substituent. Also, Tyr181 adopts opposing conformations in the two structures.
As presented in the crystal structures for RT:1 and RT:2, compounds 1 and 2 form hydrogen bonds conserved among several next generation NNRTIs [19,27] with the Lys101 residue of the NNIBP in the p66 subunit. As observed in both complexes, the protonated aniline nitrogen linker forms a hydrogen bond with the backbone carbonyl oxygen of Lys101 (Fig. 5A & 5B). Interaction distances are 2.8 Å and 3.4 Å for compounds 1 and 2, respectively. Although there are differences in spatial location between the two inhibitors, with 2 binding approximately 3.4 Å deeper, its c.a. 3° rotation towards the tunnel region of the NNIBP positions the aniline nitrogen linker within hydrogen bonding distance of the acceptor, Lys101. Though compounds 1 and 2 bind the NNIBP in different orientations, hydrogen bonding interactions critical for the low nanomolar affinities observed kinetically are maintained.
Fig. 5.

The molecular interactions of compounds 1 and 2 within the NNIBP. The aniline linker nitrogen of (A) compound 1 and (B) compound 2 form direct hydrogen bond interactions with the backbone carbonyl of Lys101 as represented with dashed lines. (A) Compound 1 also makes favorable aromatic interactions with Tyr181 while (B) Compound 2 makes favorable aromatic interactions with Tyr188. The morpholinopropoxy substituent extends into the entrance channel of the NNIBP, between Lys101 and Glu138, at the RT/solvent interface and is directed towards Ile135. The RT p66 subunit is shown in blue and the p51 subunit is shown in cyan.
In addition to hydrogen bonding, interactions at the RT/solvent exposed interface inform the low nanomolar affinities of the two inhibitors and indicate the structural consequences of appending a morpholinopropoxy substituent to the triazine core. The RT:1 structure depicts an extensive hydration shell at the solvent interface wherein the inhibitor and solvent exposed amino acids of the NNIBP coordinate nearby water molecules to form a dynamic hydrogen bond network (Fig. S2). One water molecule coordinates the N5 atom of the triazine scaffold (ca. 2.9 Å) and also mediates a hydrogen bond with the proximal carboxylate oxygen of Glu138 of the p51 subunit (ca. 2.8 Å). The carboxylate oxygen atoms of residue Glu138 (p51) participate in a bifurcated hydrogen bond system with the Nζ atom of residue Lys101 (hydrogen bonding distances are both ca. 3.2 Å). Furthermore, the Nζ atom of Lys101 makes close contacts with a second water molecule (ca. 3.1 Å) and the Lys103 Nζ atom hydrogen bonds with a third water molecule (ca. 2.6 Å). The inter-subunit interactions and extensive water molecule coordination of the residues at the solvent exposed interface of the NBBP have been previously observed [19,27] and support our observations of a dynamic hydrogen bond network in the RT:1 complex.
Considerably fewer solvent interactions are identified in the RT:2 structure owing to the extension of the morpholinopropoxy substituent into the solvent exposed interface of the NNIBP (Fig. 5B) and the lower resolution of the diffracting crystal. The morpholinopropoxy substituent is positioned between residues Glu138 (p51) and Lys101 (p66) extending towards Ile135 (p51). To prevent steric clash, side chain residues reposition to accommodate the morpholinopropoxy substituent at the NNIBP/solvent interface. Most strikingly, the carboxylate oxygen atoms of Glu138 (p51) and the Nζ atom of Lys101 in the RT:2 complex reposition lower in the NNIBP and are not within hydrogen bonding distance as observed for the RT:1 complex (Fig. 4). In this context it is important to note that the RT:2 complex diffracted to amplitudes of lower resolution compared to RT:1 (2.89 Å vs. 2.71 Å). Thus, water molecules that may be present and coordinating the morpholinopropoxy group are not apparent in the lower resolution RT:2 complex. Additionally, the lower resolution structure may be indicative of disorder of the RT:2 complex, suggesting intrinsic flexibility in binding modes [28] adopted by the morpholinopropoxy substituent. As evidenced by the spatial reorientation of 2 in the crystal structure, the optimization of DATA inhibitors with solubilizing substituents that extend into the NNIBP entrance channel is feasible and permits inhibitor binding of the NNIBP [29].
