

Abstract
Desmosomal cadherins mediate cell–cell adhesion in epithelial tissues and have been known to be altered in cancer. We have previously shown that one of the two intestinal epithelial desmosomal cadherins, desmocollin-2 (Dsc2) loss promotes colonic epithelial carcinoma cell proliferation and tumor formation. In this study we show that loss of the other intestinal desmosomal cadherin, desmoglein-2 (Dsg2) that pairs with Dsc2, results in decreased epithelial cell proliferation and suppressed xenograft tumor growth in mice. Dsg2-deficient cells demonstrated a compensatory increase in Dsc2 expression, and small interfering RNA-mediated loss of Dsc2 restored proliferation in Dsg2-deficient cells. Dsg2 downregulation inhibited epidermal growth factor receptor (EGFR) signaling and cell proliferation through altered phosphorylation of EGFR and downstream extracellular signal-regulated kinase activation in parallel with inhibited EGFR receptor internalization. Additionally, we demonstrated a central role of Dsc2 in controlling EGFR signaling and cell proliferation in intestinal epithelial cells. Consistent with these findings, analyses of human colon cancers demonstrated increased Dsg2 protein expression. Taken together, these data demonstrate that partner desmosomal cadherins Dsg2 and Dsc2 play opposing roles in controlling colonic carcinoma cell proliferation through differential effects on EGFR signaling.
Keywords: Desmosome, intestinal epithelium, EGFR, desmoglein, desmocollin, cancer
INTRODUCTION
Desmoglein and desmocollin are cadherin family members that mediate cell–cell adhesion in desmosomes of epithelial tissues and cardiac muscle. In humans, four desmoglein (Dsg1–4) and three desmocollin isoforms (Dsc1–3) are expressed in a tissue-specific and differentiation-specific manner.1 Stratified epithelia such as the epidermis express multiple isoforms of each desmosomal cadherin, whereas simple human columnar intestinal epithelium expresses only the two desmosomal cadherins, desmoglein-2 (Dsg2) and desmocollin-2 (Dsc2).1 Both Dsg and Dsc are required for desmosome function.2 Furthermore, both Dsg and Dsc are needed for the de novo formation of desmosomes in cells lacking cadherin-based junctions.3,4 Such studies suggest that these partner desmosomal cadherins have complementary roles in the maintenance of intercellular adhesion.
In addition to their function in mediating cell adhesion, desmosomal cadherins have also been implicated in the regulation of epithelial cell proliferation and tumorigenesis.5–7 Our studies have been focused on understanding the functional role of Dsg2 and Dsc2 in intestinal epithelial cells, as expression of these proteins is altered in cancers and inflammatory diseases.7–10 We recently demonstrated that loss of Dsc2 promoted colonic epithelial cell proliferation and tumor growth in vivo,7 thus supporting a role of Dsc2 as a tumor suppressor in colon cancer. However, the contribution of Dsg2 to colon cancer cell growth is not known.
In the studies described herein, we examined the effect of Dsg2 downregulation on growth of colon cancer cells. In contrast to Dsc2, loss of Dsg2 is associated with decreased epithelial cell proliferation and impaired xenograft tumor growth. We show that this effect is dependent on Dsc2 expression, as Dsc2 knockdown reversed the proliferation defect in Dsg2-deficient cells. Furthermore, we demonstrate that growth suppression correlates with a diminished epidermal growth factor receptor (EGFR) signaling in Dsg2-deficient cells and identify Dsc2 as a key mediator of EGFR inhibition. Importantly, Dsg2 protein expression is increased in human colonic adenocarcinoma tissue specimens. Taken together, these results suggest that Dsg2 and Dsc2 have distinct roles in regulating intestinal epithelial cell growth, despite their complementary cell–cell adhesive functions. These data support the notion that Dsg2 is a pro-proliferative cadherin family member, and that interfering with the expression and/or function of this protein may be a viable strategy to impair cancer cell growth.
