

Abstract
During synaptic development, presynaptic differentiation occurs as an intrinsic property of axons to form specialized areas of plasma membrane [active zones (AZs)] that regulate exocytosis and endocytosis of synaptic vesicles. Genetic and biochemical studies in vertebrate and invertebrate model systems have identified a number of proteins involved in AZ assembly. However, elucidating the molecular events of AZ assembly in a spatiotemporal manner remains a challenge. Syd-1 (synapse defective-1) and Liprin-α have been identified as two master organizers of AZ assembly. Genetic and imaging analyses in invertebrates show that Syd-1 works upstream of Liprin-α in synaptic assembly through undefined mechanisms. To understand molecular pathways downstream of Liprin-α, we performed a proteomic screen of Liprin-α-interacting proteins in Drosophila brains. We identify Drosophila protein phosphatase 2A (PP2A) regulatory subunit B′ [Wrd (Well Rounded)] as a Liprin-α-interacting protein, and we demonstrate that it mediates the interaction of Liprin-α with PP2A holoenzyme and the Liprin-α-dependent synaptic localization of PP2A. Interestingly, loss of function in syd-1, liprin-α, or wrd shares a common defect in which a portion of synaptic vesicles, dense-core vesicles, and presynaptic cytomatrix proteins ectopically accumulate at the distal, but not proximal, region of motoneuron axons. Strong genetic data show that a linear syd-1/liprin-α/wrd pathway in the motoneuron antagonizes glycogen synthase kinase-3β kinase activity to prevent the ectopic accumulation of synaptic materials. Furthermore, we provide data suggesting that the syd-1/liprin-α/wrd pathway stabilizes AZ specification at the nerve terminal and that such a novel function is independent of the roles of syd-1/liprin-α in regulating the morphology of the T-bar structural protein BRP (Bruchpilot).
Keywords: active zone assembly, Drosophila, Liprin-alpha, PP2A, presynaptic differentiation, Syd-1
Introduction
During presynaptic development, small synaptic vesicle (SV) precursors, dense-core vesicles (DCVs), and synaptic cytomatrix proteins are generated in the soma, transported along the axon, and eventually incorporated into the nerve terminal (Jin and Garner, 2008). Within the nerve terminal, active zones (AZs) are specialized areas of plasma membrane containing a group of evolutionarily conserved proteins, including ELKS (glutamine, leucine, lysine, and serine-rich protein)[also called CAST (cytomatrix at the active zone-associated structural protein), Dorsophila homologue is BRP (Bruchpilot)], Munc13 (mammalian uncoordinated homology 13), RIM (Rab3-interacting molecule), Syd-1 (synapse defective-1), and Liprin-α, in which the releasable pool of vesicles dock and are released on stimulation (Südhof, 2012). Despite intensive studies of the proteins localized at the presynaptic density, the assembly and maintenance of AZs remains enigmatic (Owald and Sigrist, 2009). Studies conducted in invertebrate model organisms suggested that Syd-1, a putative RhoGAP, and Liprin-α are two master organizers of presynaptic differentiation (Zhen and Jin, 1999; Hallam et al., 2002; Kaufmann et al., 2002; Owald et al., 2010). Genetic analyses in Caenorhabditis elegans demonstrated that Syd-1 works upstream of Liprin-α in synaptic assembly (Dai et al., 2006; Patel et al., 2006). Studies in Drosophila further confirmed this hierarchy by showing that Syd-1 regulates and retains proper localization of Liprin-α at the AZ (Owald et al., 2010, 2012). However, studies also found that Syd-1 regulates Liprin-α-independent processes, such as retention of Neurexin at the presynaptic side and glutamate receptor incorporation at the postsynaptic side(Owald et al., 2010, 2012). The morphology of the AZ is distinctly different in liprin-α and syd-1 mutants (Kaufmann et al., 2002; Owald et al., 2010). Therefore, it is unclear how Syd-1- and Liprin-α-mediated signaling collaborate to achieve the complex regulation of presynaptic differentiation. Identifying novel Liprin-α-interacting proteins at the synapse holds the key to delineating the regulatory network mediated by these two genes.
We identified protein phosphatase 2A (PP2A) as one prominent Liprin-α-interacting protein complex through an in vivo tandem affinity purification (TAP) approach (Rigaut et al., 1999; Tian et al., 2013). PP2A is an abundant heterotrimeric serine/threonine phosphatase that regulates a broad range of cellular processes. PP2A is highly enriched in neurons and is implicated in Tau-mediated neurodegeneration (Rudrabhatla and Pant, 2011; Seshacharyulu et al., 2013), regulation of long-term potentiation (Belmeguenai and Hansel, 2005; Pi and Lisman, 2008), and presynaptic and postsynaptic apposition (Viquez et al., 2009). The diverse functions of PP2A are attributed primarily to its many interchangeable regulatory subunits (B, B′, B″, or B‴), each showing specific spatial and temporal expression patterns (Sents et al., 2013). The Liprin-α-interacting PP2A holoenzyme that we identified in the fly brain contains the B′ regulatory subunit [also called Wrd (Well Rounded) in fly]. Wrd is highly expressed in synapses and regulates synaptic terminal growth at the Drosophila neuromuscular junction (NMJ) (Viquez et al., 2006). Interestingly, the Liprin-α–Wrd physical interaction may be evolutionarily conserved because PP2A B56γ, the human homolog of Wrd, can bind Liprin-α1 in HEK 293 cells (Arroyo et al., 2008). However, the function of the Liprin-α–Wrd/PP2A B56γ interaction in the nervous system is unexplored.
Here we show that Syd-1, Liprin-α, and Wrd work in a linear pathway to restrain the localization of vesicles and presynaptic cytomatrix proteins at the nerve terminal. Disruption of such a pathway results in ectopic accumulation of SVs and presynaptic proteins at the distal, but not proximal, end of axons.
Materials and Methods
TAP procedure.
To purify the Liprin-α-associated or Wrd-associated protein complex, we collected ∼30,000 adult fly heads expressing Liprin-α–TAP or TAP–Wrd in neurons (BG380–Gal4;UAS–liprin-α–CTAP or BG380–Gal4;UAS–TAP–Wrd/TM6B,Sb), respectively. TAP purification was performed as described previously (Tian et al., 2011, 2013). A portion of the final elution was analyzed by an SDS-PAGE gel (4–15%), followed by a sensitive Coomassie blue G-250 staining (Candiano et al., 2004). The rest of the purified protein mix was precipitated, digested with trypsin, and then subjected to liquid chromatography–tandem mass spectrometry (LC-MS/MS). The searching program MASCOT (Matrix Science) was used to identify the protein species.
Fly stocks.
Flies were maintained at 25°C on normal food. The following strains were used in this study: w1118, BG380–Gal4, D42–Gal4, elav–Gal4, OK6–Gal4, UAS–FYVE (Fab 1, YOTB, Vac 1, and EEA1 homology domain)–GFP, UAS–LAMP (lysosomal-associated membrane protein)–GFP, UAS–Mito–GFP, UAS–Syt (Synaptotagmin)–GFP, sggG0055, UAS–SggA81T (Bloomington Stock Center); liprin-α mutants (liprin-αEPexR60, liprin-αF3ex15) and UAS–liprin-α–GFP transgenes on the X and second chromosome (gifts from David Van Vactor, Harvard Medical School, Boston, MA; Kaufmann et al., 2002); wrd mutants (wrd104, wrd189; Viquez et al., 2006); syd-1 mutants (syd-1ex1.2, syd-1ex3.4; Owald et al., 2010); and UAS–atrial natriuretic factor (ANF)–GFP (Shakiryanova et al., 2005). The mutant genotypes for this study are the following: syd-1, syd-1ex1.2/syd-1ex3.4; liprin-α, liprin-αEPexR60/liprin-αF3ex15; and wrd, wrd104/wrd189, unless otherwise specified. We randomly chose the sex of flies for this study.
Transgenic constructs.
A full-length liprin-α cDNA [Drosophila Genomics Resource Center (DGRC) Clone LD33094] was obtained, and the coding sequence was cloned to a pUAST–CTAP vector (a derivative from a TAP construct provided by EUROSCARF; Rigaut et al., 1999; Tian et al., 2013) to generate a UAS–liprin-α–TAP construct. A full-length wrd cDNA (DGRC Clone LD47425) was obtained, and the coding sequence was subcloned into pUAST–EGFP (Parker et al., 2001), pUAST–mCherry (a gift from Richard Daniels, University of Wisconsin, Madison, WI, unpublished), or pUAST–TAP (Tian et al., 2013) to generate UAS–EGFP–wrd, UAS–mCherry–wrd, or UAS–TAP–Wrd constructs, respectively. To generate a full-length syd-1 cDNA, we cloned two overlapping cDNA fragments spanning the entire syd-1 ORF and then sequentially ligated the two fragments together. In brief, total RNA was extracted from adult Drosophila heads, and reverse transcription reactions were performed using gene-specific primers to obtain two cDNA fragments that were ligated together using the unique internal cutting site NheI: 5′ end to NheI (2437), NheI (2437) to 3′ end. NotI and XbaI sites were introduced onto the 5′ end and 3′ end, respectively, to facilitate subcloning into the pUAST–EGFP vector to generate a UAS–GFP–Syd-1 construct. All transgenic fly lines were generated by BestGene.
TAP pull-downs and Western blots.
For TAP pull-downs, ∼100 larval brains for each sample were collected by manual dissection. Larval brains were then homogenized in 0.5 ml of lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 2 mm NaF, 0.5% NP-40, and protease inhibitors). Clean lysate was then incubated with IgG Sepharose beads (25 μl; Pharmacia) for 1 h. The beads were then washed twice with the lysis buffer and twice with PBS before elution. The following primary antibodies were used: peroxidase–anti-peroxidase complex (1:4000, P2026; Sigma) for detection of TAP-tagged Liprin-α; E7 (1:1000; Developmental Studies Hybridoma Bank) for β-tubulin; a polyclonal rabbit anti-Wrd antibody (1:1000; a gift from Aaron DiAntonio, Washington University School of Medicine, St Louis, MO; Viquez et al., 2006); and rat anti-PP2A-C subunit (1:500) and rat anti-PP2A-A subunit (1:500; Shiomi et al., 1994). All secondary antibodies were used at 1:10,000.