Compounds 1 and 2 maintain favorable π-π stacking interactions of the DATA scaffold with key residues of the NNIBP but with altered conformations. In the RT:1 complex, the trimethyl substituted phenyl ring participates in aromatic, π-π stacking interactions with Tyr181, which in this structure adopts the “up” conformation observed in RT:DATA, RT:rilpivirine, and RT:etravirine crystal structures (PDB IDs: 1S9E, 2ZD1, 3M8P) [19,20,30] (Fig. 5A). In contrast, the trimethyl substituted phenyl ring of 2 is rotated towards the tunnel region as previously described and therefore participates in aromatic stacking interactions with Tyr188 rather than Tyr181. The open space created by the rotation and translation of 2 enables Tyr181 to adopt its apo “down” conformation and point facing away from the polymerase active site (Fig. 5B) and is an observation unaccounted for in the modeling of compound 2, which utilized a configuration of RT in which Tyr181 adopted the “up” orientation [6]. Instances of this “down” configuration have been observed previously (PDB IDs: 2B5J, 2BE2, 4H4M, 4H4O, 3LAL, 3DLG) [15,31–33] and highlights the conformational flexibility and adaptable binding modes of DATAs even in instances of extensive inhibitor modifications that extend into the RT/solvent interface.
Variations in the orientation of Tyr181 are commonly observed in RT:inhibitor complexes and appear inhibitor specific within a given NNRTI class. The impact of opposing conformations of the Tyr181 residue observed in RT:NNRTI complexes in interpreting structure activity relationships has been previously examined for the 1-[(2-hydroxyethoxy)-methyl]-6-(phenylthio) thymine (HEPT) class of NNRTIs. In their comparative structural analysis of NNRTI:HEPT complexes, Hopkins et al. indicate the conformational switching of Tyr181 influences the binding affinity of the inhibitor [34]. The less potent HEPT inhibitor, containing a small methyl group at the 5 position, is observed to bind the NNIBP and induce rotation of only Tyr188, while the more potent HEPT derivatives MKC-442 and TNK-651 contain larger isopropyl groups at the 5 position and induce conformational switching of both Tyr188 and Tyr181, resulting in an increase in favorable aromatic interactions and thus tighter binding. However, in their investigation of a closely related series of pyrimidinediones containing isopropyl groups at the 5 position, Mitchell et al. observe that substitution of the smaller ethyl substituents at the 1 position induce rotation of only Tyr188 without a significant loss in inhibitor potency relative to the bulkier N1 substituted inhibitors [32]. The comparative structural analysis of compounds 1 and 2 representative of the DATA class of NNRTIs presented here suggests the conformational switching of Tyr181 does not impact the binding affinity. Here compound 2 induces flipping of only Tyr188 but compound 2 is rotated and translated in the NNIBP with respect to 1. This repositioning enables key aromatic and hydrogen bond interactions to be maintained and thus the affinity of 2 is uncompromised. This lends structural support to our biochemical observations of low nanomolar inhibition constants for compounds 1 and 2. While both 1 and 2 maintain low nanomolar RT inhibition, the opposing conformations observed for the Tyr181 do have implications for the structural basis of inhibition imparted by 1 and 2 as outlined below.