RESULTS AND DISCUSSION
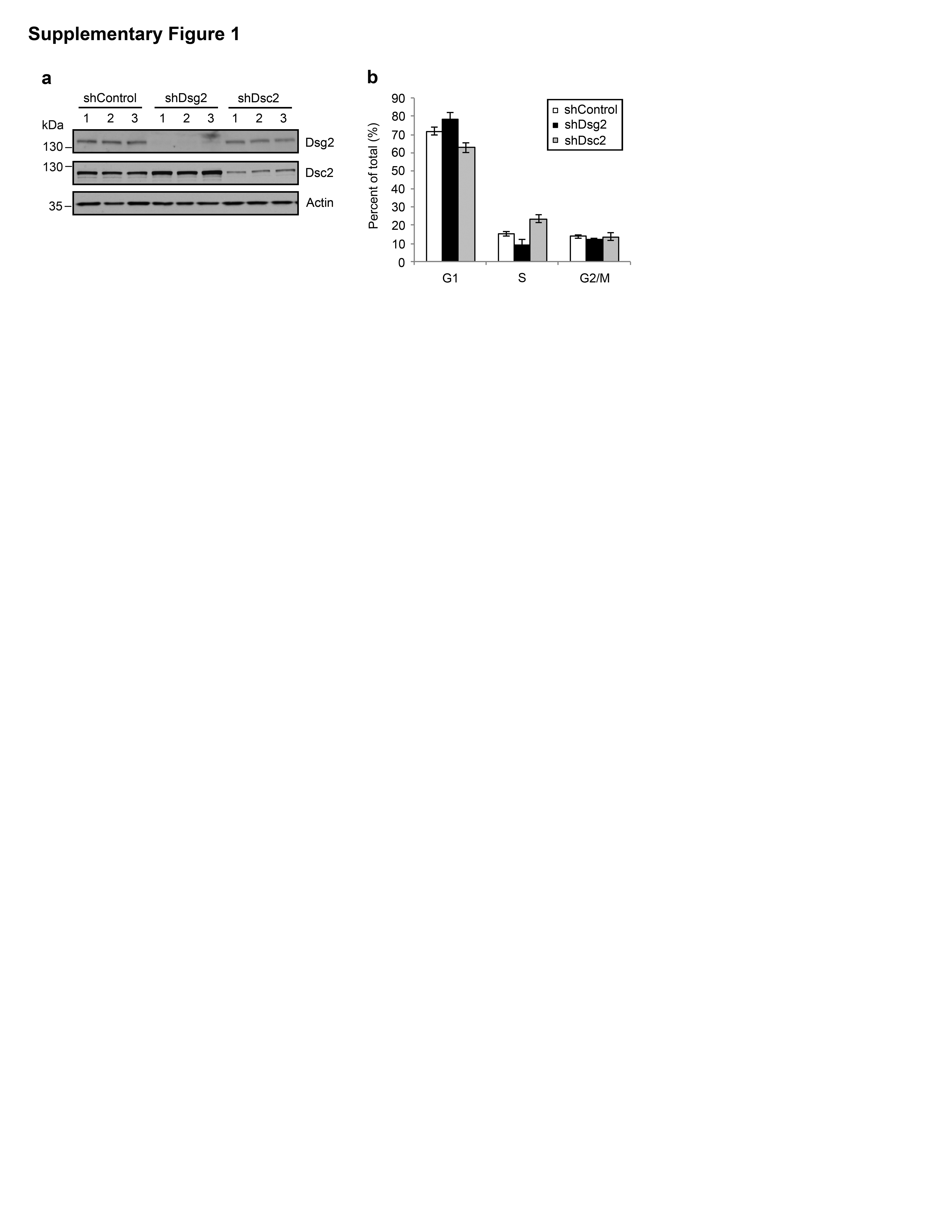
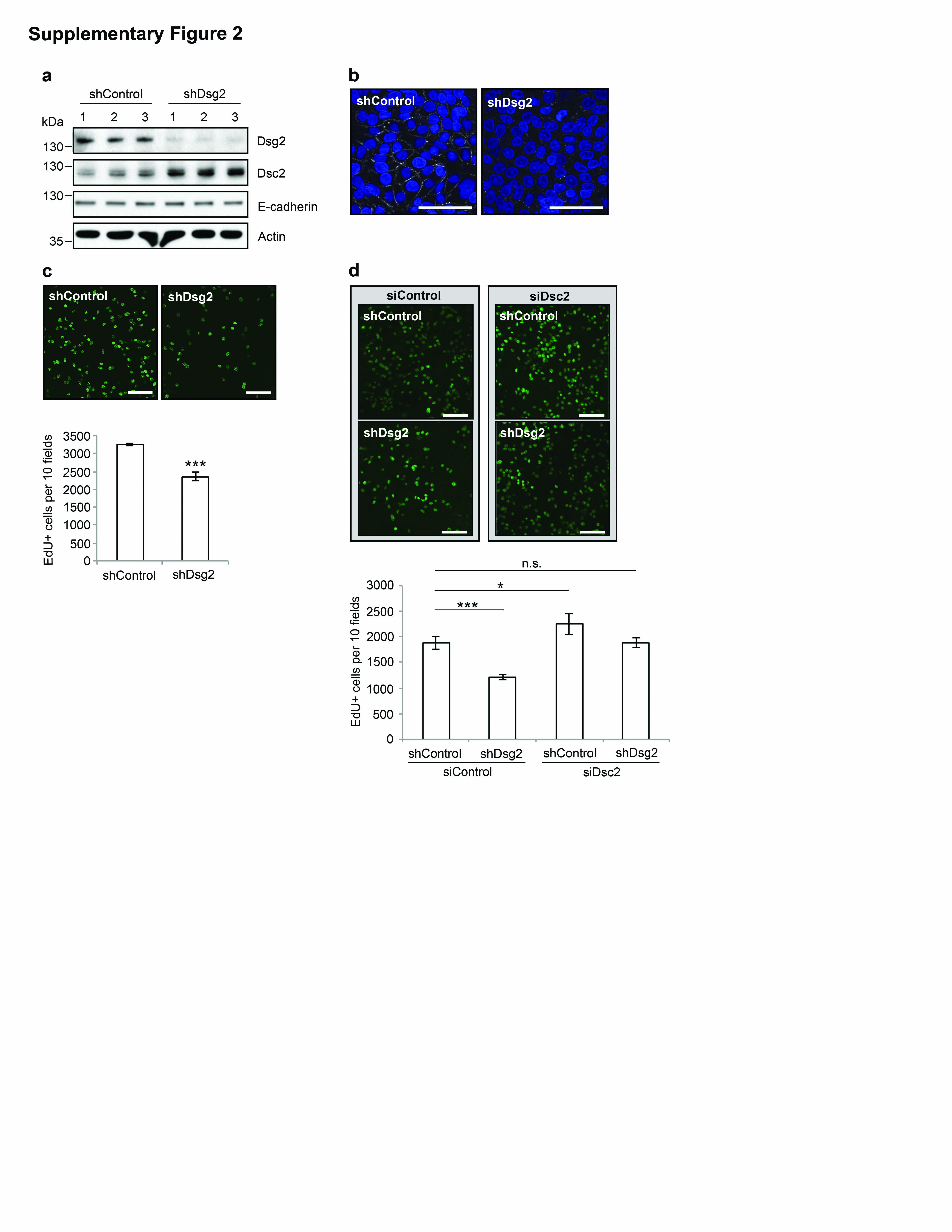
Dsg2 downregulation in colon cancer cells reduces proliferation We previously demonstrated that loss of Dsc2 in colonic adenocarcinoma cell lines increased cell proliferation and enabled a xenograft tumor growth.7 To assess whether loss of Dsg2 influences colon cancer growth, we generated stable knockdown SK-CO15 cell lines expressing the Dsg2-specific short hairpin RNA (shDsg2) via lentivirus transduction and compared the proliferation of these cells with controls expressing the non-targeting shRNA (shControl) or Dsc2 knockdown clones (shDsc2) generated in parallel. Downregulation of Dsg2 and Dsc2 protein was confirmed by immunoblotting (Figure 1a and Supplementary Figure 1a) and immunofluorescence labeling (data not shown). Three clones of each stable cell line (shControl 1–3, shDsg2 1–3 and shDsc2 1–3) were selected for the subsequent experiments. Cell cycle analysis using propidium iodide revealed that shDsc2 cells had an increased percentage of cells in S-phase compared to the controls, and is consistent with our previous report that loss of Dsc2 increased cell proliferation.7 Interestingly, downregulation of Dsg2 decreased the percentage of cells in S-phase (Supplementary Figure 1b). To confirm this observation, we also assessed cell proliferation using an EdU (5-ethynyl-2′-deoxyuridine) incorporation assay. Indeed, shDsg2 cells had significantly reduced EdU incorporation compared to their control counterparts (Figure 1b). Additionally, decreased phospho-Histone H3 (pHistone H3), a marker of cell division was observed in shDsg2 cells (Figure 1c). To determine whether loss of Dsg2 influenced cell proliferation in other colon cancer cell lines, we also assessed the effect of Dsg2 knockdown in the tumorigenic colonic adenocarcinoma cell line SW480 (Supplementary Figures 2a and b). Similarly, EdU incorporation assay revealed analogous suppression of cell proliferation in shDsg2 SW480 cells (Supplementary Figure 2c). Taken together, these data indicate that loss of Dsg2 reduces proliferation in colonic adenocarcinoma cells.