Immunocytochemistry.
Wandering third-instar larvae were dissected in ice-cold PBS and fixed in Bouin's fixative for 6 min unless otherwise specified. Fixed larvae were washed in PBT (PBS plus 0.1% Triton X-100) and then blocked in 5% normal goat serum (NGS) in PBT for 15 min. Primary antibody incubation was performed in 5% NGS in PBT ranging from 1 h to overnight. The following primary antibodies were used: rabbit anti-Drosophila vesicular glutamate transporter (DVGlut) at 1:5000 (Daniels et al., 2004); mouse anti-GFP and rabbit anti-GFP antibody (A11122; Invitrogen) at 1:1000; anti-ATP5α at 1:1000 (MS507; MitoScience); cyanine 5 (Cy5)-conjugated goat anti-HRP at 1:1000 (Jackson ImmunoResearch); and a series of mouse monoclonal antibodies (from Developmental Studies Hybridoma Bank), including anti-BRP (NC82, 1:200), anti-fasciclin II (FasII; 1D4, 1:20), anti-Syt (3H2 2D7, 1:100), anti-Synapsin (3C11, 1:100), anti-Syntaxin (8C3, 1:100), anti-Kinesin (SUK 4, 1:100), and anti-Dynein (2C11–2, 1:100). The following secondary antibodies were used: DyLight 549-conjugated goat anti-mouse IgG; and Alexa Fluor 488-conjugated anti-mouse IgG at 1:1000 (Invitrogen). A modified protocol was used for anti-Wrd antibody in which larvae were fixed in 4% formaldehyde and incubated with primary antibody for 30 min and then secondary antibody for 2 h. PBS rather than PBT was used for incubation in the secondary antibodies, as well as the final washes. For direct visualization of GFP- or mCherry-tagged transgenic proteins or markers without antibody staining, samples were briefly fixed in 4% paraformaldehyde (PFA) and then stained with Cy3 anti-HRP for 10 min or NC82 for 1 h in detergent-free solutions to preserve GFP or mCherry signals.
Confocal imaging and data analysis.
Samples were imaged on a Nikon C1 confocal microscope. For all the NMJ images, the gain was chosen as the maximum gain that did not saturate the signal for each sample. A complete Z-stack was acquired for each NMJ and rendered as a maximum projection. Quantifications for DVGlut accumulation in distal axons described in Figures 4, 5, and 8 and Table 2 were performed at motoneuron axons innervating muscle 12 in segments A2–A4, although the SV accumulation phenotype is strikingly similar among all distal axons in liprin-α, syd-1, and wrd mutants. Distal axonal areas were selected based on anti-HRP immunoreactivity. Specified intensities within each selected axonal region were then measured using NIS-Elements software. Data were described as average mean intensity (total intensity divided by defined axonal or NMJ area). The gain was chosen as the maximum gain that did not saturate the signals and was kept constant for the same batch of experiments and quantification. For the larval ventral ganglion images, complete Z-stacks were acquired for the entire ventral ganglion and rendered as a maximum projection. Statistical analysis was performed with Origin (Origin Lab) using one-way ANOVA for comparison of samples within an experimental group. All histograms and measurements are shown as mean ± SEM.
Figure 4.
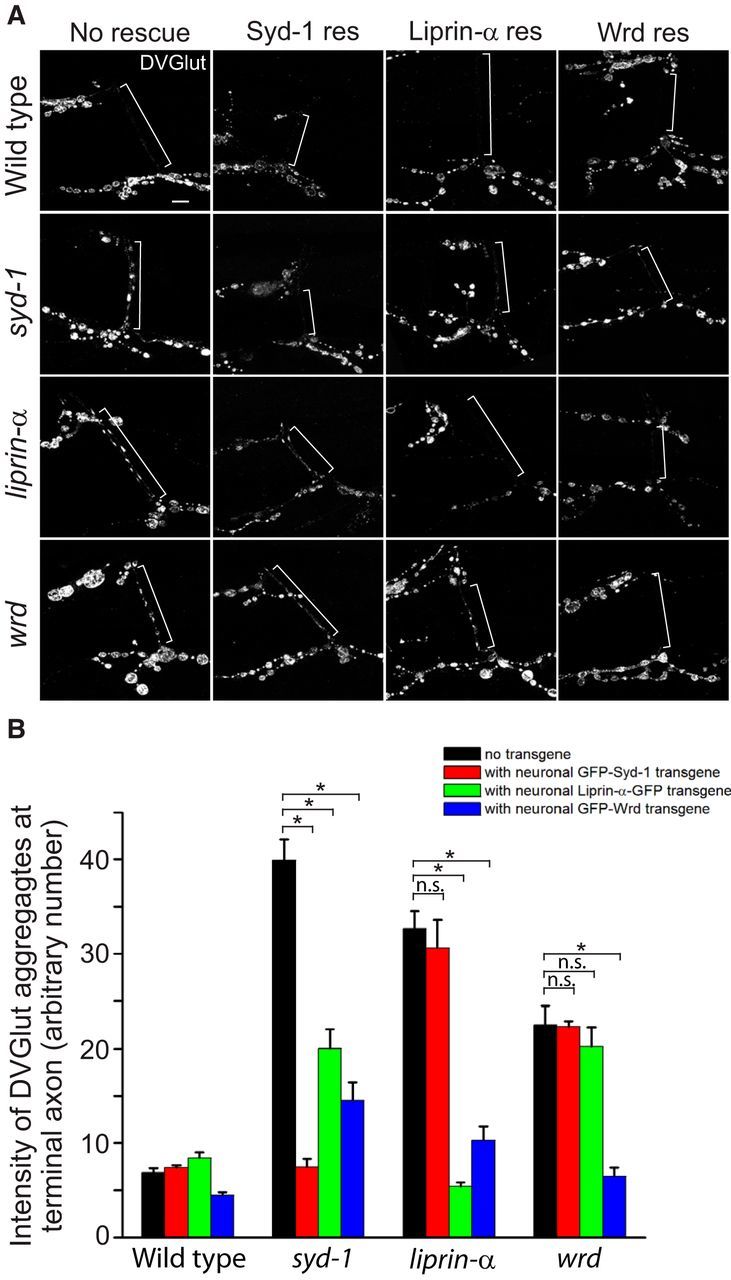
syd-1/liprin-α/wrd function in a linear pathway to prevent ectopic distribution of SV proteins at the distal axon. A, Representative confocal images of the distal axons of muscle 12/13 NMJs costained with anti-DVGlut and anti-HRP (not depicted, used as a reference for the distal axon) antibodies in third-instar larvae of wild-type, syd-1, liprin-α, or wrd mutants, as well as each of the above genotypes plus neuronal expression of each of three individual rescuing transgenes (D42–Gal4/UAS–GFP–Syd-1, D42–Gal4/UAS–liprin-α–GFP, D42–Gal4/UAS–GFP–Wrd; for full description of genotypes, see Materials and Methods). White brackets show terminal axons crossing muscle 13 to innervate muscle 12. B, Quantification of the distal axon vesicle clustering (measured by average intensity of DVGlut at muscle 12/13 terminal axons) in third-instar larvae of flies presented in A. n.s., Not significant; res, rescue. *p < 0.001. Error bars denote SEM.
Figure 5.
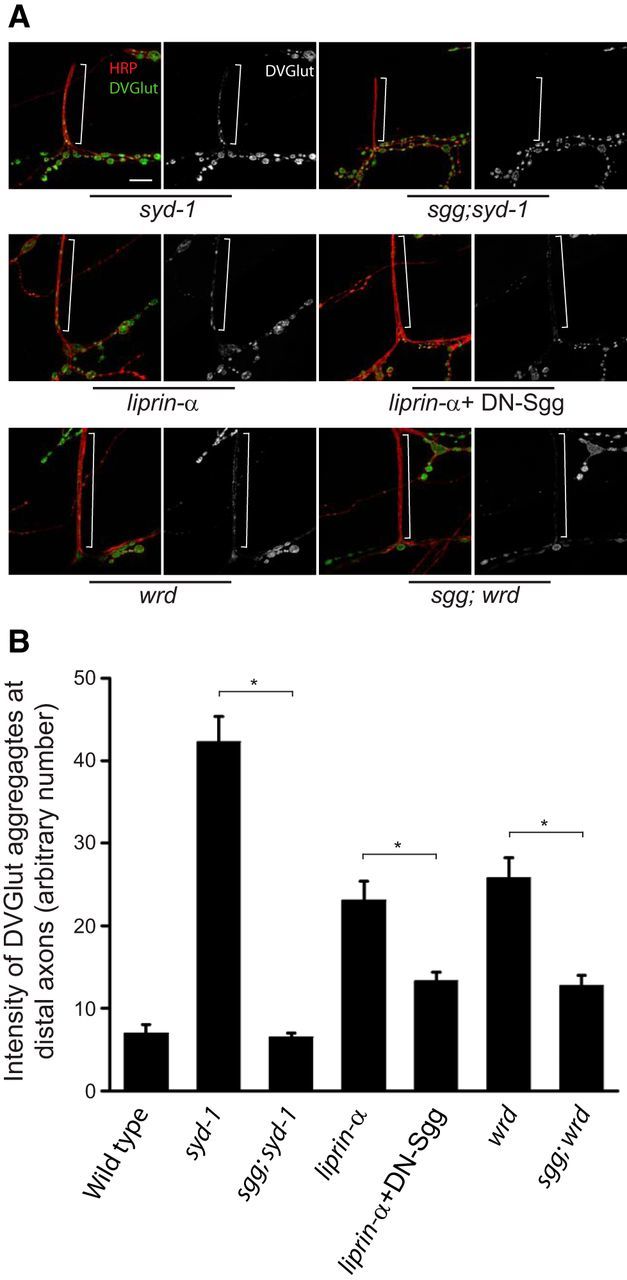
Loss of GSK-3β function suppresses the syd-1, liprin-α, and wrd mutant distal axon phenotype. A, Representative confocal images of the distal axons of muscle 12/13 NMJs costained with DVGlut and HRP in third-instar larvae of syd-1 (syd-1ex1.2/syd-1ex3.4), sgg;syd-1 (sggG0055;syd-1ex1.2/syd-1ex3.4), liprin-α (liprin-αEPexR60/liprin-αF3ex15), liprin-α + DN–sgg (BG380–Gal4;liprin-αEPexR60/liprin-αF3ex15,UAS–sggA81T), wrd (wrd104/wrd104), and sggG0055;wrd (sggG0055;wrd104/wrd104) mutants. The reason for using the dominant-negative UAS–Sgg transgene instead of the sgg mutant with liprin-α mutant is that the second-chromosome-balanced sgg mutants are lethal. White brackets show terminal axons crossing muscle 13 to innervate muscle 12. Scale bar, 10 μm. B, Quantification of the distal axon vesicle accumulation (measured by average intensity of DVGlut at muscle 12/13 terminal axons) in third-instar larvae of wild-type and flies presented in A. n = 6, 7, 12, 12, 16, 11, and 8, respectively. *p < 0.001. Error bars denote SEM.