3.3 Structural basis of inhibition for 1 and 2: Kinetic complementarity
The electron density indicates the structure of the p66/p51 RT heterodimer in the RT:1 and RT:2 adopts the “open-cleft” conformation as observed in other RT:NNRTI crystal structures [15,19,20,31] (Fig. 3 & Fig. S1). Prior structural analysis indicates that NNRTIs bind the allosteric NNIBP located 10 Å distal to the polymerase active site and induce a conformational change that prevents the formation of a catalytically competent complex for polymerization [35–37]. The conformational change induced by NNRTI binding disrupts geometry of the catalytic residues at the active site, namely Tyr183, Met184, Asp185, and Asp186 (YMDD loop) and Asp110 [35], and deforms the β12-β13-β14 strand residues comprising the primer grip (residues 227–240 of p66 subunit) of the polymerase which is critical for positioning the 3′ terminus of the primer at the active site and movements associated with translocation of the polymerase during polymerization [36,37]. As our kinetic data suggests that compounds 1 and 2 behave as conventional NNRTIs by inhibiting polymerization, we compared the structural changes in RT observed upon compound 1 and 2 binding with RT structures representing catalytically competent complexes.
To investigate the effects of 1 and 2 on the YMDD loop and primer grip regions of RT implicated in conventional NNRTI inhibition, we superimposed RT:1 and RT:2 with unliganded (PDB ID: 1DLO) [36] (Fig. 6A). The Cα superposition of RT:1 and RT:2 with 1DLO yield RMSDs of 2.813 Å and 2.784 Å, respectively, indicative of structural dissimilarity. Indeed, the binding of compound 1 or compound 2 induces the formation of the NNIBP and is accompanied by substantial shifts of the β12-β13-β14 strand residues away from the YMDD loop and polymerase active site relative to unliganded RT, in agreement with prior observations [36,38]. The primer grip regions are shifted by 2.9 and 3.5 Å as observed in all atom alignments of RT:1 and RT:2 relative to the apo structure. To understand how the substantial shifts in these key regions may disrupt RT interactions when bound with a DNA substrate, we superimposed RT:1 and RT:2 with a catalytically competent RT:DNA binary complex with dATP bound in the nucleotide binding site (PDB ID: 3KK2) [38,39] (Fig. 6B). Superposition with the catalytically competent complex yield RMSDs of 3.010 Å and 3.179 Å and indicates compounds 1 and 2 substantially but differentially perturb the YMDD loop and primer grip regions that are key to the catalytic activity of RT. Compound 1 displaces the YMDD loop by ~ 4.5 Å and the primer grip region by ~ 4.3 Å relative to 3KK2. Comparative structural analysis of the YMDD loop residue Tyr183 indicates it is shifted upwards by 4.2 Å in the RT:1 complex and clashes with the primer terminus. The upward displacement of the primer grip residues 227–240 in RT:1 complex are also observed to clash with the duplex DNA substrate. Compound 2 shifts the YMDD motif by ~ 2.9 Å and the primer grip region by ~ 4.9 Å relative to 3KK2. The Tyr183 residue in the RT:2 complex shifts up by 2.3 Å but unlike the RT:1 complex, it does not clash with the primer terminus of the bound DNA substrate. Interestingly, because compound 2 is rotated 3° relative to 1 and induces rotation of only the Tyr188 side chain into the “up” conformation to face the catalytic site, the resulting concomitant shift in the position of the YMDD loop is not as drastic as observed for compound 1, which induces flipping of both Tyr188 and Tyr181 residues towards the YMDD loop of the catalytic site. That 2 perturbs the YMDD motif to a lesser extent than 1 is compensated for when examining the primer grip residues 227–240 of the RT:2 complex. Since 2 binds 3.4 Å deeper than 1 in the NNIBP, it imparts a greater shift in the position of the primer grip residues (4.9 vs. 4.3 Å) and causes this region to clash with the DNA of the catalytically competent complex. Additionally, the trimethyl substituted phenyl ring of compound 2 itself would speculatively clash with the primer grip regions of the RT:DNA binary complex, a feature that is not observed with compound 1. We noted similar observations when overlaying RT:1 and RT:2 with other catalytically competent RT:DNA binary complexes (PDB IDs: 2HMI, 3V6D) (Fig. S3) [38,40].
Fig. 6.

