

Abstract
Epithelial ovarian cancer is the most lethal type of gynecologic malignancy. Sixty percent of women who are diagnosed with ovarian cancer present with advanced-stage disease that involves the peritoneal cavity and these patients have a 5-year survival rate of less than 30%. For more than two decades, tumor-debulking surgery followed by platinum-taxane combination chemotherapy has remained the conventional first-line treatment of ovarian cancer. Although the initial response rate is 70%-80%, most patients with advanced-stage ovarian cancer eventually relapse and succumb to recurrent chemoresistant disease. A number of molecular aberrations that drive tumor progression have been identified in ovarian cancer cells and intensive efforts have focused on developing therapeutic agents that target these aberrations. However, increasing evidence indicates that reciprocal interactions between tumor cells and various types of stromal cells also play important roles in driving ovarian tumor progression and that these stromal cells represent attractive therapeutic targets. Unlike tumor cells, stromal cells within the tumor microenvironment are in general genetically stable and are therefore less likely to become resistant to therapy. This concise review discusses the biological significance of the cross-talk between ovarian cancer cells and three major types of stromal cells (endothelial cells, fibroblasts, macrophages) and the development of new-generation therapies that target the ovarian tumor microenvironment.
Keywords: Ovarian cancer, Tumor stroma, Endothelial cells, Fibroblasts, Macrophages, Targeted therapy
Core tip: Despite advances in clinical management, advanced-stage ovarian cancer is still rarely cured by conventional chemotherapy. Substantial efforts have been directed to developing new therapies that target ovarian cancer cells. However, recent studies have revealed important roles of a variety of stromal cells in driving the aggressive behavior of ovarian cancer. Here, we discuss: (1) the significance of three major types of stromal cells in the progression of ovarian cancer; (2) how receptor/ligand-mediated interactions between ovarian cancer cells and stromal cells serve as focal points for therapeutic intervention; and (3) key examples of new-generation agents that target stromal cells.
INTRODUCTION
Epithelial ovarian cancer is the fifth leading cause of cancer death in women and the most lethal form of gynecologic malignancy[1]. The high morbidity and mortality caused by ovarian cancer primarily stems from late diagnosis. Sixty percent of women who are diagnosed with ovarian cancer present with extensive peritoneal carcinomatosis and these patients have a 5-year survival rate of less than 30%[1]. For more than 20 years, tumor-debulking surgery followed by platinum-taxane combination chemotherapy has remained the standard first-line treatment[2]. Although the initial response rate is 70%-80%, most patients with advanced-stage ovarian cancer relapse within 18 mo and eventually die from the disease[2]. Substantial efforts have been directed to developing new-generation agents that target functionally relevant molecular aberrations in ovarian cancer cells[3]. Inhibitors of poly (ADP-ribose) polymerase, a DNA repair enzyme, have been undergoing clinical trials in patients with BRCA-deficient ovarian cancer and have attracted considerable attention[4]. In addition to agents that target pathways in ovarian cancer cells, agents that target the tumor vasculature have been the focus of intensive clinical investigation[5,6]. Increasing evidence indicates that ovarian tumor progression is driven not only by dynamic interplay between tumor cells and endothelial cells but also by other types of stromal cells that are “educated” by tumors to acquire properties that are permissive for tumor growth. In this article, we provide an overview of the cross-talk between ovarian cancer cells, endothelial cells and two other key constituents of the tumor microenvironment, specifically, fibroblasts and macrophages, and discuss examples of clinically used and emerging experimental agents that target these stromal cells.
ENDOTHELIAL CELLS
Of the cell types that comprise the ovarian tumor microenvironment, the endothelial cell has been the most extensively studied in terms of its clinical significance. A number of independent studies have identified that increased tumor angiogenesis as manifested by high microvessel density is predictive of poor outcomes in ovarian cancer patients[7-9]. Angiogenesis is a dynamic process that involves the recruitment of endothelial progenitors, growth and maturation of endothelial cells and vessel formation, and is orchestrated by a repertoire of pro-angiogenic and anti-angiogenic factors[10,11]. Key pro-angiogenic factors include the vascular endothelial growth factors (VEGF), platelet-derived growth factor (PDGF), fibroblast growth factor-2 (FGF-2), angiopoietin, interleukin (IL)-6 and IL-8. Of these factors, VEGF-A has emerged as the predominant pro-angiogenic factor that is highly expressed in ovarian cancers[5,6]. VEGF-A has also been identified to be the causative factor of ascites formation by inducing vascular permeability[12].