Figure 8.

The presence of BRP in distal axons is not required for the ectopic vesicle accumulation. A, Representative confocal images of muscle 4 NMJs stained with anti-BRP antibody (NC82) in third-instar larvae of wild-type, wrd (wrd104/wrd189), syd-1 (syd-1ex1.2/syd-1ex3.4), liprin-α (liprin-α EPexR60/liprin-α F3ex15), syd-1 Wrd Res (syd-1ex3.4, D42–Gal4/syd-1ex1.2, UAS–GFP–Wrd) syd-1 Liprin-α Res (syd-1ex3.4, D42–Gal4/syd-1ex1.2, UAS–liprin-α–GFP), or liprin-α Wrd Res (liprin-αR60/liprin-αF3ex15;D42–Gal4/UAS–GFP–Wrd). Arrows indicate elongated and abnormally shaped BRP puncta in syd-1 and syd-1-rescuing NMJs. Arrowheads indicated enlarged BRP puncta in liprin-α and liprin-α-rescuing NMJs. Scale bar, 5 μm. B, Representative confocal images of the distal axons of muscle 12/13 NMJs costained with DVGlut (green), BRP (red), and HRP (blue) in third-instar larvae of syd-1 (BG380; syd-1ex1.2/syd-1ex3.4), syd-1 + brp RNAi (BG380;syd-1ex1.2, UAS–brp–RNAi/syd-1ex3.4), liprin-α (BG380;liprin-αEPexR60/liprin-αF3ex15), liprin-α + brp RNAi (BG380;liprin-αEPexR60/liprin-αF3ex15;UAS–brp–RNAi/+), wrd (BG380;wrd104/wrd189), and wrd + brp RNAi (BG380;wrd189, UAS–brp–RNAi/ wrd104). White brackets show terminal axons crossing muscle 13 to innervate muscle 12. Scale bar, 20 μm. C, Quantification of the effect of BRP knockdown on distal axon accumulation of SVs. Data are presented as relative average mean intensity of DVGlut and BRP at muscle 12/13 distal axons, as well as BRP at muscle 12 NMJs, normalized to the corresponding mutants without brp knockdown. n = 13, 13, 10, 10, 10, and 10, respectively. *p < 0.001. n.s., Not significant. Error bars denote SEM.
Table 2.
Intensities of various markers at distal axons of wild-type, syd-1, liprin-α, or wrd mutant larvae
| Markers | Wild-type | syd-1 | liprin-α | wrd |
|---|---|---|---|---|
| Syt | 1 ± 0.08 | 4.98 ± 0.72** | 3.86 ± 0.47** | 2.37 ± 0.50* |
| BRP | 1 ± 0.42 | 26.82 ± 3.17** | 23.27 ± 2.32** | 10.37 ± 1.72** |
| FYVE–GFP | 1 ± 0.04 | 0.98 ± 0.06 | 0.80 ± 0.08 | 0.84 ± 0.06 |
| LAMP–GFP | 1 ± 0.15 | 0.81 ± 0.10 | 1.23 ± 0.11 | 0.84 ± 0.10 |
| AT5α (Mito) | 1 ± 0.17 | 0.99 ± 0.18 | 1.01 ± 0.09 | 0.93 ± 0.12 |
| Kinesin | 1 ± 0.09 | 0.82 ± 0.06 | 0.97 ± 0.03 | 0.87 ± 0.12 |
| Dynein | 1 ± 0.04 | 0.92 ± 0.03 | 0.92 ± 0.04 | 0.91 ± 0.07 |
Note that all quantifications are done at the distal axons of muscle 12/13. For each individual marker, intensities are normalized to wild-type levels. n = 6 for each data point.
*p < 0.05,
**p < 0.001 when compared with wild type. The remainder are not significantly different from wild type.
Stimulated emission depletion microscopy.
Larvae (UAS–liprin–GFP/+;elav–Gal4/UAS–mCherry–wrd) were fixed for 10 min with 4% PFA in 0.1 mm PBS. Primary antibodies were used at the following concentrations: mouse anti-GFP monoclonal antibody 3E6 (Invitrogen) at 1:500; and rabbit anti-dsRed (Clontech) at 1:500. Secondary antibodies for stimulated emission depletion (STED) were used at the following concentrations: goat anti-mouse Atto590 (ATTO-TEC) at 1:100; and goat anti-rabbit star635 (Abberior) at 1:100. For STED imaging, larvae were mounted in Mowiol (the group of Stefan Hell, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany). Images from fixed samples were taken from third-instar larval NMJs. Two-color STED images were recorded on a custom-built STED microscope that combines two pairs of excitation laser beams of 595 and 640 nm wavelength with one STED fiber laser beam at 775 nm (Göttfert et al., 2013). STED images were processed using a linear deconvolution function integrated into Imspector Software (Max Planck Innovation). The point spread function for deconvolution was generated by using a 2D Lorentz function with its FWHM fitted to the resolution estimate of each individual image. Images for figures were processed with NIH ImageJ software to remove background.
Electron microscopy.
Transmission electron microscopy (EM) images were obtained from dissected preparations of third-instar larvae (muscle 6/7 NMJs, segments A2/A3) following a standard protocol as described previously (Owald et al., 2010).
Live imaging.
A live imaging study was performed as described previously (Shakiryanova et al., 2005; Wong et al., 2012). Briefly, third-instar larvae were dissected in room temperature PBS and then maintained in calcium-free HL3 solution supplemented with 10 mm glutamate to desensitize postsynaptic receptors and minimize spontaneous muscle contraction while preserving vesicle motility. Images were acquired on a Nikon C1 epifluorescence upright microscope equipped with a Nikon 60× water-immersion objective and a CoolSNAP HQ2 CCD camera (Photometrics). For the simultaneous photobleaching and imaging (SPAIM) experiments, selected distal axons were first photobleached with repeated scans at maximal power to quench the florescence of preexisting vesicles. The axons were let to recover for ∼30 min while the 488 nm laser was used to continuously scan the area connecting distal axons with nerve terminals. At the end of the recovery, time-lapse images of the recovered distal axons were captured with NIS-Elements software at 500 ms intervals for a duration of 5 min. The movie presented is replayed at 6× speed.
Analysis of ANF–GFP release.
Wide-field imaging of the distal axon and type Ib boutons on muscle 4 of third-instar larvae was performed in glutamate-supplemented HL3 (in mm: 70 NaCl, 5 KCl, 1.5 CaCl2, 20 MgCl2, 10 NaHCO3, 5 trehalose, 115 sucrose, 10 sodium glutamate, and 5 sodium HEPES, pH 7.2) with an Olympus 60× water-immersion objective as described previously (Shakiryanova et al., 2005). Synaptic boutons were stimulated via intact segmental motor nerves with a suction electrode at 70 Hz for 1 min. Peptide secretion was measured as the loss of GFP fluorescence.
FM 1-43 dye labeling.
FM 1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] dye loading experiments were conducted by following the protocol described by Verstreken et al. (2008). Larvae were stimulated by applying a KCl/Ca2+ solution (in mm: 25 NaCl, 90 KCl, 2 CaCl2, 10 NaHCO3 HEPES, 30 sucrose, 5 trehalose, and 10 MgCl2, pH 7.2) containing 4 μm fixable FM 1-43 (F-35355; Invitrogen) for 30 min, washed for 10 min, and then fixed and stained for HRP.
Results
Liprin-α interacts with a specific subtype of PP2A through its B′ regulatory subunit Wrd
To identify Liprin-α-interacting proteins in neurons, we generated a UAS–liprin-α–TAP transgene that, when expressed in neurons, is able to completely rescue the AZ morphological defects caused by loss of liprin-α function (data not shown). The UAS–liprin-α–TAP transgene was then expressed in the nervous system, the Liprin-α-associated protein complexes were purified from Drosophila adult brains using a two-step TAP purification method, and the molecular identity of proteins were then determined by LC-MS/MS (Rigaut et al., 1999; Tian et al., 2013). The results are shown in Figure 1A as the separation of the protein complex in a SDS-PAGE gel and in Table 1 as the list of proteins identified by MS. These experiments identified the fly homologs of two known Liprin-α binding partners, BRP and RIM (Ohtsuka et al., 2002; Schoch et al., 2002; Ko et al., 2003). In addition, a specific subtype of PP2A that contains the B′ regulatory subunit Wrd, the structural subunit A (PP2A-A), and the catalytic subunit C [also called Microtubule Star (MTS)] was also identified with high scores (Table 1).
Figure 1.