Comparative structural analysis of RT:inhibitor complexes with unliganded, a catalytically competent RT:DNA binary complex, and an RT:DNA:NNRTI ternary complex. (A) Superposition of RT:1(green), RT:2(orange), and the unliganded RT(purple) (PDB ID: 1DLO) complexes illustrating YMDD loop and β12-β13-β14 shifts upon 1 and 2 binding. (B) Structural overlay of RT:1(green), RT:2(orange) and the catalytically competent binary RT:DNA complex with dATP bound in the nucleotide binding site (blue) (PDB ID: 3KK2). Compound 1 imparts a greater shift on the YMDD loop relative to compound 2 (4.5 Å vs. 2.9 Å), and Tyr183(green) is observed to clash with the primer terminus of the substrate DNA. However, compound 2 perturbs the primer grip region to a greater extent than compound 1 (4.9 Å vs. 4.3 Å) and the trimethyl substituted phenyl ring(orange) of the inhibitor itself is observed to clash with the primer grip residues of the catalytically competent complex. (C) Structural overlay of RT:1(green), RT:2(orange), and the ternary RT:DNA:NVP(tan) complex (PDB ID: 3V81) illustrating a similar mechanism of action.
To compare the binding modes of compounds 1 and 2 with a ternary RT:DNA complex containing a bound NNRTI, we superimposed the RT:DNA:NNRTI ternary complex containing nevirapine (NVP) (PDB ID: 3V81) [38] with RT:1 and RT:2 complexes (Fig. 6C). The Cα superposition yields RMSDs of 1.255 Å and 1.577 Å for RT:1 and RT:2, respectively, illustrating overall structural similarity. The YMDD loop and primer grip movements observed in the RT:1 and RT:2 complexes closely resemble those present in the RT:DNA:NVP ternary complex, suggesting a similar mechanism of action and that inhibitor binding disrupts positioning of the primer template relative to the active site to prevent interactions of the YMDD motif with the primer terminus necessary for catalysis. Furthermore, we observe that the YMDD loop including the Tyr183 residue, the primer grip residues, and NVP of the 3V81 structure are positioned between the analogous regions of the RT:1 and RT:2 complexes, in support of our observations that compound 1 and compound 2 differentially perturb key regions of RT critical for polymerization.
The structural interactions observed for RT:1 and RT:2 compliment our understanding of the molecular mechanism of inhibition. Though they exhibit different binding modes, high affinity molecular interactions of compounds 1 and 2 with the NNIBP are preserved and inform the low nanomolar inhibition constants obtained in vitro. The reduction in burst phase amplitude with increasing concentrations of 1 or 2 reflects a reduced number of active enzyme sites and indicates that compounds 1 and 2 inhibit the chemistry step of the polymerization pathway. To inhibit the chemistry step of polymerization, compounds 1 and 2 differentially disrupt the arrangement and orientation of the catalytic and primer grip regions of RT and prevent formation of a catalytically competent complex. The mechanistic and structural understanding presented here suggests that DATAs can be modified with solubilizing substituents to achieve profound improvements in solubility and preserve key interactions with residues of the NNIBP to maintain binding affinity and inhibitory activity towards RT.
3.4 Implications for drug resistance
The development of drug resistance is a major hurdle in the effective treatment and management of HIV-1 infection and is the primary cause of treatment failure [28]. For this reason, second-generation NNRTIs etravirine and rilpivirine were developed to possess higher genetic barriers for the development of drug resistance. Integral to furthering prevention and treatment of drug-resistant viral variants is the development of NNRTIs with improved physiochemical properties to facilitate ease in formulation, enhance bioavailability, optimize dose and dosing regimen, and minimize adverse effects [8]. An advancement to this end is our development of compound 2, with 63- to 700-fold greater aqueous solubility than compound 1 and both FDA-approved NNRTIs etravirine and rilpivirine [6]. The comparative kinetic and structural analysis of compounds 1 and 2 presented here indicates carefully selected structural modifications that improve physiochemical properties yields inhibitors with potent inhibitory activity against RT and positional adaptability necessary to maintain inhibitor-RT interactions. Additionally, our observation that compounds 1 and 2 adopt differing binding modes, with 2 translated and rotated relative to 1 in the NNIBP, and exhibit the torsional flexibility of the aniline nitrogen linker connecting the triazine and cyanophenyl rings may inherently be advantageous for accommodating resistance mutations [19,28].