Intensive clinical efforts have focused on evaluating agents that inhibit VEGF signaling. These agents fall into two categories: (1) those that inhibit the ligand; and (2) those that inhibit tyrosine kinase activity of the VEGF receptors (VEGFR) (Figure 1). Of the former group, bevacizumab has been the most extensively evaluated agent in ovarian cancer. Bevacizumab is a humanized monoclonal antibody (mAb) that neutralizes all forms of VEGF and was originally Food and Drug Administration-approved in 2004 for treatment of metastatic colorectal cancer. Bevacizumab has been evaluated as a single agent in the treatment of patients with recurrent ovarian cancer in two pivotal phase II trials. In one of these studies (AVF 2949g), the response rate was 15.9% and median overall survival (OS) was 10.7 mo[13]. This study was terminated early due to a high rate of gastrointestinal perforations (5 of 44 patients, 11.4%). In the other study [Gynecologic Oncology Group (GOG) 170D], the response rate was 21.0%, median OS was 16.9 mo, and no bowel perforations were observed[14]. One possible explanation for the differences in results of these trials is that the GOG study was limited to patients who had received no more than two prior lines of therapy, whereas 21 of the 44 patients in the AVF 2949g study (including the five patients who developed bowel perforations) had received three prior regimens. Bevacizumab has also been evaluated in combination with carboplatin and paclitaxel. In the first-line setting, two phase III trials (GOG 218 and ICON7) reported that progression-free survival (PFS) was increased (by 3.8 and 1.7 mo, respectively) with the combination of bevacizumab and standard chemotherapy followed by bevacizumab maintenance, as compared to standard chemotherapy alone[15,16]. In the recurrent setting, two other phase III studies have found that PFS was increased by approximately 3.6 mo when bevacizumab was combined with standard chemotherapy[17,18]. Another ligand-inhibitory agent is aflibercept, a fusion protein that acts as a soluble VEGFR decoy. In a phase II study of aflibercept in patients with recurrent ovarian cancer, the rate of gastrointestinal perforations was found to be low (1.4%) but the primary endpoint of a response rate of greater than 5% was not achieved[19].
Figure 1.
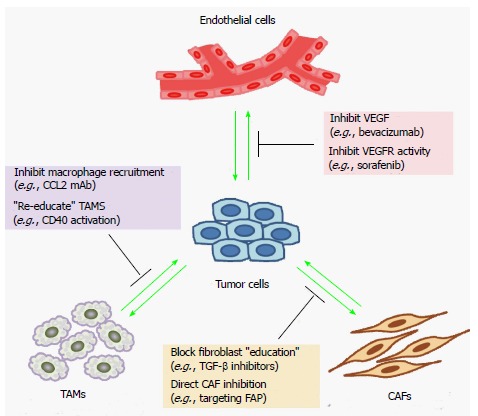
Therapeutic strategies to target the tumor microenvironment. Shown are examples of different strategies and agents that inhibit the regulation of a specific type of stromal cell or its functional properties. Several of these agents are in clinical use, whereas others are at different stages of clinical development. VEGF: Vascular endothelial growth factors; TAMs: Tumor-associated macrophages; CAFs: Cancer-associated fibroblasts; TGF-β: Transforming growth factor-β; FAP: Fibroblast activation protein; CCL2: chemokine (C-C motif) ligand 2; VEGFR: Vascular endothelial growth factor receptor.