Liprin-α interacts with the PP2A B′ regulatory subunit Wrd in Drosophila brain. A, The Liprin-α-associated complex was purified from adult fly brains through TAP and was analyzed by one-dimensional SDS-PAGE gel, followed by Coomassie blue G-250 staining. MS identifies BRP, Liprin-α, Wrd, PP2A-A, and PP2A-C (MTS), indicated by arrows. B, Larval brain lysate of wild-type (WT), wrd null mutant (wrd104) flies that express UAS–liprin-α–TAP transgene in neurons was subject to TAP pull-down (PD). Both the input and the pull-down complexes were analyzed by Western blots with the anti-TAP (peroxidase anti-peroxidase), the anti-Wrd (* marks the specific Wrd band), the anti-PP2A-A, or the anti-MTS antibody. C, The Wrd-associated proteins were purified from adult fly brains through TAP and analyzed by one-dimensional SDS-PAGE, followed by silver staining. MS identified Liprin-α, Wrd, PP2A-A, and PP2A-C (MTS), indicated by arrows. D, Single-layer STED images showing the association of transgenic mCherry–Wrd and Liprin-α–GFP proteins expressed at wild-type synaptic boutons (genotype: UAS–liprin–GFP/+;elav-Gal4/UAS–mCherry–wrd). Scale bars: orange, 120 nm; pink, 500 nm; white, 1 μm.
Table 1.
Proteins identified through TAP
| Name of protein | n peptides identified | Mascot score |
|---|---|---|
| TAP–Liprin-α | ||
| BRP | 76 | 4808 |
| PP2A-A | 36 | 4238 |
| PP2A-B′ (Wrd) | 37 | 3129 |
| PP2A-C (MTS) | 19 | 2762 |
| Rim | 5 | 71 |
| TAP–Wrd | ||
| Liprin-α | 59 | 3523 |
| PP2A-A | 28 | 1871 |
| PP2A-C (MTS) | 16 | 856 |
| Fat body protein 1 | 14 | 492 |
| α-Tubulin | 4 | 269 |
| HSP cognate 72 | 3 | 196 |
| CaMKII | 3 | 154 |
The specific interaction between Liprin-α and Wrd (B′), but not the other PP2A regulatory subunits, suggests that Wrd mediated the Liprin-α–PP2A interaction in neurons. To test this notion and to confirm the Liprin-α–Wrd interaction, we performed Liprin-α pull-down experiments using larval brains. In wild-type brains, Liprin-α pulled down Wrd and the other two PP2A subunits PP2A-A and MTS. However, in wrd null larval brains, Liprin-α could no longer pull down PP2A-A and MTS (Fig. 1B), suggesting that Wrd is required for the association between Liprin-α and the PP2A holoenzyme. To further confirm the physical interaction, we performed a reciprocal TAP proteomic experiment following a similar procedure as Liprin-α–TAP but expressing a UAS–TAP–Wrd transgene as the bait. Remarkably, endogenous Liprin-α was identified as the strongest binding partner of Wrd in the fly brains (Fig. 1C, Table 1). These data suggest that Wrd is a bona fide binding partner of Liprin-α.
To analyze the molecular organization of Liprin-α and Wrd at the fly NMJ, we generated a UAS–mCherry–Wrd transgene and coexpressed it with a UAS–liprin-α–GFP transgene (Fouquet et al., 2009) in Drosophila brains. STED microscopy of third-instar larval NMJs was performed to obtain improved optical resolution. As shown in single-layer scans in Figure 1D, the two fluorescently tagged proteins are substantially colocalized in puncta within the NMJ bouton. These data strongly suggest the presence of a Liprin-α–Wrd complex at the NMJ.
Liprin-α is required for normal synaptic localization of Wrd
To determine whether Wrd is a downstream effector of Liprin-α, we first tested whether Liprin-α is required for the synaptic localization of Wrd. In the wild-type larval CNS, the majority of Wrd proteins are enriched at the synapse-dense neuropil region (Viquez et al., 2006). However, when Liprin-α is absent, the preferential neuropil localization of Wrd is lost (Fig. 2A). This is not attributable to a decrease in Wrd protein level, because Wrd total protein levels in the CNS remain unchanged in liprin-α mutants when assessed by Western blot (Fig. 2B). Similarly, Liprin-α also regulates Wrd localization at the larval NMJs. In the wild-type animals, motoneuron-expressed mCherry–Wrd proteins show a modest but homogeneous distribution among all boutons of the NMJs (Fig. 2C1). This pattern is disrupted in liprin-α mutant NMJs, in which mCherry–Wrd localization is significantly reduced in proximal boutons and is enriched at the terminal boutons (Fig. 2C2). Moreover, coexpression of liprin-α–GFP with mCherry–Wrd in motoneurons is able to significantly boost the levels of mCherry–Wrd localization at the NMJ (Fig. 2, compare C1 with C3). Hence, Liprin-α is both necessary and sufficient to localize Wrd to the synaptic region.
Figure 2.
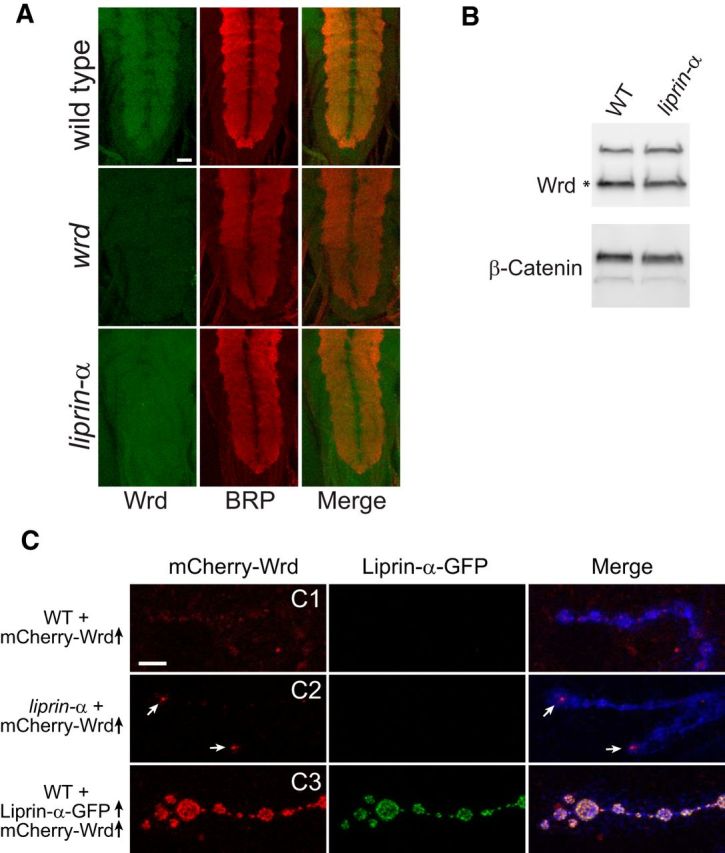
Liprin-α localizes Wrd to the synapse. A, Representative confocal images of ventral nerve cords costained with Wrd (green) and BRP (red) in third-instar larvae of wild-type, wrd, or liprin-α mutants. Images representing the Wrd channel (green), the BRP channel (red), and the merge are shown. Scale bar, 25 μm. B, Western blot analysis of endogenous Wrd protein (marked by *) levels in the larval brains of wild-type or liprin-α mutant flies. C, A UAS–mCherry–Wrd transgene was expressed in neurons of wild-type or liprin-α mutant flies using the pan-neuronal driver BG380–Gal4. The same transgene was also coexpressed with a UAS–liprin-α–GFP transgene in neurons of wild-type flies. Third-instar larvae were dissected and fixed with 4% PFA. Representative confocal images of muscle 4 NMJs emitting green (GFP) and red (mCherry) fluorescence are shown. Arrows point to mCherry–Wrd enriched in terminal boutons. Confocal scans of all the mCherry fluorescence were performed with the same gain. Scale bar, 5 μm.
Loss of function in syd-1, liprin-α, or wrd causes ectopic accumulation of a subset of presynaptic materials in the distal, but not proximal, axon
To determine whether Liprin-α–Wrd-mediated signaling events function downstream of Syd-1, we performed phenotypic analysis on synaptogenic defects in syd-1, liprin-α, and wrd mutants aimed at identifying common processes regulated by all three genes. To our surprise, a unique vesicle localization phenotype is shared by syd-1, liprin-α, and wrd mutant larvae: significant ectopic aggregates of DVGlut are present at distal axon regions just proximal to the synaptic nerve terminal (Fig. 3A). This phenotype is not limited to axons innervating muscle 12/13 (as shown in Fig. 3A); axons innervating all other body wall muscles, including muscle 4 and muscle 1/9, show the same phenotype (data not shown). In contrast to the readily identifiable DVGlut aggregates in the distal axon region, no observable accumulation of DVGlut is evident at the segmental nerves (bundled proximal axons connecting the ventral nerve cord with body wall muscles; Fig. 3B). This ectopic DVGlut accumulation is distinct from the typical axon transport defect seen in kinesin and sunday driver mutants, in which massive accumulation of DVGlut is present at the segmental nerves attributable to focal axon swelling, with little synaptic materials reaching the nerve terminal (Fig. 3B). Furthermore, ectopic clustering of DVGlut in the distal axon in wrd, liprin-α, or syd-1 mutants can be clearly identified in early first-instar larvae (data not shown), suggesting that the ectopic accumulation near synaptic termini occurs early in the synaptic development and is not attributable to a gradual accumulation of SVs during synaptic terminal growth.
Figure 3.