Figure 1.

Downregulation of Dsg2 in colon cancer cells reduces cell proliferation. (a) Immunoblot analysis demonstrating Dsg2, Dsc2 and E-cadherin levels in lentiviral-transduced desmoglein-2-specific shRNA (shDsg2, Open Biosystems, Huntsville, AL, USA) and control nonsilencing shRNA (shControl, Addgene, Cambridge, MA, USA) SK-CO15 cells. Cell lysates were subjected to immunoblot analysis against Dsg2 clone AH12.2 (generated in-house),18,21 Dsc2 clone 7G6 (Invitrogen, Carlsbad, CA, USA) and E-cadherin clone HECD-1.22 (b) Cell proliferation was measured by evaluating EdU incorporation. shControl and shDsg2 SK-CO15 cells were pulse labeled with EdU (Click-iT EdU, Invitrogen) for 1 h and processed to detect incorporated EdU. Images were obtained by confocal microscopy, and the number of EdU-positive nuclei was counted. ***P < 0.0001, t-test. (c) Immunoblot analysis of pHistone H3 levels in shControl and shDsg2 SK-CO15 cells. Total cell lysates of shControl and shDsg2 SK-CO15 cells were subjected to immunoblot analysis against pHistone H3 (Ser10, Cell Signaling, Beverly, MA, USA). (d) Immunoblot analysis confirming downregulation of Dsg2 in shDsg2 HeLa cells compared to shControl HeLa cells. (e) Proliferation of shControl and shDsg2 HeLa cells was determined by EdU incorporation. n.s., not significant (P = 0.2402, t-test). (f) Immunoblot analysis demonstrating pHistone H3 level in shControl and shDsg2 HeLa cells. (g) Immunoblot analysis confirming knockdown of Dsc2 in shControl and shDsg2 SK-CO15 cells after transfection with Dsc2-specific siRNA (siDsc2) and control siRNA (siControl) (Sigma-Aldrich, St Louis, MO, USA). (h) Proliferation of shControl and shDsg2 SK-CO15 cells transfected with siDsc2 was determined by EdU Incorporation. ***P < 0.0001, *P = 0.0499, n.s., not significant (P = 0.0694), t-test. Actin (Sigma-Aldrich) in a, c, d, f and g was used as a loading control. The histograms in b, e and h represent the mean number of EdU-positive nuclei from at least 10 different fields ± s.e.m.