Tyrosine kinase inhibitors (TKIs) represent another important class of anti-angiogenic agents. Sorafenib is an oral multi-kinase inhibitor that targets several receptor tyrosine kinases including VEGFR-2, VEGFR-3, platelet-derived growth factor receptor-β (PDGFR-β) and c-kit, and also the RAF family of serine/threonine kinases[20]. In a phase II trial of sorafenib monotherapy in patients with recurrent ovarian cancer, two of the 59 evaluable patients had partial responses whereas 20 had stable disease and 30 had progressive disease[21]. Another phase II study found that sorafenib did not improve efficacy of first-line carboplatin/paclitaxel treatment and resulted in additional toxicity[22]. Several TKIs that inhibit all three VEGFRs and both PDGFRs have been developed such as sunitinib, cediranib and pazopanib. Sunitinib has been found to have only modest activity as a single agent in patients with recurrent ovarian cancer[23,24]. Clinical trials are ongoing to evaluate cediranib[25] for treatment of recurrent ovarian cancer and pazopanib[26] as maintenance therapy for patients in remission following first-line platinum-taxane chemotherapy.
CANCER-ASSOCIATED FIBROBLASTS
Cancer-associated fibroblasts (CAFs) constitute the cellular fibrotic component of the tumor stroma that is commonly described as “reactive” or desmoplastic stroma. CAFs are often distinguished from normal quiescent fibroblasts by their expression of markers of myofibroblasts and activated fibroblasts such as α-smooth muscle actin (αSMA) and fibroblast activation protein (FAP)[27,28]. CAFs derive from multiple cell types. Two important sources are mesenchymal stem cells (MSCs) and tissue-resident fibroblasts. MSCs are abundant in white adipose tissues such as the omentum[29], the most commonly involved site in ovarian cancer. It has been demonstrated that ovarian cancer cell-derived factors, such as transforming growth factor-β (TGF-β) and lysophosphatidic acid, induce normal omental fibroblasts and adipose MSCs to acquire features of CAFs[30,31]. Studies of other types of tumors have shown that CAFs can also derive from bone marrow MSCs that are recruited to tumors[32,33]. There is evidence in breast cancer that some CAFs derive from tumor cells that have undergone epithelial-to-mesenchymal transition[34]. However, a study of ovarian cancer xenograft models found that stromal αSMA+ cells did not derive from tumor cells, suggesting that ovarian cancer cells are not a major source of CAFs[31].
Substantial evidence indicates that CAFs contribute to poor survival of cancer patients by promoting tumor cell proliferation, angiogenesis and metastasis[27,28]. In a study of gene expression profiles of clinical specimens of ovarian cancer, Tothill et al[35] identified that the subset of cases with the poorest outcomes was characterized by a desmoplastic gene signature. As compared to normal omental fibroblasts, CAFs more highly express IL-6, chemokine (C-X-C motif) ligand 12 (CXCL12) and VEGF-A, and are more effective in stimulating growth of ovarian cancer cells and endothelial cells[31]. The abundance of CAFs in ovarian cancers has been found to correlate with microvessel density[36]. CAFs also highly express TGF-β, matrix metalloproteinases (MMPs) and numerous extracellular matrix proteins[27,28], and stimulate invasiveness of ovarian cancer cells[36]. Furthermore, McLean and colleagues identified that propagating ovarian cancer cells with MSCs derived from ovarian cancer specimens increased the number of cancer stem cells[37]. These findings suggest that another mechanism by which CAFs drive tumorigenesis is by expanding the sub-population of tumor-initiating cells.