Disruption of the syd-1, liprin-α, or wrd results in ectopic clustering of SVs, DCVs, and presynaptic matrix proteins at distal, but not proximal, axons. A, Representative confocal images of distal axons of muscle 12/13 NMJs costained with DVGlut (green) and FasII (red) in third-instar larvae of wild-type, syd-1, liprin-α, or wrd mutants. White brackets show distal axons crossing muscle 13 to innervate muscle 12, in which DVGlut-positive aggregates are identified in the mutant but not wild-type animals. B, Representative confocal images of segmental nerves ∼100 μm away from the tip of the ventral nerve cord, costained with DVGlut (green) and HRP (red), in third-instar larvae of wild-type, syd-1, liprin-α, wrd, khc [khc6/Df(2R)JP6], or sunday driver (syd2H/sydz4) mutants. C, Representative confocal images of muscle 12/13 NMJs costained with HRP (blue) and a series of markers (all in green), including Syt, Rab3, Synapsin, BRP, FYVE–GFP (early endosome marker), LAMP–GFP (late endosome/lysosome marker), Mito-GFP, and Syntaxin in third-instar larvae of wild-type and liprin-α mutants. Scale bar, 10 μm. D, Costaining of BRP (NC82) and Cac–GFP (anti-GFP antibody) at the distal axons of muscle 12/13 NMJs of wild-type, syd-1, or wrd mutant larvae. Arrowheads point to Cac–GFP puncta that are colocalized with BRP puncta. Scale bar, 5 μm. E, Costaining of BRP (NC82) and ANF–GFP (anti-GFP antibody) at the distal axons of muscle 1/9 NMJs of wild-type, syd-1, liprin-α, or wrd mutant larvae. Scale bar, 5 μm.
To further characterize this unique DVGlut distribution defect, we analyzed a series of vesicle, organelle, and AZ markers in syd-1, liprin-α, and wrd mutant larvae. Interestingly, three other tested SV-associated proteins (Syt, Rab3, synapsin) and the DCV marker ANF–GFP (Fig. 3C,D) were ectopically accumulated in the three mutant distal axons. In contrast, the distribution of mitochondria, lysosomes, early and late endosomes, and Kinesin and Dynein motors along the entire axon showed no significant difference between wild type and the three mutants (Fig. 3C, Table 2; data not shown). These data suggest that syd-1, liprin-α, and wrd are all required to restrict the localization of two classes of vesicles to the nerve terminal, whereas their function are dispensable for axonal trafficking of other cellular organelles and the transport machinery.
The fact that only presynaptically vesicles are ectopically located at the syd-1, liprin-α, and wrd mutant distal axons prompted us to examine whether an AZ-like structure is present along with the ectopically localized vesicles. Indeed, we detected ectopic accumulation of endogenous BRP, the T-bar structure protein, and motoneuron-expressed transgenic voltage-gated Ca2+ channel [Cacophony (Cac)–GFP] and GFP–RIM at the mutant distal axon regions, although the number of Cac–GFP puncta was much fewer than the number of BRP puncta (Fig. 3C–E, Table 2; data not shown). The fact that all the Cac–GFP puncta are colocalized with BRP puncta suggests that Cac may be mislocalized because of its physical interaction with BRP (Fig. 3D; Fouquet et al., 2009). Confocal imaging analysis of distal axons double stained with neuronal membrane marker (HRP) and individual synaptic markers showed no membrane association of ectopically localized synaptic materials (data not shown). These data and EM analysis (see below) suggest the limited presence of the transmembrane protein Cac–GFP in this region may not indicate distal axonal synapse formation. The presence of the AZ components inside the distal axons suggests possible mislocalization of an AZ-like structure, which in turn may serve as a structural basis for ectopic vesicle accumulation.
syd-1/liprin-α/wrd function as a linear pathway in presynaptic neurons to promote normal SV localization
Given the similar ectopic accumulation defects in syd-1, liprin-α, and wrd mutant distal axons, a series of classic genetic rescue experiments were performed to define the genetic hierarchy among the three genes. The principle of these rescue experiments is that overexpression of a downstream gene can restore the mutant phenotypes of its upstream genes, whereas overexpression of the upstream genes cannot bypass the defects caused by mutations of its downstream genes. Three transgenes, UAS–GFP–syd-1, UAS–liprin-α–GFP, and UAS–GFP–Wrd, when expressed in the motoneurons, completely rescued the distal axon DVGlut accumulation in syd-1, liprin-α, and wrd mutants, respectively (Fig. 4A,B), suggesting that syd-1, liprin-α, and wrd work in the presynaptic motoneuron to promote proper localization of SVs. We then expressed each transgene in the other two mutants and examined its ability to rescue the ectopic vesicle accumulation. Neuronal expression of Wrd substantially cleared DVGlut accumulation at the distal axons of syd-1 or liprin-α mutants, but the latter two proteins did not rescue the wrd mutant (Fig. 4A,B) and neuronal expression of Liprin-α significantly suppressed the distal axon phenotype of syd-1 mutants but not vice versa (Fig. 4A,B). Therefore, these results establish the hierarchy of a linear pathway, from syd-1 to liprin-α to wrd, that promotes normal SV localization.
We showed previously that Liprin-α is required for the proper synaptic localization of Wrd and that the homogenous distribution of mCherry–Wrd at the NMJ is significantly disrupted in liprin-α null mutants (Fig. 2). Because of the high-level expression of the mCherry–Wrd transgene driven by the Gal4–UAS system, we still detected residual mCherry–Wrd expression in boutons under this condition (Fig. 2C). Because neuronal expression of this mCherry–Wrd transgene rescued the wrd and liprin-α mutant distal axon phenotype (data not shown), we think that a small amount of Wrd transgenic proteins may possibly diffuse to the synaptic sites and rescue the liprin-α mutant phenotype.
The syd-1/liprin-α/wrd pathway antagonizes glycogen synthase kinase-3β kinase activity at the NMJ
PP2A shares substrates with a number of serine-threonine kinases, including ERK/MAPK, CaMKII and glycogen synthase kinase-3β (GSK-3β; Janssens and Goris, 2001; Viquez et al., 2009). Therefore, we tested for genetic interactions between these kinases and the syd-1/liprin-α/wrd pathway. Blocking GSK-3β, but not ERK/MAPK or CaMKII, suppressed the distal axon vesicle clustering defect in syd-1, liprin-α, or wrd mutants (Fig. 5A,B; data not shown). These data confirm the linear relationship of the syd-1/liprin-α/wrd pathway and also suggest that dephosphorylation by Wrd-containing PP2A phosphatase and phosphorylation by GSK-3β converge to prevent the ectopic distribution of SVs at distal axons. Collectively, our data establish that Wrd is an effector for Syd-1 and Liprin-α that antagonizes GSK-3β phosphorylation to prevent ectopic SV and DCV accumulation at the distal axon region.
Ultrastructure and live imaging analysis of ectopic accumulation of synaptic materials in syd-1/liprin-α/wrd mutant distal axons
We showed above that an syd-1/liprin-α/wrd pathway functions to prevent the ectopic accumulation of synaptic materials at the distal axon. To directly analyze the ultrastructure of the ectopically accumulated materials, we performed EM experiments. In wild-type boutons, high-electron-dense materials reside exclusively at the presynaptic or postsynaptic plasma membrane. However, in syd-1 mutant boutons, in addition to AZs at the presynaptic membrane, floating dense materials surrounded by vesicles were detected inside the synaptic boutons, some of which were very close to the plasma membrane (Fig. 6). We also captured images containing tips of axonal regions that are connected to the synaptic boutons, in which floating high-electron-dense materials surrounded by vesicles were located near the axonal membrane (Fig. 6E,F). Similar data, including high-electron-dense materials partially touching the plasma membrane of nerve terminal in syd-1 mutant NMJs, were reported previously (Owald et al., 2010). These data suggest that ectopically accumulated synaptic materials—detected by immunocytochemistry—at syd-1 mutant distal axons are likely parts of these floating high-electron-dense materials.
Figure 6.
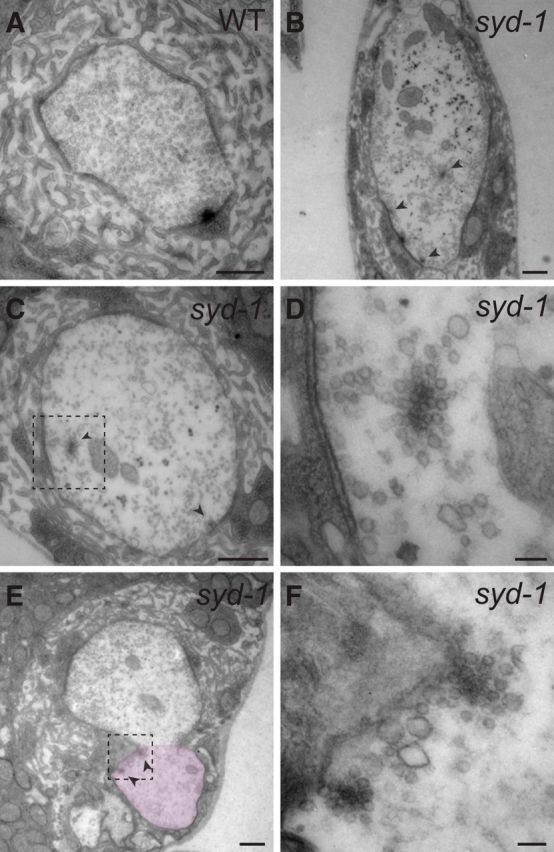
EM analysis of the ectopic vesicle accumulation. A–D, Electron micrographs of wild-type (WT) and syd-1 (syd-1ex1.2/syd-1ex3.4) mutant NMJ boutons. E, F, Electron micrographs of syd-1, a mutant NMJ bouton, and its connected axonal tip (pink-marked area in E). Identification of the axonal tip was based on the absence of subsynaptic reticulum (SSR) and the presence of glial wrapping. Arrowheads mark floating high-electron-dense materials surrounded by vesicles in syd-1 mutant samples. Some of the floating high-electron-dense materials are very close to or almost touching the plasma membrane. D and F show high-magnification images of dashed boxes in C and E, respectively. Scale bars: A, B, C, E, 0.5 μm; D, F, 80 nm.