As we have successfully improved solubility for a potent class of anti-HIV compounds, further optimization of the series will aim to improve potency for several clinically relevant, resistant variants of RT such as Lys103Asn and Tyr181Cys. The antiviral activity of compounds 1 and 2 in cell culture [6] against prevalent NNRTI resistant variants that we have profiled are Tyr181Cys with EC50 values of 47 nM and 310 nM, respectively, and the double mutant Tyr181Cys/Lys103Asn with EC50 of 90 nM and 31 nM, respectively [41]. This suggests inhibitors possessing solubilizing modifications extending into the solvent exposed entrance channel maintain antiviral potency against the common resistance mutations profiled, but can be optimized further. Thus, analogous application of the present strategy to increase the solubility of DAPY inhibitors like etravirine and rilpivirine, which possess single-digit nanomolar and picomolar antiviral activity against the Tyr181Cys and Tyr181Cys/Lys103Asn [6] mutations, will hopefully yield improvements in solubility while maintaining their potent antiviral activity against these resistance mutations.
Crystal structures of RT:1 and RT:2 reveal that Lys101 (p66 subunit) and Glu138 (p51 subunit) are important residues for compound binding, and mutations at these residues will most likely effect binding of DAPYs and DATAs with solubilizing morpholinopropoxy sidechains. In this context it will be important to perform structure-activity investigations of DAPY derivatives containing solubilizing modifications against resistance mutations arising in patients treated with etravirine and rilpivirine, particularly in understanding how the substitutions of Glu138 and/or Lys101 within the entrance channel influence inhibitor potency [42,43]. One mechanism of resistance imparted by mutations of Glu138 and/or Lys101 is electrostatic repulsion resulting in disruption of the salt bridge between the two residues and presumably altered dynamics of inhibitor binding and access to the NNIBP [44,45]. Though we observe disruption of the salt bridge in the RT:2 complex as the morpholinopropoxy substituent extends between Glu138 and Lys101, separating the carboxylate oxygen atoms of Glu138 from the Nζ atom of Lys101 by 4.7 Å, perhaps the morpholinopropoxy substituent forms favorable electrostatic contacts to retain antiviral activity [6]. Thus, the potency of these modified derivatives against emerging second-generation resistance mutations merits further exploration. Nonetheless, the vast improvements in inhibitor solubility afforded by solubilizing modifications that extend into the entrance channel warrant further optimization and examination with particular attention to viral variants possessing drug resistant mutations in response to etravirine and rilpivirine, particularly those lining the entrance channel.