Given the profound negative impact of CAFs on outcomes, there have been intensive efforts to develop strategies to target this cell population (Figure 1). Several approaches to inhibit CAFs have been directed to targeting FAP. A humanized mAb to FAP has been found to be well-tolerated, but failed to show efficacy in a clinical trial of patients with metastatic colorectal cancer[38]. In a preclinical study, a DNA vaccine against FAP inhibited tumor growth and increased survival in a mouse colon cancer model[39]. A study by Brennen and colleagues exploited both the expression of FAP on CAFs and its proteolytic activity. These authors generated a prodrug that consisted of a FAP-specific peptide coupled to a thapsigargin analog as the cytotoxic moiety, and demonstrated that the compound induced stromal cell death and inhibited growth of breast and prostate tumor xenografts[40]. Another potential approach to inhibit CAFs is to prevent normal MSCs and fibroblasts from transitioning into CAFs by blocking TGF-β signaling. A number of agents that inhibit TGF-β signaling have been developed including TGF-β-ligand traps, TGF-β antisense oligonucleotides and small molecule inhibitors of the TGF-β type I receptor kinase, and several of these agents have been evaluated in clinical trials[41,42]. The utility of TGF-β inhibitors has been little-explored in ovarian cancer. In one study, treatment of mice with the TGF-β type I receptor inhibitor A83-01 reduced the fibrotic component of ovarian tumor xenografts but did not increase survival times[43]. Unlike TGF-β, PDGF does not induce myofibroblastic differentiation but instead stimulates fibroblasts to produce mitogenic factors for tumor cells and pro-angiogenic factors. Blockade of PDGFR signaling in a mouse model of cervical cancer has been found to inhibit tumor growth and angiogenesis in part by inhibiting FGF-2 production by CAFs[44]. As discussed earlier, several TKIs that block VEGFR signaling also inhibit the PDGFRs. The impact of these TKIs on the desmoplastic stroma warrants further study as the PDGFRs are often highly expressed in CAFs.
TUMOR-ASSOCIATED MACROPHAGES
Macrophages are normally present in the peritoneal cavity of healthy women and are abundant in ascites of ovarian cancer patients[45]. Tumor-associated macrophages (TAMs) are the major immune component of the tumor stroma[46,47]. Macrophages exhibit polarized phenotypes in response to different microenvironmental cues. Macrophages that are stimulated with microbial agents and interferon-γ exhibit an immunostimulatory M1 phenotype. In contrast, TAMs exhibit an immunosuppressive M2 macrophage phenotype[46,47]. Polarization of macrophages towards an M2 phenotype is induced by stimulation with various cytokines such as IL-6, IL-10 and leukemia inhibitory factor (LIF) that are present at elevated levels in ascites of ovarian cancer patients[48,49]. Chemokine (C-C motif) ligand 2 (CCL2) and TGF-β2 are also expressed in ovarian cancer cells and in CAFs, and these ligands have been recently shown to induce normal peritoneal macrophages to acquire an M2 phenotype[50]. CCL2 is also a key chemotactic factor that is responsible for macrophage infiltration into tumors[47].
TAMS are strongly associated with poor outcomes in cancer patients[46]. A principal mechanism by which TAMs promote tumor progression is by suppressing adaptive immunity. The M2 macrophage phenotype is characterized by high expression of immunosuppressive cytokines and chemokines such as CCL17, CCL18, CCL22, IL-10 and TGF-β1[47]. IL-10 and TGF-β1 inhibit T cell proliferation and dendritic cell maturation[47]. CCL18 induces naïve T cell anergy and has been identified to be the most abundant chemokine present in ovarian cancer patient ascites[51]. CCL17 and CCL22 promote recruitment of T regulatory cells (Treg) cells[52,53]. Treg cells suppress activity of effector T cells and have been found to promote ovarian tumor growth and to be predictive of poor survival in ovarian cancer patients[52]. In addition to expressing factors that suppress adaptive immunity, TAMs express MMPs, VEGF-A and other growth factors that stimulate metastasis and angiogenesis[46,47]. Depletion of peritoneal macrophages has been found to inhibit ascites and peritoneal spread of ovarian cancer in xenograft models[54].