Dsg2-deficient cells have increased Dsc2 protein
Unlike Dsg2 loss, Dsc2 downregulation increased colonic epithelial proliferation.7 To determine whether the distinct functional effect of Dsg2 downregulation was due to changes in expression of its partner cadherin, we also assessed Dsc2 protein levels in shDsg2 cells by immunoblotting. As shown in Figure 1a and Supplementary Figure 2a, downregulation of Dsg2 was associated with increased Dsc2 protein levels in SK-CO15 and SW480 stable cell lines. To assess whether Dsg2 knockdown influences another key cadherin, we examined E-cadherin protein expression in shControl and shDsg2 cells. However, loss of Dsg2 did not significantly alter the E-cadherin protein in either SK-CO15 or SW480 cells (Figure 1a and Supplementary Figure 2a). As above, changes in cadherin composition following an altered expression of a specific cadherin have also been reported.11 However, the underlying mechanisms that mediate such compensation are not well understood. The potential mechanisms for increased Dsc2 protein following Dsg2 downregulation include transcriptional regulation and/or Dsc2 protein stabilization that could be mediated by scaffold proteins.
In the absence of Dsc2, downregulation of Dsg2 has no effect on cell proliferation
Given that shDsg2 cells have an increased Dsc2 protein, we hypothesized that Dsc2 contributed to growth suppression in these cells. We first examined whether knockdown of Dsg2 influenced proliferation in cancer cells that lack Dsc2 expression. As the human carcinoma cell line HeLa expresses Dsg2 but not Dsc2 or E-cadherin,12 we assessed the effect of shRNA-induced Dsg2 downregulation on HeLa cell proliferation (Figure 1d). EdU incorporation assay and immunoblotting for pHistone H3 demonstrated that downregulation of Dsg2 did not alter cell proliferation in these cells (Figures 1e and f). These data indicate that Dsc2 expression may be required for growth suppression following Dsg2 downregulation.
In order to specifically test whether Dsc2 was required for growth inhibition in shDsg2 cells, we next performed dual knockdown experiments in SK-CO15 and SW480 stable cell lines using Dsc2-specific small interfering RNA (siDsc2). Dsc2 expression was significantly downregulated in shControl and shDsg2 cells treated with siDsc2 compared to those treated with non-targeting control siRNA (siControl) in SK-CO15 (Figure 1g) and SW480 (data not shown) cell lines. To determine the effect of dual Dsg2/Dsc2 knockdown on cell proliferation, we performed EdU incorporation assays. As described above, shDsg2 cells had reduced proliferation compared to the control counterparts. In addition, downregulation of Dsc2 enhanced proliferation in both SK-CO15 and SW480 cells as previously reported.7 Finally, dual knockdown of Dsg2 and Dsc2 rescued the growth suppressive effect of Dsg2 downregulation in SK-CO15 and SW480 stable cell lines (Figure 1h and Supplementary Figure 2d). Thus, these data indicate that Dsc2 expression is required for the growth suppressive effects of Dsg2 downregulation.
EGFR signaling is diminished in cells lacking Dsg2
We next investigated the mechanism of growth suppression in Dsg2-deficient cells. A previous study reported that another isoform of Dsg, Dsg1, is expressed in suprabasal differentiated keratinocytes and is required for suppressing EGFR-extracellular signal-regulated kinase (Erk) 1/2 signaling to allow for normal differentiation.13 However, cell proliferation was not measured in this study. As we had observed that EGFR signaling promotes proliferation in Dsc2-deficient colonic epithelial cells,7 we assessed whether EGFR signaling was altered in SK-CO15 cells lacking Dsg2.
Given that activation of cells by epidermal growth factor (EGF) leads to internalization and degradation of EGFR, we first localized the receptor in shControl and shDsg2 cells following treatment with the ligand. As shown in Figure 2a, EGF exposure led to the internalization of EGFR within 1 h in shControl cells with diminishing protein levels at later time points. In contrast, this response was delayed in shDsg2 cells. Additionally, EGF treatment decreased the EGFR protein levels in shControl cells, consistent with degradation of the receptor in response to EGF stimulation (Figure 2b). However, EGFR protein remained stable in shDsg2 cells despite 1 h of EGF treatment. Taken together, these data indicate that shDsg2 cells are less responsive to EGF stimulation.
Figure 2.