4. Conclusions
Current antiviral therapies targeting HIV-1 RT are limited by poor physiochemical properties that limit dosing regimens and bioavailability. Thus there is need to develop inhibitors with improved physiochemical properties that maintain potent antiviral activity. To this end, we previously reported significant enhancements in aqueous solubility for the DATA class of NNRTIs by appending solubilizing substituents to the azine scaffold. Importantly, this modification did not impact the antiviral activity of the DATAs, which was maintained at low nanomolar potencies. We sought to understand the molecular basis of inhibition with transient kinetic analysis and the binding interactions by performing comparative structural analysis for two analogues of the DATA compound series, 1 and its morpholinopropoxy derivative, 2. We demonstrated that both inhibitors behave as conventional NNRTIs and inhibit polymerase catalysis, with HIV-1 RT inhibition constants of 9 ± 2 nM and 14 ± 4 nM for 1 and 2, respectively. Comparative structural analysis indicates that both inhibitors distort the catalytic YMDD loop and primer grip region geometry and maintain key interactions with the residues of the NNIBP but with differing binding modes. Compounds 1 and 2 participate in hydrogen bonding with the residues of the NNIBP, specifically the carbonyl oxygen of Lys101. The RT:1 complex has an extensive hydration shell that forms a hydrogen bond network and coordinates the inhibitor which is not readily apparent in the RT:2 complex. Instead the morpholinopropoxy substituent of compound 2 is accommodated by shifting of the residues lining the NNIBP entrance channel and extending into the RT/solvent interface. With respect to compound 1, compound 2 binds 3.4 Å deeper in the NNIBP and is rotated ~3°. This positional difference of 2 in the NNIBP enables it to maintain favorable π-π stacking interactions with Tyr188, while compound 1 makes aromatic interactions with Tyr181. It also lends compound 2 to clash with DNA template of the catalytically competent binary RT:DNA structure and is reflected in 2 distorting the primer grip region to a greater extent than compound 1, shifting residues 227–240 by 4.9 Å vs. 4.3 Å. This analysis suggests the flexibility and positional adaptability of DATAs and their morpholine derivatives enable significant enhancements to physiological properties while maintaining antiviral HIV-1 RT activity by preventing formation of a catalytically competent complex. These studies will aid in the further development of novel soluble and potent antiviral agents targeting HIV-1 RT.
Supplementary Material
Highlights.
Novel DATA NNRTI with improved solubility is a potent inhibitor of HIV RT.
The novel morpholinopropoxy substituent extends into the RT/solvent interface.
Derivatives adopt differential binding modes in the NNBP to inhibit polymerization.
Implications of the altered binding modes observed for novel DATAs are described.
Acknowledgments
Gratitude is expressed to the National Institutes of Health (GM49551, AI44616, GM32136) for research support, (GM007324) for training support of ACM, and (AI104334) for fellowship support of KMF. We also thank the National Synchrotron Light Source at Brookhaven National Laboratory for beam time on X29A and training in their RapiData 2013 course.
Footnotes
Abbreviations HIV-1, human immunodeficiency virus type 1; RT, reverse transcriptase; NNRTI, non-nucleoside reverse transcriptase inhibitor; DAPY, diarylpyrimidine; DATA, diaryltriazine; NNIBP, non-nucleoside inhibitor binding pocket; PDB, protein data bank;
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arts EJ, Hazuda DJ. HIV-1 Antiretroviral Drug Therapy. Cold Spring Harbor Perspectives in Medicine. 2012;2:a007161–a007161. doi: 10.1101/cshperspect.a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts JD, Bebenek K, Kunkel TA. The accuracy of reverse transcriptase from HIV-1. Science. 1988;242:1171–1173. doi: 10.1126/science.2460925. [DOI] [PubMed] [Google Scholar]