The recruitment of macrophages and their polarization towards a tumor-promoting M2 phenotype represent two candidate focal points for therapeutic intervention (Figure 1). Several approaches have been identified that “re-educate” TAMs towards a more tumoricidal M1 phenotype. Inhibition of the colony stimulating factor-1 receptor has been found to inhibit M2 macrophage polarization and to block glioma progression in animal models[55]. Inhibition of nuclear factor κB signaling in TAMs also induced an M2-to-M1 switch, increased tumoricidal activity of macrophages and led to regression of ovarian tumor xenografts[56]. Activation of CD40, a member of the tumor necrosis factor receptor superfamily, induced tumoricidal activity of macrophages in mouse models of pancreatic adenocarcinoma[57]. The combination of agonistic CD40 mAb and gemcitabine chemotherapy has been found to be well-tolerated and to have anti-tumor activity in a phase I study of patients with advanced pancreatic adenocarcinoma[58]. Zoledronic acid is clinically used to prevent bone fractures and also impairs M2 polarization of macrophages[59]. CCL2 is an attractive target because of its ability to stimulate monocyte chemotaxis as well as M2 polarization. Neutralization of CCL2 induced regression of prostate cancer xenografts[60]. A mAb to CCL2 has recently undergone clinical evaluation[61]. Bindarit, an anti-inflammatory compound that inhibits CCL2 synthesis, has been found to inhibit growth of breast and prostate tumor xenografts[62]. Trabectedin is an alkaloid that binds the minor groove of DNA and disrupts the cell cycle[63]. In a phase III study of patients with recurrent ovarian cancer, the combination of Trabectedin and pegylated liposomal doxorubicin (PLD) was found to increase PFS by 1.5 mo as compared to PLD alone[64]. Trabectedin also inhibits production of CCL2 and IL-6 and inhibits the differentiation of monocytes into macrophages[65]. Germano et al[66] recently demonstrated the selective toxicity of Trabectedin for macrophages in xenograft models of ovarian cancer and several other solid tumors. In another recent study, Cieslewicz et al[67] identified a peptide (M2pep) that selectively binds to M2 macrophages. Administration of a fusion peptide comprising M2pep and a proapoptotic moiety improved survival rates of xenograft-bearing mice[67], raising the possibility that the M2pep peptide could be used as a vehicle for delivering cytotoxic agents to TAMs.
CONCLUSION
Over the past decade, a wealth of insight has been gained into the biology of ovarian cancer, the fertile nature of the peritoneal cavity for carcinomatosis, and the complex networks of receptor/ligand-mediated interactions between tumor cells and stromal cells. Several of the key receptors and ligands serve as molecular targets against which new-generation therapeutic agents have been developed and evaluated. Although several studies have yielded promising results, the efficacy of most stromal-targeting drugs as single agents seems limited. Several challenges remain such as identifying the most effective combinations of these drugs with conventional chemotherapy or with other targeted therapies, minimizing toxicity, and determining the appropriate clinical setting for their use.
ACKNOWLEDGMENTS
The authors apologize for the inability to cite all contributing primary literature due to space constraints.
Footnotes
P- Reviewers: Cavallaro U, Sherbini MAHE S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
Supported by Cancer and Prevention Research Institute of Texas grant, NO. RP120390 (HN); and United States National Institutes of Health grant, NO. CA141078 (HN)
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502–516. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 3.Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9:167–181. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- 4.Garber K. PARP inhibitors bounce back. Nat Rev Drug Discov. 2013;12:725–727. doi: 10.1038/nrd4147. [DOI] [PubMed] [Google Scholar]
- 5.Spannuth WA, Sood AK, Coleman RL. Angiogenesis as a strategic target for ovarian cancer therapy. Nat Clin Pract Oncol. 2008;5:194–204. doi: 10.1038/ncponc1051. [DOI] [PubMed] [Google Scholar]