We then asked whether the floating high-electron-dense materials at the mutant distal axons may possess AZ functions, i.e., whether they are able to capture vesicles that are transported either anterogradely or retrogradely. To address this question and to directly observe axonal vesicle trafficking in distal axons, we visualized the transport of Syt–GFP (SVs) and ANF–GFP (DCVs) along the axons by live imaging in dissected third-instar larvae. Again, neither SVs nor DCVs in segmental nerves showed a significant difference among wild-type, syd-1, liprin-α, and wrd mutants. However, syd-1, liprin-α, and wrd mutant distal axons show a significant amount of statically clustered SVs and DCVs, despite the normal movement of many vesicles in both anterograde and retrograde directions (data not shown). In addition, vesicles joining or leaving the static clusters were occasionally identified in the three mutants (data not shown). These data confirmed that a portion of SV precursors and DCVs are indeed accumulated at some static spots on the mutant distal axons.
To test whether anterogradely transported DCVs can be attracted to distal axon regions, we performed ANF–GFP SPAIM (Wong et al., 2012) experiments, in which a segment of the distal axon was photobleached and then allowed to recover while at the same time constantly photobleaching the area that connects the distal axon with the nerve terminal (i.e., to block retrograde transport from contributing to recovery of the ANF–GFP signal). Within 30 min, static DCV accumulation occurred in distal axons in wrd, liprin-α, and syd-1 mutants but not in the wild type (Fig. 7A). Some of the puncta occur at approximately the existing accumulation sites that are detectable before the photobleaching. The ectopic accumulations of DCVs in the mutants were confirmed by 5 min live time-lapse recording of the distal axons (see Movie 1 and data not shown). These data suggest that the ectopic DCV accumulation is a relatively dynamic process and that anterogradely transporting DCVs are capable of accumulating at the mutant distal axons.
Figure 7.
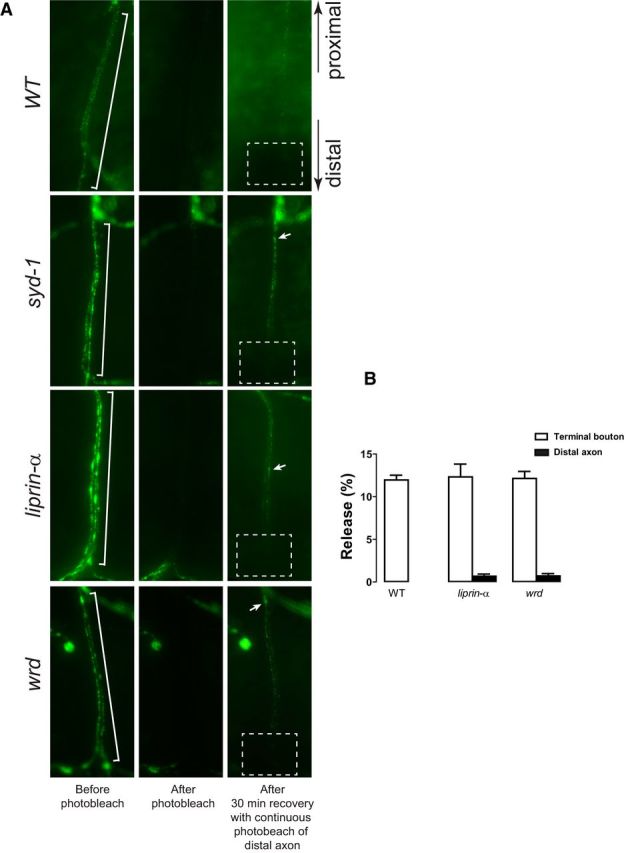
Live imaging analysis of the ectopic vesicle accumulation. A, Snapshot images taken by a fluorescence camera through a water-immersion objective show ANF–GFP-positive vesicle clustering at the distal axons targeting muscle 1/9 in dissected live wild-type (WT: UAS–ANF–GFP/D42–Gal4), syd-1 (syd-1ex1.2, UAS–ANF–GFP/syd-1ex3.4, D42–Gal4), liprin-α (liprin-αEPexR60/liprin-αF3ex15;D42–Gal4/UAS–ANF–GFP), and wrd (wrd104, UAS–ANF–GFP/wrd189, D42–Gal4) mutant third-instar larvae. For each genotype, images representing the same distal axon before and after photobleaching, as well as 30 min recovery after photobleaching, are shown. White brackets show the distal axons that were photobleached. Dashed boxes show the areas that were continuously photobleached during the 30 min recovery. Arrows show newly formed ANF–GFP cluster after photobleaching. B, Segmental nerves were stimulated at 70 Hz for 30 s. Release of ANF–GFP-positive DCVs during stimulation was measured as a percentage of prestimulation ANF–GFP intensity. DCVs release at the terminal boutons and/or distal axons of wild-type (WT: UAS–ANF–GFP/D42–Gal4), liprin-α (liprin-αEPexR60/liprin-αF3ex15;D42–Gal4/UAS–ANF–GFP), and wrd (wrd104, UAS–ANF–GFP/wrd189, D42–Gal4) are presented.
Live recording of ANF–GFP-positive vesicles at the distal axon targeting muscle 1/9 in dissected live syd-1 mutant (syd-1ex1.2, UAS–ANF–GFP/syd-1ex3.4, D42–Gal4) third- instar larvae. This movie shows the dynamics of ANF–GFP-positive dense-core vesicles after photobleaching and 30 min recovery (described in Fig. 7A). Static accumulation of dense-core vesicles occurs in multiple spots along the distal axon; some of them are in the same locations before photobleaching.
We next asked whether the ectopically accumulated vesicles participate in recycling. We first tested the release of ANF–GFP after stimulating the nerve at 70 Hz for 30 s. Although the release of ANF–GFP was similar at the terminal boutons of wild-type, liprin-α, and wrd NMJs, no release of ANF–GFP was detected at liprin-α or wrd mutant distal axons (Fig. 7B). We then performed FM 1-43 dye loading experiments. Wild-type and syd-1/liprin-α/wrd mutants show comparable levels of FM 1-43 dye uptake at NMJ boutons. Even after prolonged stimulation (30 min) of the NMJs, we failed to detect FM 1-43-labeled vesicles at the distal axons of the three mutants (data not shown). Together, these data suggest that ectopically accumulated vesicles are localized in a region that does not support release and endocytosis, consistent with the notion that some trafficking vesicles ectopically accumulate in the floating high-electron-dense AZ materials in the distal axon but not in the axonal plasma membrane.
Wrd is not required for Liprin-α- and Syd-1-mediated organization of BRP at the synaptic AZ
Previous studies using BRP as an AZ marker suggested important roles for Liprin-α and Syd-1 in preserving the normal presynaptic AZ morphology at the fly larval NMJ (Kaufmann et al., 2002; Owald et al., 2010). However, when we examined the AZ morphology in wrd mutants, we found that the size and shape of BRP clusters were normal in wrd null mutant NMJs (Fig. 8A). This result suggests that Wrd is not required for the normal organization of BRP-based T-bar structures at the nerve terminal, and Liprin-α and syd-1 organize AZ BRP via other molecules. Consistent with this notion, although transgenic Wrd protein rescues the syd-1 or liprin-α mutant distal axon phenotype, it fails to rescue the AZ morphological defects in syd-1 or liprin-α mutant NMJs (Fig. 8A).
Although previous studies demonstrated that both syd-1 and liprin-α mutants show a reduced number of release sites (BRP cluster; Owald et al., 2010), the mutant phenotypes of the two genes differ regarding individual AZ morphology. syd-1 mutant NMJs show interconnected and abnormally shaped T-bars, whereas liprin-α mutant NMJs show striking heterogeneous AZ size distribution with a wide range of size in AZ areas and BRP puncta (Kaufmann et al., 2002; Fig. 8A). Again, in a rescue experiment, neuronally expressed Liprin-α does not rescue AZ defects in syd-1 mutants, but it can prevent ectopic vesicle clustering in the distal axon of syd-1 mutants (Figs. 3, 8A). These data suggest that, although syd-1/liprin-α/wrd work in a linear pathway to promote normal vesicle distribution at the nerve terminal, Syd-1 and Liprin-α may each independently regulate different aspects of AZ morphology, a process that does not require Wrd function.
The presence of BRP in the distal axon is not required for ectopic vesicle accumulation in syd-1, liprin-α, or wrd mutants
Although the results above suggest that there is no correlation between distal axon vesicle accumulation and abnormal BRP morphology at the synapse, they do not determine whether the ectopic BRP proteins present at the distal axon area are responsible for attracting SVs. Double immunostaining revealed that, in syd-1, liprin-α, and wrd mutant distal axons, the ectopic BRP puncta do not always cocluster with SVs and DCVs (Figs. 3E, 8B), suggesting that ectopic vesicle accumulation occurs at spots lacking BRP. To directly test the requirement of BRP in the ectopic accumulation, we used previously reported transgenic BRP RNAi lines (C8 and B3; Wagh et al., 2006) to knock down BRP levels in the motoneurons of syd-1, liprin-α, or wrd mutants. BRP protein levels at the NMJs were reduced by >60%, whereas BRP puncta were almost eliminated from distal axons (Fig. 8B,C). In the absence of BRP at the distal axon region, the ectopic accumulation of DVGlut in the mutant distal axons showed no significant change compared with mutant animals without RNAi knockdown (Fig. 8B,C). Together, these data demonstrate that the presence of BRP at the distal axonal region is not essential for the ectopic accumulation of SV precursors in syd-1, liprin-α, or wrd mutant axons, suggesting that the floating high-electron-dense materials at the distal axons retain the ability to attract SVs in the absence of BRP. This is consistent with the fact that BRP is not required for the clustering of SVs at the AZs in the motoneuron nerve terminal (Kittel et al., 2006).
Presynaptic cytomatrix proteins such as BRP are believed to be transported along the axon as large dense-core transport vesicles (Shapira et al., 2003; Maas et al., 2012). Therefore, the ectopically accumulated BRP puncta in the syd-1, liprin-α, or wrd mutant distal axons could be in the forms of transporting vesicles, ectopically localized AZ components from the nerve terminal, or both. Our data indicate that, when BRP levels were reduced in motoneurons of syd-1, liprin-α, or wrd mutants, the remaining BRP proteins were preferentially localized to the nerve terminal, but were not present in the distal axons (Fig. 8B,C). This nonproportional reduction in BRP levels in the NMJs versus the distal axons supports the notion that BRP proteins in the mutant distal axons (when BRP protein levels are normal) are not in the form of transport vesicles but rather are structurally integrated into the AZ-like protein aggregates. Again, these data suggest that ectopically localized AZ materials selectively attract the accumulation of DCVs and SVs at the mutant distal axons.
