

Abstract
The motor and learning functions of the striatum are critically dependent on synaptic transmission from midbrain dopamine neurons and striatal cholinergic interneurons (CINs). Both neural populations alter their discharge in vivo in response to salient sensory stimuli, albeit in opposite directions. Whereas midbrain dopamine neurons respond to salient stimuli with a brief burst of activity, CINs exhibit a distinct pause in firing that is often followed by a period of increased excitability. Although this “pause–rebound” sensory response requires dopaminergic signaling, the precise mechanisms underlying the modulation of CIN firing by dopaminergic afferents remain unclear. Here, we show that phasic activation of nigrostriatal afferents in a mouse striatal slice preparation is sufficient to evoke a pause–rebound response in CINs. Using a combination of optogenetic, electrophysiological, and pharmacological approaches, we demonstrate that synaptically released dopamine inhibits CINs through type 2 dopamine receptors, while another unidentified transmitter mediates the delayed excitation. These findings imply that, in addition to their direct effects on striatal projection neurons, midbrain dopamine neurons indirectly modulate striatal output by dynamically controlling cholinergic tone. In addition, our data suggest that phasic dopaminergic activity may directly participate in the characteristic pause–rebound sensory response that CINs exhibit in vivo in response to salient and conditioned stimuli.
Keywords: acetylcholine, basal ganglia, dopamine
Introduction
As the principal input structure to the basal ganglia, the striatum plays an important role in selecting and reinforcing motor actions (Grillner et al., 2005). Its activity is controlled by glutamatergic afferents from cortex and thalamus, by dopaminergic inputs from the ventral midbrain, and by local interneurons. Cholinergic interneurons (CINs) provide the main source of acetylcholine (ACh) to the striatum (Bolam, 1984). Although CINs account for <3% of all striatal neurons (Woolf and Butcher, 1981), they powerfully influence locomotion and procedural learning by modulating the excitability of striatal projection neurons (SPNs), the plasticity of corticostriatal synapses, and the local release of dopamine (Kaneko et al., 2000; Pakhotin and Bracci, 2007; Shen et al., 2007; Witten et al., 2010; English et al., 2012; Threlfell et al., 2012). Importantly, their dysfunction is implicated in Parkinson's disease and other movement disorders (Pisani et al., 2007). Understanding the cellular mechanisms regulating the activity of CINs is therefore central to elucidating striatal function in health and disease.
CINs receive synaptic inputs from midbrain dopamine neurons (Pickel and Chan, 1990; Dimova et al., 1993) and express both Gαs-coupled dopamine D5 receptors (of the D1 receptor family) and Gαi-coupled dopamine D2 receptors (Yan et al., 1997; Yan and Surmeier, 1997). Indeed, pharmacological activation of D1 and D2 receptor families in vivo respectively increases and decreases ACh levels in striatum (Stoof et al., 1992; DeBoer and Abercrombie, 1996). In slice, D1 receptor agonists depolarize CINs and promote spiking (Aosaki et al., 1998; Pisani et al., 2000; Centonze et al., 2003), whereas D2 receptor signaling slows CIN pacemaking (Maurice et al., 2004; Ding et al., 2010; Tozzi et al., 2011). The net effect of synaptically released dopamine on CIN excitability is unclear. Moreover, midbrain dopamine neurons release several neurotransmitters (Stuber et al., 2010; Tecuapetla et al., 2010; Tritsch et al., 2012), and their effects might not be limited to the actions of dopamine.
Extracellular recordings from putative CINs (“tonically active neurons”) in the dorsal striatum of monkeys have revealed a characteristic response to salient sensory events, consisting of a prominent suppression of tonic firing followed by a transient increase in activity. This “pause–rebound” response is occasionally preceded by a brief burst. The cellular mechanisms underlying this firing pattern in vivo are unknown, but several models have been proposed (Schulz and Reynolds, 2013). The prevailing view is that inputs from thalamus drive initial excitation of CINs, with a pause in firing then resulting from an intrinsically generated afterhyperpolarizing potential. However, this model does not explain how rebound excitation arises, nor does it account for pauses that occur in the absence of a preceding burst.