- 6.Burger RA. Overview of anti-angiogenic agents in development for ovarian cancer. Gynecol Oncol. 2011;121:230–238. doi: 10.1016/j.ygyno.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 7.Hollingsworth HC, Kohn EC, Steinberg SM, Rothenberg ML, Merino MJ. Tumor angiogenesis in advanced stage ovarian carcinoma. Am J Pathol. 1995;147:33–41. [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez AA, Krigman HR, Whitaker RS, Dodge RK, Rodriguez GC. The prognostic significance of angiogenesis in epithelial ovarian carcinoma. Clin Cancer Res. 1999;5:587–591. [PubMed] [Google Scholar]
- 9.Stone PJ, Goodheart MJ, Rose SL, Smith BJ, DeYoung BR, Buller RE. The influence of microvessel density on ovarian carcinogenesis. Gynecol Oncol. 2003;90:566–571. doi: 10.1016/s0090-8258(03)00367-6. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 11.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 12.Byrne AT, Ross L, Holash J, Nakanishi M, Hu L, Hofmann JI, Yancopoulos GD, Jaffe RB. Vascular endothelial growth factor-trap decreases tumor burden, inhibits ascites, and causes dramatic vascular remodeling in an ovarian cancer model. Clin Cancer Res. 2003;9:5721–5728. [PubMed] [Google Scholar]
- 13.Cannistra SA, Matulonis UA, Penson RT, Hambleton J, Dupont J, Mackey H, Douglas J, Burger RA, Armstrong D, Wenham R, et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol. 2007;25:5180–5186. doi: 10.1200/JCO.2007.12.0782. [DOI] [PubMed] [Google Scholar]
- 14.Burger RA, Sill MW, Monk BJ, Greer BE, Sorosky JI. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a Gynecologic Oncology Group Study. J Clin Oncol. 2007;25:5165–5171. doi: 10.1200/JCO.2007.11.5345. [DOI] [PubMed] [Google Scholar]
- 15.Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, Mannel RS, Homesley HD, Fowler J, Greer BE, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365:2473–2483. doi: 10.1056/NEJMoa1104390. [DOI] [PubMed] [Google Scholar]
- 16.Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, Carey MS, Beale P, Cervantes A, Kurzeder C, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365:2484–2496. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- 17.Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, Sorio R, Vergote IB, Witteveen P, Bamias A, et al. AURELIA: A randomized phase III trial evaluating bevacizumab (BEV) plus chemotherapy (CT) for platinum (PT)-resistant recurrent ovarian cancer (OC) J Clin Oncol. 2012;30(suppl; abstr):LBA5002. doi: 10.1200/JCO.2013.51.4489. [DOI] [PubMed] [Google Scholar]
- 18.Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, Sovak MA, Yi J, Nycum LR. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30:2039–2045. doi: 10.1200/JCO.2012.42.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tew WP, Colombo N, Ray-Coquard I, Del Campo JM, Oza A, Pereira D, Mammoliti S, Matei D, Scambia G, Tonkin K, et al. Intravenous aflibercept in patients with platinum-resistant, advanced ovarian cancer: results of a randomized, double-blind, phase 2, parallel-arm study. Cancer. 2014;120:335–343. doi: 10.1002/cncr.28406. [DOI] [PubMed] [Google Scholar]
- 20.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 21.Matei D, Sill MW, Lankes HA, DeGeest K, Bristow RE, Mutch D, Yamada SD, Cohn D, Calvert V, Farley J, et al. Activity of sorafenib in recurrent ovarian cancer and primary peritoneal carcinomatosis: a gynecologic oncology group trial. J Clin Oncol. 2011;29:69–75. doi: 10.1200/JCO.2009.26.7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson DS, Dudley BS, Bismayer JA, Gian VG, Merritt WM, Whorf RC, Burris HA, Hainsworth JD. Paclitaxel/carboplatin with or without sorafenib in the first-line treatment of patients with stage III/IV epithelial ovarian cancer: A randomized phase II study of the Sarah Cannon Research Institute. J Clin Oncol. 2013;31:5513. doi: 10.1002/cam4.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biagi JJ, Oza AM, Chalchal HI, Grimshaw R, Ellard SL, Lee U, Hirte H, Sederias J, Ivy SP, Eisenhauer EA. A phase II study of sunitinib in patients with recurrent epithelial ovarian and primary peritoneal carcinoma: an NCIC Clinical Trials Group Study. Ann Oncol. 2011;22:335–340. doi: 10.1093/annonc/mdq357. [DOI] [PubMed] [Google Scholar]