Discussion
Much progress toward understanding presynaptic differentiation has been made through unbiased forward genetic screens in invertebrates. These studies have led to the identification of several key factors for AZ formation, including two evolutionarily conserved master organizer proteins of AZ assembly: syd-1 and syd-2/liprin-α (Zhen and Jin, 1999; Hallam et al., 2002; Kaufmann et al., 2002; Miller et al., 2005; Dai et al., 2006; Patel et al., 2006; Fouquet et al., 2009). However, how Syd-1/Liprin-α organize presynaptic sites remains unclear. Here we identify a new synaptic player, the PP2A B′ regulatory subunit, that is localized to the synapse by Liprin-α and mediates Syd-1/Liprin-α signaling in stabilizing AZs and their associated vesicles at the nerve terminal.
Wrd is a strong binding partner and a downstream effector of Liprin-α
Liprin-α was first identified as a protein interacting with the LAR (leukocyte antigen-related-like) family of phosphatases (Serra-Pagès et al., 1995, 1998). Studies during the past two decades demonstrate that Liprin-α regulates presynaptic and postsynaptic development, as well as neurotransmitter release through protein–protein interactions with a range of molecules, including CAST/ELKS/BRP, RIM, CASK (calcium/calmodulin-dependent serine protein kinase), GIT (G-protein-coupled receptor kinase-interacting ArfGAP), GRIP (glutamate receptor interacting protein), LAR, CaMKII, and Liprin-β (Spangler and Hoogenraad, 2007). Our proteomic data confirmed the interaction between Liprin-α and BRP/RIM in Drosophila. We also identified another important Liprin-α binding partner at the presynaptic sites, the B′ regulatory subunit of PP2A (Wrd), which depends on Liprin-α for it proper synaptic localization.
Phenotypic analysis of syd-1, liprin-α, and wrd mutants demonstrate that they share a unique trafficking defect, in which SVs, DCVs, presynaptic scaffolding proteins, and voltage-gated Ca2+ channels ectopically accumulate at the distal, but not the proximal, region of the axon. Genetic rescue experiments define a linear pathway, from syd-1 to liprin-α to wrd, that works cell autonomously in the presynaptic neuron to ensure proper localization of presynaptic materials to the nerve terminal and prevents ectopic accumulation. Together, our biochemical and genetic data suggest that Wrd mediates a novel Syd-1/Liprin-α function at the presynaptic site. Such a Syd-1/Liprin-α function is likely independent of their well established roles in regulating the T-bar structure protein BRP/ELKS.
A dephosphorylation event mediated by Wrd regulates proper localization of presynaptic materials
Two lines of evidence suggest that a Wrd-containing PP2A mediates the function of Syd-1/Liprin-α in regulating AZ stability. First, two rounds of in vivo biochemical purification using either Liprin-α or Wrd as the bait copurified Liprin-α with Wrd and the other two core subunits of PP2A, indicating the presence of a Liprin-α/Wrd/PP2A protein complex in neurons. Second, loss of GSK-3β kinase [sgg (shaggy)] function suppresses the syd-1, liprin-α, and wrd mutant distal axon phenotype, suggesting that a Wrd/PP2A-mediated phosphatase activity normally functions to antagonize a GSK-3β kinase activity in neurons to stabilize AZ and clustering of SVs at the nerve terminal.
Molecular action downstream of the syd-1/liprin-α/wrd pathway
What is the primary cause for the unique distal axon phenotype in syd-1/liprin-α/wrd mutant larvae? Liprin-α was shown to interact with KIF1A (kinesin family member 1A)/Unc-104 (Shin et al., 2003; Wagner et al., 2009), a neuron-specific kinesin motor known to transport SV precursors containing synaptophysin, Syt, and Rab1A. It was reported that Drosophila Liprin-α regulates the trafficking of SVs through its interaction with Kinesin-1 and that liprin-α mutant peripheral nerves show accumulation of clear-core vesicles similar to kinesin heavy chain (khc) mutants (Miller et al., 2005). However, when we focused on the location of the phenotypes relative to the entire axonal length, we found that liprin-α mutant accumulation of clear-core vesicles is present exclusively in the distal end (the ventrolateral peripheral nerve bundles, as well as axonal regions proximal to NMJs), whereas khc mutant larvae show massive aggregation of SV-associated proteins in the proximal end (segmental nerve bundles), and very few SV precursors reach the distal of axon. The distribution pattern of the vesicle accumulation in syd-1 and wrd mutants is the same as liprin-α mutants. Such a pattern is distinct from that of typical trafficking defects induced by mutations in vesicle-transporting motors or cargos.
Although we could not completely exclude a unique vesicle trafficking defect as the primary cause for the syd-1/liprin-α/wrd mutant axonal phenotype, we have a number of lines of evidence that suggest a plausible explanation: AZ materials at the nerve terminal become destabilized when the syd-1/liprin-α/wrd pathway is impaired, and the floating AZ materials diffuse back to the adjacent axonal regions as ectopic docking sites for vesicles. First, Syd-1, Liprin-α, and Wrd show clear synaptic localization, with little or no axonal localization detected, consistent with a collaborative function of the three at the AZs. Second, our EM analysis detected floating AZ materials in the synaptic boutons and the connected axonal regions in syd-1 mutants. Some of the floating materials are very close to or touching the bouton plasma membrane, indicating a possible defect in AZ stabilization and subsequent back-diffusion of detached AZ materials to axonal regions. Third, AZ components such as BRP, RIM, and voltage-gated Ca2+ channels are identified in the mutant distal axons along with vesicles, including SVs and DCVs, but not vesicles that transport AZ scaffolding proteins, or other synaptically localized organelles, or transport machineries. This is consistent with an ectopic accumulation of vesicles attracted by ectopic floating AZ components. Fourth, our live imaging analysis found that anterogradely transported DCVs accumulate at preferred spots at the mutant distal axons, consistent with the existence of static docking sites at these axonal regions. Fifth, ectopically accumulated vesicles do not participate in release or recycling, consistent with the notion that the vesicles do not dock on the axonal plasma membrane.
The fact that knockdown of a kinase (GSK-3β) rescues the distal axonal defects of syd-1/liprin-α/wrd mutants indirectly suggests that a Wrd-dependent dephosphorylation event is antagonized by a phosphorylation event (mediated by GSK-3β) to regulate AZ stability. However, these data cannot exclude the possibility that PP2A-independent functions of Wrd are involved. One way to seek direct evidence that Wrd-containing PP2A is involved in regulating AZ stability is to study the loss of function of PP2A; however, this approach has its own set of complications. As a ubiquitous heterotrimetric enzyme, the substrate specificity and subcellular localization of PP2A are greatly dependent on its regulatory subunit (such as Wrd). Mutating the catalytic or structural domain blocks overall PP2A action mediated by all regulatory subunits, which precludes analysis of Wrd-specific PP2A action. For example, mutations in MTS (the PP2A catalytic subunit) cause early lethality (Snaith et al., 1996). Overexpression of a dominant MTS protein causes massive axonal transport defects in the entire axon, as well as defects in AZ development (Viquez et al., 2009). Therefore, identifying common substrates shared by Wrd/PP2A and GSK-3β and studying how their phosphorylation status regulates AZ stability and/or vesicle trafficking will ultimately unravel the mechanism by which a PP2A-dependent pathway regulates presynaptic development. In this context, we set up a model to study how synapse scaffolding proteins can regulate localized phosphorylation/dephosphorylation through recruitment of specific phosphatases or kinases.
A mammalian homolog of Syd-1 was identified recently as an important regulator of presynaptic differentiation at central synapses, at least partially through its interaction with mammalian Liprin-α2 (Wentzel et al., 2013). Given that Liprin-α1 interacts with PP2A B56γ (mammalian homolog of Wrd) in HEK 293 cells (Arroyo et al., 2008), it will be of interest to investigate whether the function of Drosophila Liprin-α in mediating the signaling from Syd-1 to the PP2A B′ subunit is also evolutionarily conserved during vertebrate synapse development.
Footnotes
S.J.S. is partially supported by A6/Collaborative Research Centre 958 and NeuroCure Cluster of Excellence Grant Exc 257. This work is supported by National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke Grant NS070962 (C.Wu) and a COBRE Pilot Project (C.Wu) funded by NIH/National Institute of General Medical Sciences Grant GM103340 (Program director: Dr. Nicolas G. Bazan). We thank Aaron DiAntonio, David Van Vactor, Hongyan Wang, Tadashi Uemura, Bloomington Stock Centre, and Developmental Studies Hybridoma Bank for antibodies and fly stocks. We thank Xia Li and Jing Li for technical support and Ryan Labadens for editorial assistance. We also thank Matthew Whim for reviewing our manuscript before submission.
The authors declare no competing financial interests.