- 3.Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54:355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 4.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 5.Ekkati AR, Bollini M, Domaoal RA, Spasov KA, Anderson KS, Jorgensen WL. Discovery of dimeric inhibitors by extension into the entrance channel of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2012;22:1565–1568. doi: 10.1016/j.bmcl.2011.12.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bollini M, Cisneros JA, Spasov KA, Anderson KS, Jorgensen WL. Optimization of diarylazines as anti-HIV agents with dramatically enhanced solubility. Bioorg Med Chem Lett. 2013;23:5213–5216. doi: 10.1016/j.bmcl.2013.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weuts I, Van Dycke F, Voorspoels J, De Cort S, Stokbroekx S, Leemans R, et al. Physicochemical properties of the amorphous drug, cast films, and spray dried powders to predict formulation probability of success for solid dispersions: etravirine. J Pharm Sci. 2011;100:260–274. doi: 10.1002/jps.22242. [DOI] [PubMed] [Google Scholar]
- 8.Janssen PAJP, Lewi PJP, Arnold EE, Daeyaert FF, de Jonge MM, Heeres JJ, et al. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R278474, rilpivirine) J Med Chem. 2005;48:1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- 9.Sun LQ, Qin B, Huang L, Qian K, Chen CH, Lee KH, et al. Optimization of 2,4-diarylanilines as non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg Med Chem Lett. 2012;22:2376–2379. doi: 10.1016/j.bmcl.2012.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Zhan P, Liu X, Cheng Z, Meng C, Shao S, et al. Design, synthesis, anti-HIV evaluation and molecular modeling of piperidine-linked amino-triazine derivatives as potent non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem. 2012;20:3856–3864. doi: 10.1016/j.bmc.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 11.Kerr SG, Anderson KS. Pre-steady-state kinetic characterization of wild type and 3″-azido-3-″deoxythymidine (AZT) resistant human immunodeficiency virus type 1 reverse transcriptase: implication of RNA directed DNA polymerization in the mechanism of AZT resistance. Biochemistry. 1997;36:14064–14070. doi: 10.1021/bi9713862. [DOI] [PubMed] [Google Scholar]
- 12.Kati WM, Johnson KA, Jerva LF, Anderson KS. Mechanism and fidelity of HIV reverse transcriptase. J Biol Chem. 1992;267:25988–25997. [PubMed] [Google Scholar]
- 13.Ray AS, Murakami E, Basavapathruni A, Vaccaro JA, Ulrich D, Chu CK, et al. Probing the molecular mechanisms of AZT drug resistance mediated by HIV-1 reverse transcriptase using a transient kinetic analysis. Biochemistry. 2003;42:8831–8841. doi: 10.1021/bi034435l. [DOI] [PubMed] [Google Scholar]
- 14.Bailey CM, Sullivan TJ, Iyidogan P, Tirado-Rives J, Chung R, Ruiz-Caro J, et al. Bifunctional inhibition of human immunodeficiency virus type 1 reverse transcriptase: mechanism and proof-of-concept as a novel therapeutic design strategy. J Med Chem. 2013;56:3959–3968. doi: 10.1021/jm400160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frey KM, Bollini M, Mislak AC, Cisneros JA, Gallardo-Macias R, Jorgensen WL, et al. Crystal structures of HIV-1 reverse transcriptase with picomolar inhibitors reveal key interactions for drug design. J Am Chem Soc. 2012;134:19501–19503. doi: 10.1021/ja3092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 17.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das K, Bauman JD, Clark AD, Frenkel YV, Lewi PJ, Shatkin AJ, et al. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci USa. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das K, Clark AD, Lewi PJ, Heeres J, De Jonge MR, Koymans LMH, et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhat TN, Cohen GH. OMITMAP: An electron density map suitable for the examination of errors in a macromolecular model. J Appl Crystallogr. 1984;17:244–248. [Google Scholar]
- 24.The Pymol Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; [Google Scholar]
- 25.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludovici DW, Kavash RW, Kukla MJ, Ho CY, Ye H, De Corte BL, et al. Evolution of anti-HIV drug candidates. Part 2: Diaryltriazine (DATA) analogues. Bioorg Med Chem Lett. 2001;11:2229–2234. doi: 10.1016/s0960-894x(01)00411-5. [DOI] [PubMed] [Google Scholar]
- 27.Lansdon EB, Brendza KM, Hung M, Wang R, Mukund S, Jin D, et al. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): implications for drug design. J Med Chem. 2010;53:4295–4299. doi: 10.1021/jm1002233. [DOI] [PubMed] [Google Scholar]