- 24.Campos SM, Penson RT, Matulonis U, Horowitz NS, Whalen C, Pereira L, Tyburski K, Roche M, Szymonifka J, Berlin S. A phase II trial of Sunitinib malate in recurrent and refractory ovarian, fallopian tube and peritoneal carcinoma. Gynecol Oncol. 2013;128:215–220. doi: 10.1016/j.ygyno.2012.07.126. [DOI] [PubMed] [Google Scholar]
- 25.Ledermann JA, Perren TJ, Raja FA, Embleton A, Rustin GJS, Jayson G, Kaye SB, Swart AM, Vaughan M, Hirte H. Randomised double-blind phase III trial of cediranib (AZD 2171) in relapsed platinum sensitive ovarian cancer: Results of the ICON6 trial. The European Cancer Congress. 2013:Abstract. [Google Scholar]
- 26.Du Bois A, Floquet A, Kim JW, Rau J, Del Campo JM, Friedlander M, Pignata S, Fujiwara K, Vergote I, Colombo N, Mirza MR, Monk BJ, Wimberger P, Ray-Coquard I, Zang R, Diaz-Padilla I, Baumann KH, Kim JH, Harter P, on behalf of an Intergroup consortium. Randomized, double-blind, phase III trial of pazopanib versus placebo in women who have not progressed after first-line chemotherapy for advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (AEOC): Results of an international intergroup trial (AGO-OVAR16) J Clin Oncol. 2013;31:LBA5503. [Google Scholar]
- 27.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 28.Marsh T, Pietras K, McAllister SS. Fibroblasts as architects of cancer pathogenesis. Biochim Biophys Acta. 2013;1832:1070–1078. doi: 10.1016/j.bbadis.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13:4279–4295. doi: 10.1091/mbc.E02-02-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeon ES, Moon HJ, Lee MJ, Song HY, Kim YM, Cho M, Suh DS, Yoon MS, Chang CL, Jung JS, et al. Cancer-derived lysophosphatidic acid stimulates differentiation of human mesenchymal stem cells to myofibroblast-like cells. Stem Cells. 2008;26:789–797. doi: 10.1634/stemcells.2007-0742. [DOI] [PubMed] [Google Scholar]
- 31.Ko SY, Barengo N, Ladanyi A, Lee JS, Marini F, Lengyel E, Naora H. HOXA9 promotes ovarian cancer growth by stimulating cancer-associated fibroblasts. J Clin Invest. 2012;122:3603–3617. doi: 10.1172/JCI62229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, Alison MR, Wright NA. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 33.Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petersen OW, Nielsen HL, Gudjonsson T, Villadsen R, Rank F, Niebuhr E, Bissell MJ, Rønnov-Jessen L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162:391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, Traficante N, Fereday S, Hung JA, Chiew YE, Haviv I, Australian Ovarian Cancer Study Group, Gertig D, DeFazio A, Bowtell DD. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Tang H, Cai J, Zhang T, Guo J, Feng D, Wang Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011;303:47–55. doi: 10.1016/j.canlet.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 37.McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, Cabrera L, Keller E, McCauley L, Cho KR, et al. Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest. 2011;121:3206–3219. doi: 10.1172/JCI45273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jäger D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26:44–48. doi: 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 39.Wen Y, Wang CT, Ma TT, Li ZY, Zhou LN, Mu B, Leng F, Shi HS, Li YO, Wei YQ. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010;101:2325–2332. doi: 10.1111/j.1349-7006.2010.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J Natl Cancer Inst. 2012;104:1320–1334. doi: 10.1093/jnci/djs336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonafoux D, Lee WC. Strategies for TGF-beta modulation: a review of recent patents. Expert Opin Ther Pat. 2009;19:1759–1769. doi: 10.1517/13543770903397400. [DOI] [PubMed] [Google Scholar]
- 42.Connolly EC, Freimuth J, Akhurst RJ. Complexities of TGF-β targeted cancer therapy. Int J Biol Sci. 2012;8:964–978. doi: 10.7150/ijbs.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai J, Tang H, Xu L, Wang X, Yang C, Ruan S, Guo J, Hu S, Wang Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis. 2012;33:20–29. doi: 10.1093/carcin/bgr230. [DOI] [PubMed] [Google Scholar]