Loss of Dsg2 suppresses EGFR signaling. (a) Immunofluorescence labeling confocal microscopy demonstrating EGF-induced EGFR internalization. shControl and shDsg2 SK-CO15 cells were treated with 20 ng/ml EGF (BD Biosciences, San Jose, CA, USA) at different time course. Green, EGFR (Cell Signaling); scale bar is 5 µm. (b) Immunoblot analysis of EGFR and Dsg2 in shControl and shDsg2 SK-CO15 cells at steady state and after the addition of EGF. The cells were incubated with 20 ng/ml EGF 5–120 min. The graph displays the relative EGFR expression normalized to actin as determined by densitometric analysis. Mean ± s.e.m. The data were averaged from three independent experiments. (c) Immunoblot analysis against pEGFR Y845, pEGFR S1142 and EGFR levels in shControl and shDsg2 SK-CO15 cells. Cell lysates were subjected to immunoblot analysis against Dsg2 (AH12.2), pEGFR Y845 (Cell Signaling), pEGFR S1142 (ECM Biosciences, Versailles, KY, USA) and EGFR. (d) Immunoblot analysis of EGFR signaling cascade in shControl and shDsg2 SK-CO15 cells. Cell lysates were subjected to immunoblot analysis against Dsg2 (AH12.2), pErk1/2 T202/Y204 (Cell Signaling), Erk1/2 (Cell Signaling), pSrc Y416 clone 9A6 (Millipore, Billerica, MA, USA) and Src clone M259 (ECM Biosciences). (e) Immunoblot analysis demonstrating EGFR signaling cascade and pHistone H3 in Dsc2 overexpressing SK-CO15 cells. Cells were transfected with 2.5 µg of pcDNA3.1 empty vector (control) or 2.5 µg of the pcDNA3.1-Dsc2-myc expression vector using Lipofectamine 2000 (Invitrogen) in Opti-MEM medium (Invitrogen). The cells were collected 48 h post transfection. Total cell lysates were subjected to immunoblot analysis against Myc Tag clone 9B11 (Cell Signaling), pEGFR Y845, pEGFR S1142, EGFR, pErk1/2, Erk1/2 and pHistone H3. (f) A model for how Dsg2 regulates cell proliferation. Loss of Dsg2 inhibits intestinal epithelial cell proliferation and tumor growth through inactivation of the EGFR, Erk and Src signaling pathway. Knockdown of Dsg2 results in increased Dsc2 protein that compensates for Dsg2 loss, which leads to a decrease active (Y845) EGFR and increase in inactive (S1142) EGFR resulting in less cell proliferation. Our model is further supported by a decrease in Src and downstream Erk activation, which are involved in regulated cell proliferation through further effectors. Downregulation of Dsg2 negatively regulates cell proliferation and tumor growth in vitro and in vivo. Actin in b, c and e was used as a loading control.
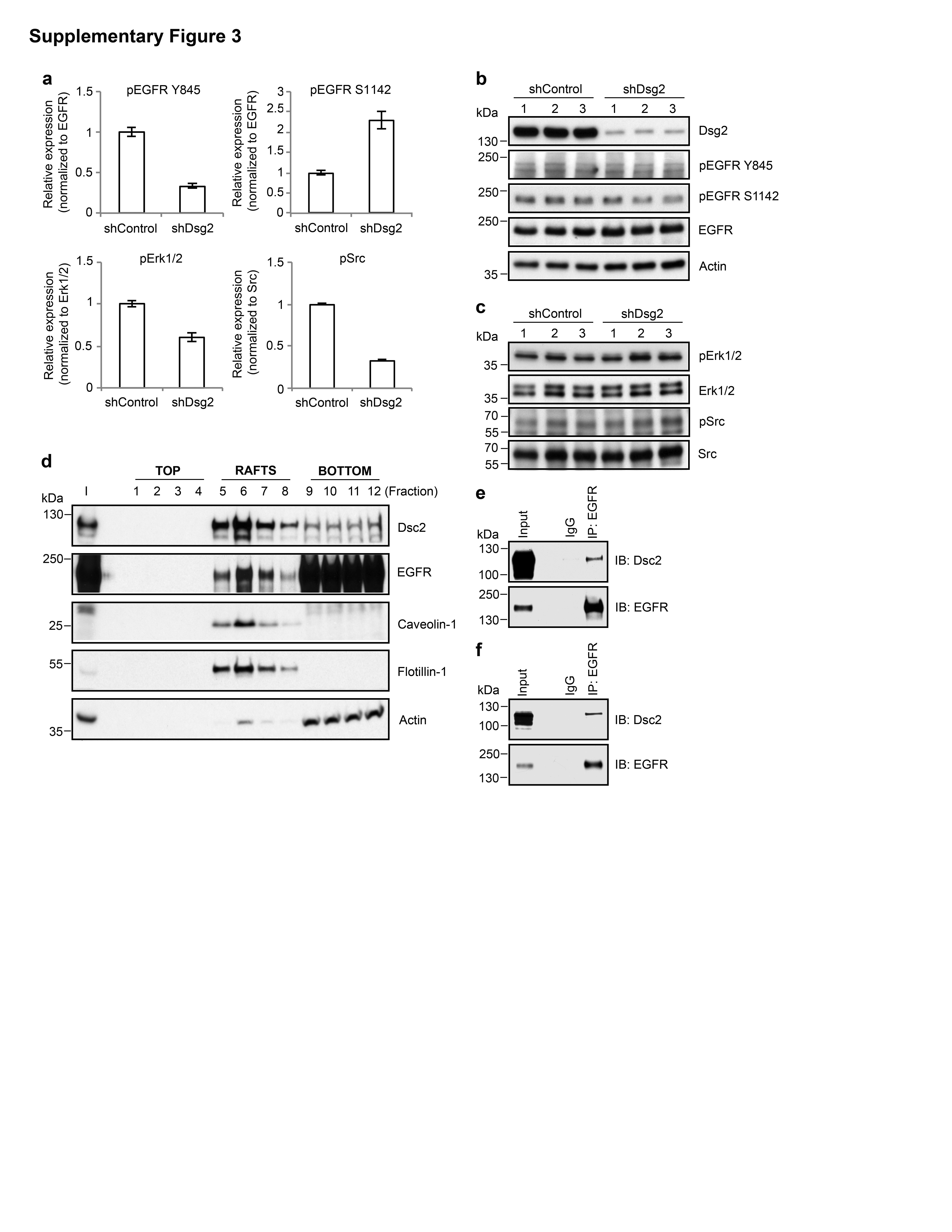
To assess EGFR activity, we examined the EGFR phosphorylation by immunoblotting for phospho-EGFR Tyr845 (pEGFR Y845) that has been linked to receptor activation.14 As shown in Figure 2c and Supplementary Figure 3a, shDsg2 cells had decreased pEGFR Y845 compared to shControl cells. We also assessed levels of phospho-EGFR Ser1142 (pEGFR S1142) that is inversely correlated with receptor internalization and degradation.15 Indeed, increased pEGFR S1142 was observed in shDsg2 cells. These findings suggest that there is decreased EGFR activity in shDsg2 cells, and that the inactive receptor is more stable at the cell surface.