References
- Arroyo JD, Lee GM, Hahn WC. Liprin alpha1 interacts with PP2A B56gamma. Cell Cycle. 2008;7:525–532. doi: 10.4161/cc.7.4.5390. [DOI] [PubMed] [Google Scholar]
- Belmeguenai A, Hansel C. A role for protein phosphatases 1, 2A, and 2B in cerebellar long-term potentiation. J Neurosci. 2005;25:10768–10772. doi: 10.1523/JNEUROSCI.2876-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiano G, Bruschi M, Musante L, Santucci L, Ghiggeri GM, Carnemolla B, Orecchia P, Zardi L, Righetti PG. Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis. 2004;25:1327–1333. doi: 10.1002/elps.200305844. [DOI] [PubMed] [Google Scholar]
- Dai Y, Taru H, Deken SL, Grill B, Ackley B, Nonet ML, Jin Y. SYD-2 Liprin-alpha organizes presynaptic active zone formation through ELKS. Nat Neurosci. 2006;9:1479–1487. doi: 10.1038/nn1808. [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouquet W, Owald D, Wichmann C, Mertel S, Depner H, Dyba M, Hallermann S, Kittel RJ, Eimer S, Sigrist SJ. Maturation of active zone assembly by Drosophila Bruchpilot. J Cell Biol. 2009;186:129–145. doi: 10.1083/jcb.200812150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttfert F, Wurm CA, Mueller V, Berning S, Cordes VC, Honigmann A, Hell SW. Coaligned dual-channel STED nanoscopy and molecular diffusion analysis at 20 nm resolution. Biophys J. 2013;105:L01–L03. doi: 10.1016/j.bpj.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam SJ, Goncharov A, McEwen J, Baran R, Jin Y. SYD-1, a presynaptic protein with PDZ, C2 and rhoGAP-like domains, specifies axon identity in C. elegans. Nat Neurosci. 2002;5:1137–1146. doi: 10.1038/nn959. [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Garner CC. Molecular mechanisms of presynaptic differentiation. Annu Rev Cell Dev Biol. 2008;24:237–262. doi: 10.1146/annurev.cellbio.23.090506.123417. [DOI] [PubMed] [Google Scholar]
- Kaufmann N, DeProto J, Ranjan R, Wan H, Van Vactor D. Drosophila liprin-alpha and the receptor phosphatase Dlar control synapse morphogenesis. Neuron. 2002;34:27–38. doi: 10.1016/S0896-6273(02)00643-8. [DOI] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science. 2006;312:1051–1054. doi: 10.1126/science.1126308. [DOI] [PubMed] [Google Scholar]
- Ko J, Na M, Kim S, Lee JR, Kim E. Interaction of the ERC family of RIM-binding proteins with the liprin-alpha family of multidomain proteins. J Biol Chem. 2003;278:42377–42385. doi: 10.1074/jbc.M307561200. [DOI] [PubMed] [Google Scholar]
- Maas C, Torres VI, Altrock WD, Leal-Ortiz S, Wagh D, Terry-Lorenzo RT, Fejtova A, Gundelfinger ED, Ziv NE, Garner CC. Formation of Golgi-derived active zone precursor vesicles. J Neurosci. 2012;32:11095–11108. doi: 10.1523/JNEUROSCI.0195-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, DeProto J, Kaufmann N, Patel BN, Duckworth A, Van Vactor D. Direct observation demonstrates that Liprin-alpha is required for trafficking of synaptic vesicles. Curr Biol. 2005;15:684–689. doi: 10.1016/j.cub.2005.02.061. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Takao-Rikitsu E, Inoue E, Inoue M, Takeuchi M, Matsubara K, Deguchi-Tawarada M, Satoh K, Morimoto K, Nakanishi H, Takai Y. Cast: a novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13–1. J Cell Biol. 2002;158:577–590. doi: 10.1083/jcb.200202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owald D, Sigrist SJ. Assembling the presynaptic active zone. Curr Opin Neurobiol. 2009;19:311–318. doi: 10.1016/j.conb.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Owald D, Fouquet W, Schmidt M, Wichmann C, Mertel S, Depner H, Christiansen F, Zube C, Quentin C, Körner J, Urlaub H, Mechtler K, Sigrist SJ. A Syd-1 homologue regulates pre- and postsynaptic maturation in Drosophila. J Cell Biol. 2010;188:565–579. doi: 10.1083/jcb.200908055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owald D, Khorramshahi O, Gupta VK, Banovic D, Depner H, Fouquet W, Wichmann C, Mertel S, Eimer S, Reynolds E, Holt M, Aberle H, Sigrist SJ. Cooperation of Syd-1 with Neurexin synchronizes pre- with postsynaptic assembly. Nat Neurosci. 2012;15:1219–1226. doi: 10.1038/nn.3183. [DOI] [PubMed] [Google Scholar]
- Parker L, Gross S, Alphey L. Vectors for the expression of tagged proteins in Drosophila. Biotechniques. 2001;31:1280–1282. 1284–1286. doi: 10.2144/01316st01. [DOI] [PubMed] [Google Scholar]
- Patel MR, Lehrman EK, Poon VY, Crump JG, Zhen M, Bargmann CI, Shen K. Hierarchical assembly of presynaptic components in defined C. elegans synapses. Nat Neurosci. 2006;9:1488–1498. doi: 10.1038/nn1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Lisman JE. Coupled phosphatase and kinase switches produce the tristability required for long-term potentiation and long-term depression. J Neurosci. 2008;28:13132–13138. doi: 10.1523/JNEUROSCI.2348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Rudrabhatla P, Pant HC. Role of protein phosphatase 2A in Alzheimer's disease. Curr Alzheimer Res. 2011;8:623–632. doi: 10.2174/156720511796717168. [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- Sents W, Ivanova E, Lambrecht C, Haesen D, Janssens V. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. FEBS J. 2013;280:644–661. doi: 10.1111/j.1742-4658.2012.08579.x. [DOI] [PubMed] [Google Scholar]
- Serra-Pagès C, Kedersha NL, Fazikas L, Medley Q, Debant A, Streuli M. The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. EMBO J. 1995;14:2827–2838. doi: 10.1002/j.1460-2075.1995.tb07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Pagès C, Medley QG, Tang M, Hart A, Streuli M. Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J Biol Chem. 1998;273:15611–15620. doi: 10.1074/jbc.273.25.15611. [DOI] [PubMed] [Google Scholar]
- Seshacharyulu P, Pandey P, Datta K, Batra SK. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013;335:9–18. doi: 10.1016/j.canlet.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiryanova D, Tully A, Hewes RS, Deitcher DL, Levitan ES. Activity-dependent liberation of synaptic neuropeptide vesicles. Nat Neurosci. 2005;8:173–178. doi: 10.1038/nn1377. [DOI] [PubMed] [Google Scholar]
- Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger ED, Ziv NE, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron. 2003;38:237–252. doi: 10.1016/S0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Shin H, Wyszynski M, Huh KH, Valtschanoff JG, Lee JR, Ko J, Streuli M, Weinberg RJ, Sheng M, Kim E. Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J Biol Chem. 2003;278:11393–11401. doi: 10.1074/jbc.M211874200. [DOI] [PubMed] [Google Scholar]
- Shiomi K, Takeichi M, Nishida Y, Nishi Y, Uemura T. Alternative cell fate choice induced by low-level expression of a regulator of protein phosphatase 2A in the Drosophila peripheral nervous system. Development. 1994;120:1591–1599. doi: 10.1242/dev.120.6.1591. [DOI] [PubMed] [Google Scholar]
- Snaith HA, Armstrong CG, Guo Y, Kaiser K, Cohen PT. Deficiency of protein phosphatase 2A uncouples the nuclear and centrosome cycles and prevents attachment of microtubules to the kinetochore in Drosophila microtubule star (mts) embryos. J Cell Sci. 1996;109:3001–3012. doi: 10.1242/jcs.109.13.3001. [DOI] [PubMed] [Google Scholar]
- Spangler SA, Hoogenraad CC. Liprin-alpha proteins: scaffold molecules for synapse maturation. Biochem Soc Trans. 2007;35:1278–1282. doi: 10.1042/BST0351278. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The presynaptic active zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Li J, Valakh V, DiAntonio A, Wu C. Drosophila Rae1 controls the abundance of the ubiquitin ligase Highwire in post-mitotic neurons. Nat Neurosci. 2011;14:1267–1275. doi: 10.1038/nn.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Zhu M, Li L, Wu C. Identifying protein-protein interaction in Drosophila adult heads by tandem affinity purification (TAP) J Vis Exp. 2013;(82):50968. doi: 10.3791/50968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ohyama T, Bellen HJ. FM 1-43 labeling of synaptic vesicle pools at the Drosophila neuromuscular junction. Methods Mol Biol. 2008;440:349–369. doi: 10.1007/978-1-59745-178-9_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez NM, Li CR, Wairkar YP, DiAntonio A. The B′ protein phosphatase 2A regulatory subunit well-rounded regulates synaptic growth and cytoskeletal stability at the Drosophila neuromuscular junction. J Neurosci. 2006;26:9293–9303. doi: 10.1523/JNEUROSCI.1740-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez NM, Füger P, Valakh V, Daniels RW, Rasse TM, DiAntonio A. PP2A and GSK-3beta act antagonistically to regulate active zone development. J Neurosci. 2009;29:11484–11494. doi: 10.1523/JNEUROSCI.5584-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Dürrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, Wichmann C, Kittel R, Sigrist SJ, Buchner E. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Wagner OI, Esposito A, Köhler B, Chen CW, Shen CP, Wu GH, Butkevich E, Mandalapu S, Wenzel D, Wouters FS, Klopfenstein DR. Synaptic scaffolding protein SYD-2 clusters and activates kinesin-3 UNC-104 in C. elegans. Proc Natl Acad Sci U S A. 2009;106:19605–19610. doi: 10.1073/pnas.0902949106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel C, Sommer JE, Nair R, Stiefvater A, Sibarita JB, Scheiffele P. mSYD1A, a mammalian synapse-defective-1 protein, regulates synaptogenic signaling and vesicle docking. Neuron. 2013;78:1012–1023. doi: 10.1016/j.neuron.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MY, Zhou C, Shakiryanova D, Lloyd TE, Deitcher DL, Levitan ES. Neuropeptide delivery to synapses by long-range vesicle circulation and sporadic capture. Cell. 2012;148:1029–1038. doi: 10.1016/j.cell.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M, Jin Y. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature. 1999;401:371–375. doi: 10.1038/43886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live recording of ANF–GFP-positive vesicles at the distal axon targeting muscle 1/9 in dissected live syd-1 mutant (syd-1ex1.2, UAS–ANF–GFP/syd-1ex3.4, D42–Gal4) third- instar larvae. This movie shows the dynamics of ANF–GFP-positive dense-core vesicles after photobleaching and 30 min recovery (described in Fig. 7A). Static accumulation of dense-core vesicles occurs in multiple spots along the distal axon; some of them are in the same locations before photobleaching.