- 44.Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haskill S, Becker S, Fowler W, Walton L. Mononuclear-cell infiltration in ovarian cancer. I. Inflammatory-cell infiltrates from tumour and ascites material. Br J Cancer. 1982;45:728–736. doi: 10.1038/bjc.1982.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 47.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, Preisser L, Anegon I, Catala L, Ifrah N, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319–4330. doi: 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- 49.Matte I, Lane D, Laplante C, Rancourt C, Piché A. Profiling of cytokines in human epithelial ovarian cancer ascites. Am J Cancer Res. 2012;2:566–580. [PMC free article] [PubMed] [Google Scholar]
- 50.Ko SY, Ladanyi A, Lengyel E, Naora H. Expression of the homeobox gene HOXA9 in ovarian cancer induces peritoneal macrophages to acquire an M2 tumor-promoting phenotype. Am J Pathol. 2014;184:271–281. doi: 10.1016/j.ajpath.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schutyser E, Struyf S, Proost P, Opdenakker G, Laureys G, Verhasselt B, Peperstraete L, Van de Putte I, Saccani A, Allavena P, et al. Identification of biologically active chemokine isoforms from ascitic fluid and elevated levels of CCL18/pulmonary and activation-regulated chemokine in ovarian carcinoma. J Biol Chem. 2002;277:24584–24593. doi: 10.1074/jbc.M112275200. [DOI] [PubMed] [Google Scholar]
- 52.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 53.Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, Fujii H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. 2008;122:2286–2293. doi: 10.1002/ijc.23392. [DOI] [PubMed] [Google Scholar]
- 54.Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N, McFarland-Mancini MM, Drew AF. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res. 2007;67:5708–5716. doi: 10.1158/0008-5472.CAN-06-4375. [DOI] [PubMed] [Google Scholar]
- 55.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, Troxel AB, Sun W, Teitelbaum UR, Vonderheide RH, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–6295. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsagozis P, Eriksson F, Pisa P. Zoledronic acid modulates antitumoral responses of prostate cancer-tumor associated macrophages. Cancer Immunol Immunother. 2008;57:1451–1459. doi: 10.1007/s00262-008-0482-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, Wojno K, Snyder LA, Yan L, Pienta KJ. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67:9417–9424. doi: 10.1158/0008-5472.CAN-07-1286. [DOI] [PubMed] [Google Scholar]
- 61.Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31:760–768. doi: 10.1007/s10637-012-9869-8. [DOI] [PubMed] [Google Scholar]
- 62.Zollo M, Di Dato V, Spano D, De Martino D, Liguori L, Marino N, Vastolo V, Navas L, Garrone B, Mangano G, et al. Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin Exp Metastasis. 2012;29:585–601. doi: 10.1007/s10585-012-9473-5. [DOI] [PubMed] [Google Scholar]
- 63.D’Incalci M, Galmarini CM. A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther. 2010;9:2157–2163. doi: 10.1158/1535-7163.MCT-10-0263. [DOI] [PubMed] [Google Scholar]
- 64.Monk BJ, Herzog TJ, Kaye SB, Krasner CN, Vermorken JB, Muggia FM, Pujade-Lauraine E, Lisyanskaya AS, Makhson AN, Rolski J, et al. Trabectedin plus pegylated liposomal Doxorubicin in recurrent ovarian cancer. J Clin Oncol. 2010;28:3107–3114. doi: 10.1200/JCO.2009.25.4037. [DOI] [PubMed] [Google Scholar]
- 65.Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, Olimpio CO, Bonardi C, Garbi A, Lissoni A, et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005;65:2964–2971. doi: 10.1158/0008-5472.CAN-04-4037. [DOI] [PubMed] [Google Scholar]
- 66.Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 67.Cieslewicz M, Tang J, Yu JL, Cao H, Zavaljevski M, Motoyama K, Lieber A, Raines EW, Pun SH. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc Natl Acad Sci U S A. 2013;110:15919–15924. doi: 10.1073/pnas.1312197110. [DOI] [PMC free article] [PubMed] [Google Scholar]