EGFR interacts with the tyrosine kinase Src and recruits downstream signaling proteins such as Erk to influence cell proliferation.16 To further evaluate EGFR signaling, we determined Erk and Src activation using phospho-specific antibodies. As shown in Figure 2d and Supplementary Figure 3a, shDsg2 cells had decreased phospho-Erk1/2 Thr202/Tyr204 (pErk1/2), and phospho-Src Tyr416 (pSrc). These data link decreased proliferation following Dsg2 downregulation to EGFR-mediated signaling events.
Finally, we tested whether downregulation of Dsg2 in cells lacking Dsc2 altered EGFR signaling in HeLa cells. As shown in Supplementary Figure 3b, Dsg2 downregulation did not influence EGFR phosphorylation. Furthermore, there is no significant change of pErk1/2 and pSrc in shDsg2 cells (Supplementary Figure 3c). The absence of EGFR signaling following Dsg2 downregulation paralleled the lack of effects on cell proliferation in these cells. Taken together, these results demonstrate that EGFR signaling is decreased in SK-CO15 cells that lack Dsg2 but still express Dsc2.
Dsc2 overexpression suppresses EGFR activation
To examine whether Dsc2 upregulation was sufficient for suppression of EGFR signaling, we overexpressed full-length Dsc2 in SK-CO15 cells and assessed the activity of EGFR. A myc-tagged Dsc2 plasmid was transfected into cells as described previously,7,17 and the expression was confirmed by immunoblotting (Figure 2e) and immunofluorescence labeling (data not shown). Overexpression of Dsc2 decreased pHistone H3 and suppressed EGFR signaling, as determined by decreased pEGFR Y845 and an increased pEGFR S1142, which mimicked the influence of Dsg2 downregulation (Figure 2e).
To further determine the relationship between Dsc2 and EGFR, we examined the subcellular localization of these proteins in SK-CO15 cells. We and others have previously demonstrated that the desmosomal cadherins localize in the epithelial lipid rafts,18,19 which suggests that Dsc2 may interact with the EGFR within this compartment. To examine this possibility, we isolated the lipid raft fraction of SK-CO15 cells by floatation in isopycnic sucrose density gradients. Immuoblotting of gradient fractions revealed that Dsc2 co-fractionated with lipid raft-resident proteins, caveolin-1 and flotillin-1. Additionally, a pool of EGFR was also identified in the raft fractions (Supplementary Figure 3d), suggesting that Dsc2 may interact with the EGFR within this compartment. We next asked whether Dsc2 and EGFR interact within cells, as has been demonstrated with other desmosomal cadherin family members.13 To address this question, we performed coimmunoprecipitation experiments in SK-CO15 and T84 colonic adenocarcinoma lines as shown in Supplementary Figures 3e and f. Indeed, Dsc2 co-immunoprecipited with EGFR in two colonic adenocarcinoma cell lines, SK-CO15 and T84. Together, these data indicate that an increased Dsc2 protein inhibits EGFR signaling in shDsg2 cells, thereby inhibiting cell proliferation (Figure 2f).