- 28.Das K, Lewi PJ, Hughes SH, Arnold E. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Bollini M, Frey KM, Cisneros JA, Spasov KA, Das K, Bauman JD, et al. Extension into the entrance channel of HIV-1 reverse transcriptase-Crystallography and enhanced solubility. Bioorg Med Chem Lett. 2013;23:5209–5212. doi: 10.1016/j.bmcl.2013.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kertesz DJ, Brotherton-Pleiss C, Yang M, Wang Z, Lin X, Qiu Z, et al. Discovery of piperidin-4-yl-aminopyrimidines as HIV-1 reverse transcriptase inhibitors. N-benzyl derivatives with broad potency against resistant mutant viruses. Bioorg Med Chem Lett. 2010;20:4215–4218. doi: 10.1016/j.bmcl.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 31.Himmel DM, Das K, Clark AD, Hughes SH, Benjahad A, Oumouch S, et al. Crystal structures for HIV-1 reverse transcriptase in complexes with three pyridinone derivatives: a new class of non-nucleoside inhibitors effective against a broad range of drug-resistant strains. J Med Chem. 2005;48:7582–7591. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell ML, Son JC, Guo H, Im YA, Cho EJ, Wang J, et al. N1-Alkyl pyrimidinediones as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2010;20:1589–1592. doi: 10.1016/j.bmcl.2010.01.085. [DOI] [PubMed] [Google Scholar]
- 33.Ren J, Chamberlain PP, Stamp A, Short SA, Weaver KL, Romines KR, et al. Structural basis for the improved drug resistance profile of new generation benzophenone non-nucleoside HIV-1 reverse transcriptase inhibitors. J Med Chem. 2008;51:5000–5008. doi: 10.1021/jm8004493. [DOI] [PubMed] [Google Scholar]
- 34.Hopkins AL, Ren J, Esnouf RM, Willcox BE, Jones EY, Ross C, et al. Complexes of HIV-1 reverse transcriptase with inhibitors of the HEPT series reveal conformational changes relevant to the design of potent non-nucleoside inhibitors. J Med Chem. 1996;39:1589–1600. doi: 10.1021/jm960056x. [DOI] [PubMed] [Google Scholar]
- 35.Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- 36.Hsiou Y, Ding J, Das K, Clark AD, Hughes SH, Arnold E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996;4:853–860. doi: 10.1016/s0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- 37.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 38.Das K, Martinez SE, Bauman JD, Arnold E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat Struct Mol Biol. 2012;19:253–259. doi: 10.1038/nsmb.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lansdon EB, Samuel D, Lagpacan L, Brendza KM, White KL, Hung M, et al. Visualizing the molecular interactions of a nucleotide analog, GS-9148, with HIV-1 reverse transcriptase-DNA complex. J Mol Biol. 2010;397:967–978. doi: 10.1016/j.jmb.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 40.Ding J, Das K, Hsiou Y, Sarafianos SG, Clark AD, Jacobo-Molina A, et al. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J Mol Biol. 1998;284:1095–1111. doi: 10.1006/jmbi.1998.2208. [DOI] [PubMed] [Google Scholar]
- 41.De Béthune MP. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009) Antiviral Res. 2010;85:75–90. doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Tambuyzer L, Vingerhoets J, Azijn H, Daems B, Nijs S, De Béthune MP, et al. Characterization of genotypic and phenotypic changes in HIV-1-infected patients with virologic failure on an etravirine-containing regimen in the DUET-1 and DUET-2 clinical studies. AIDS Res Hum Retroviruses. 2010;26:1197–1205. doi: 10.1089/aid.2009.0302. [DOI] [PubMed] [Google Scholar]
- 43.Johnson VA, Calvez V, Günthard HF, Paredes R, Pillay D, Shafer R, et al. 2011 update of the drug resistance mutations in HIV-1. Top Antivir Med. 2011;19:156–164. [PMC free article] [PubMed] [Google Scholar]
- 44.Ren J, Nichols CE, Stamp A, Chamberlain PP, Ferris R, Weaver KL, et al. Structural insights into mechanisms of non-nucleoside drug resistance for HIV-1 reverse transcriptases mutated at codons 101 or 138. Febs J. 2006;273:3850–3860. doi: 10.1111/j.1742-4658.2006.05392.x. [DOI] [PubMed] [Google Scholar]
- 45.Singh K, Marchand B, Rai DK, Sharma B, Michailidis E, Ryan EM, et al. Biochemical mechanism of HIV-1 resistance to rilpivirine. J Biol Chem. 2012;287:38110–38123. doi: 10.1074/jbc.M112.398180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.