Dsg2-deficient SW480 colon cancer cells fail to grow as tumors in vivo
Our in vitro data indicate that loss of Dsg2 led to growth suppression via EGFR signaling. The influence of Dsg2 loss on xenograft tumor growth was evaluated using shDsg2 SW480 cells.7 As shown in Figure 3a, mice injected with shControl cells readily formed tumors. Histological analysis of the tumors from shControl-injected mice demonstrated growth of cells with a high mitotic activity, consistent with a poorly differentiated adenocarcinoma (Figure 3b). Remarkably, no tumors were detected in mice injected with shDsg2 SW480 cells (Figure 3a). Additionally, we evaluated xenograft tumor growth using shControl and shDsg2 HeLa cells that do not express Dsc2. Consistent with the proliferative profile of these cells, we did not observe any difference in xenograft tumors in shControl versus shDsg2 HeLa cells (data not shown). Taken together, these data demonstrate that stable downregulation of Dsg2 inhibits xenograft tumor formation in mice and Dsc2 is required for growth suppression.
Figure 3.

Dsg2-deficient tumorgenic SW480 colon cancer cells fail to grow as tumors in vivo. (a) In vivo xenograft tumor growth for shControl versus shDsg2 cells in Rag1−/− mice. Eight-week-old male mice were injected subcutaneously with 1 × 106 shControl or shDsg2 SW480 cells. Animals were killed at 21 days post injection and the extent of tumor development was assessed macroscopically. All animal experiments were performed in accordance with protocols approved by the Emory University IACUC. (b) Histological analysis of shControl-derived tumors stained with hematoxylin and eosin (Sigma-Aldrich). The insert is a high magnification image of the tumor. Scale bar is 20 µm. (c) Immunoblot analysis of Dsg2 protein in human colonic adenocarcinoma tumors and corresponding normal colonic tissue samples (Protein Biotechnologies Inc., Ramona, CA, USA). The samples were subjected to immunoblot analysis against Dsg2 clone DG3.10 (PROGEN Biotechnik, Heidelberg, Germany). Ponceau staining (Sigma-Aldrich) was used to show equal loading of protein. The histogram shows the relative Dsg2 expression normalized to Ponceau as determined by densitometric analysis. N, normal; T, tumor. (d) Immunofluorescence labeling to localize Dsg2 in normal human colon and human colonic adenocarcinomas (Tumor). Nuclei were stained using To-Pro-3 iodide (TOPRO, Invitrogen). Green, Dsg2 (AH12.2); blue, TOPRO. Consecutive sections of human colonic adenocarcinoma and normal colonic tissue were stained with hematoxylin and eosin. Scale bar is 100 µm.
Dsg2 expression is increased in human colonic adenocarcinomas
Our results demonstrate that Dsg2 expression in colon cancer cell lines promotes proliferation and tumor growth. Additionally, increased Dsg2 expression has been observed in malignant skin carcinoma.9 However, expression of Dsg2 in human colonic adenocarcinoma tissue specimens has not been evaluated. To assess Dsg2 expression in human colon cancers, we obtained matched samples of normal colon and colonic adenocarcinoma from individual patients with colon cancer (Supplementary Table 3) and assessed the Dsg2 protein by immunoblotting. As shown in Figure 3c, Dsg2 expression is increased in the carcinoma sample compared to normal tissue. Similarly, the increased Dsg2 protein was detected in colon adenocarcinoma compared to the normal colon by immunofluorescence labeling (Figure 3d). These findings are consistent with earlier studies demonstrating an increased expression of Dsg isoforms in other carcinomas5,9,20 and provides further evidence that Dsg2 expression may promote proliferation in colon cancers.
In summary, we provide evidence that Dsg2 exerts a proliferative, pro-tumorigenic function in colon cancer cells. Our data demonstrate that downregulation of Dsg2 expression in colonic adenocarcinoma cell lines leads to growth inhibition both in vitro and in vivo, thereby highlighting the essential role of this cell adhesion protein in control of cancer cell growth. In addition, we show that loss of Dsg2 does not influence colon cancer cell proliferation in the absence of Dsc2, further supporting the tumor suppressive role of Dsc2 in colonic epithelium. Finally, the data presented herein provide the first direct evidence that Dsg2 and Dsc2 play opposing roles in the regulation of cell proliferation, and highlight an important mechanistic interplay between these partner desmosomal cadherins.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
This work was supported by the 12th Glaxo-SmithKline International Award from Japan Society of Immunology and Allergology in Otolaryngology (RK), an American Gastroenterological Association Research Scholar Award (PN), a Crohn’s and Colitis Foundation of America Career Development Award (PN), a CONACyT grant 175854 (PN) and National Institutes of Health grants DK061379, DK072564 (CAP), DK055679, DK059888 (AN) and DDRDC DK064399.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
REFERENCES
- 1.Holthofer B, Windoffer R, Troyanovsky S, Leube RE. Structure and function of desmosomes. Int Rev Cytol. 2007;264:65–163. doi: 10.1016/S0074-7696(07)64003-0. [DOI] [PubMed] [Google Scholar]
- 2.Getsios S, Amargo EV, Dusek RL, Ishii K, Sheu L, Godsel LM, et al. Coordinated expression of desmoglein 1 and desmocollin 1 regulates intercellular adhesion. Differentiation. 2004;72:419–433. doi: 10.1111/j.1432-0436.2004.07208008.x. [DOI] [PubMed] [Google Scholar]
- 3.Tselepis C, Chidgey M, North A, Garrod D. Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci USA. 1998;95:8064–8069. doi: 10.1073/pnas.95.14.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlegel N, Meir M, Heupel WM, Holthofer B, Leube RE, Waschke J. Desmoglein 2-mediated adhesion is required for intestinal epithelial barrier integrity. Am J Physiol Gastrointest Liver Physiol. 2010;298:G774–G783. doi: 10.1152/ajpgi.00239.2009. [DOI] [PubMed] [Google Scholar]
- 5.Brennan D, Hu Y, Joubeh S, Choi YW, Whitaker-Menezes D, O’Brien T, et al. Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J Cell Sci. 2007;120:758–771. doi: 10.1242/jcs.03392. [DOI] [PubMed] [Google Scholar]
- 6.Mannan T, Jing S, Foroushania SH, Fortune F, Wan H. RNAi-mediated inhibition of the desmosomal cadherin (desmoglein 3) impairs epithelial cell proliferation. Cell Prolif. 2011;44:301–310. doi: 10.1111/j.1365-2184.2011.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolegraff K, Nava P, Helms MN, Parkos CA, Nusrat A. Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/β-catenin signaling. Mol Biol Cell. 2011;22:1121–1134. doi: 10.1091/mbc.E10-10-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biedermann K, Vogelsang H, Becker I, Plaschke S, Siewert JR, Hofler H, et al. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J Pathol. 2005;207:199–206. doi: 10.1002/path.1821. [DOI] [PubMed] [Google Scholar]
- 9.Brennan D, Mahoney MG. Increased expression of Dsg2 in malignant skin carcinomas: a tissue-microarray based study. Cell Adh Migr. 2009;3:148–154. doi: 10.4161/cam.3.2.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wex T, Kuester D, Monkemuller K, Stahr A, Fry LC, Kandulski A, et al. Assessment of desmosomal components (desmoglein 1–3, plakoglobin) in cardia mucosa in relation to gastroesophageal reflux disease and Helicobacter pylori infection. Hum Pathol. 2012;43:1745–1754. doi: 10.1016/j.humpath.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 11.Baron S, Hoang A, Vogel H, Attardi LD. Unimpaired skin carcinogenesis in Desmoglein 3 knockout mice. PLoS One. 2012;7:e50024. doi: 10.1371/journal.pone.0050024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lock JG, Stow JL. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol Biol Cell. 2005;16:1744–1755. doi: 10.1091/mbc.E04-10-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, et al. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol. 2009;185:1243–1258. doi: 10.1083/jcb.200809044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 15.Oksvold MP, Thien CB, Widerberg J, Chantry A, Huitfeldt HS, Langdon WY. Serine mutations that abrogate ligand-induced ubiquitination and internalization of the EGF receptor do not affect c-Cbl association with the receptor. Oncogene. 2003;22:8509–8518. doi: 10.1038/sj.onc.1207117. [DOI] [PubMed] [Google Scholar]
- 16.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 17.Ishii K, Norvell SM, Bannon LJ, Amargo EV, Pascoe LT, Green KJ. Assembly of desmosomal cadherins into desmosomes is isoform dependent. J Invest Dermatol. 2001;117:26–35. doi: 10.1046/j.0022-202x.2001.01400.x. [DOI] [PubMed] [Google Scholar]
- 18.Nava P, Laukoetter MG, Hopkins AM, Laur O, Gerner-Smidt K, Green KJ, et al. Desmoglein-2: a novel regulator of apoptosis in the intestinal epithelium. Mol Biol Cell. 2007;18:4565–4578. doi: 10.1091/mbc.E07-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resnik N, Sepcic K, Plemenitas A, Windoffer R, Leube R, Veranic P. Desmosome assembly and cell-cell adhesion are membrane raft-dependent processes. J Biol Chem. 2011;286:1499–1507. doi: 10.1074/jbc.M110.189464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen YJ, Chang JT, Lee L, Wang HM, Liao CT, Chiu CC, et al. DSG3 is overexpressed in head neck cancer and is a potential molecular target for inhibition of oncogenesis. Oncogene. 2007;26:467–476. doi: 10.1038/sj.onc.1209802. [DOI] [PubMed] [Google Scholar]
- 21.Kolegraff K, Nava P, Laur O, Parkos CA, Nusrat A. Characterization of full-length and proteolytic cleavage fragments of desmoglein-2 in native human colon and colonic epithelial cell lines. Cell Adh Migr. 2011;5:306–314. doi: 10.4161/cam.5.4.16911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimoyama Y, Hirohashi S, Hirano S, Noguchi M, Shimosato Y, Takeichi M, et al. Cadherin cell-adhesion molecules in human epithelial tissues and carcinomas. Cancer Res. 1989;49:2128–2133. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.