Interestingly, the response of CINs to environmental cues coincides with phasic activation of midbrain dopamine neurons (Morris et al., 2004) and requires dopaminergic signaling (Aosaki et al., 1994), raising the possibility that dopamine neurons may directly participate in generating the characteristic sensory response of CINs. Here we investigate whether phasic activation of midbrain dopamine neurons affects the discharge of CINs in dorsal striatum.
Materials and Methods
Experimental animals.
Knock-in mice expressing Cre-recombinase under the control of Slc6a3 (encoding the plasma membrane dopamine transporter) were obtained from the Jackson Laboratory (stock #006660) and are referred to here as DatIRES-Cre mice (Bäckman et al., 2006). To allow optogenetic stimulation of nigrostriatal afferents, these mice were either virally injected with channelrhodopsin (ChR2) in the substantia nigra pars compacta (SNc; see below) or crossed with Ai32 mice (Jackson Laboratory, stock #012569), which contain a floxed allele of ChR2(H134R) under a ubiquitous promoter, thereby permitting genetic expression of ChR2 in all dopaminergic neurons (Madisen et al., 2012). In some experiments, the identification of striatal cholinergic interneurons was facilitated by crossing DatIRES-Cre mice with ChatBAC-GFP mice (Jackson Laboratory, stock #007902), which express enhanced GFP from a bacterial artificial chromosome (BAC) under the control of the endogenous promoter for choline acetyltransferase (ChAT; Tallini et al., 2006). In DatIRES-Cre;ChatBAC-GFP mice, ChR2 was exclusively delivered virally. To visualize Cre expression, DatIRES-Cre mice were crossed with mice bearing a Cre-dependent tdTomato reporter transgene (Ai14; Jackson Laboratory, stock #007914), referred to here as Rosa26lsl-tdtomato mice (Madisen et al., 2010). All strains were maintained on a C57BL/6 background, and experiments were performed in animals hemizygous for all transgenes. Experimental manipulations were performed in accordance with protocols approved by the Harvard Standing Committee on Animal Care following guidelines described in the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Stereotaxic intracranial Injections.
Viral delivery of ChR2 in DatIRES-Cre mice was performed as described previously (Tritsch et al., 2012). In short, 1 μl of an adeno-associated virus (AAV; serotype 2/8) encoding double-floxed inverted ChR2(H134R)-mCherry (>1012 genomic copies per milliliter; University of North Carolina Vector Core) was injected into the SNc of male and female mice [postnatal day (P) 18–22] maintained under deep isoflurane anesthesia. Injection coordinates were 0.9 mm anterior from lambda, 1.3 mm lateral, and 4.4 mm below pia, and expression time was ≥21 d. To visualize striatal afferents, 50 nl of fluorescent latex microspheres (Green RetroBeads, Lumafluor) were injected into dorsal striatum (1.1 mm anterior from bregma, 1.7 mm lateral, 2.5 mm below pia), and mice were processed for histochemistry 14 d later.
Slice preparation and electrophysiology.
Acute sagittal brain slices and electrophysiological recordings were obtained from the dorsal striatum as described before (Tritsch et al., 2012), with the following variations: cholinergic interneurons were identified using either morphological and electrophysiological features (Bolam et al., 1984; Kawaguchi, 1992, 1993; Bennett and Wilson, 1999) or GFP fluorescence in ChatBAC-GFP mice. Unless otherwise noted, all recordings were performed in drug-free perfusate at 32–34°C. For cell-attached recordings, pipettes (2–2.5 MΩ) were filled with artificial CSF composed of (in mm) the following: 125 NaCl, 2.5 KCl, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, and 11 glucose (295 mOsm · kg−1). Action potential firing was monitored in the cell-attached recording configuration in the voltage-clamp mode (Vhold = 0 mV). For whole-cell voltage-clamp and current-clamp recordings, pipettes were filled with a potassium-based, low-chloride pipette solution, containing (in mm) the following: 135 KMeSO3, 3 KCl, 10 HEPES, 1 EGTA, 0.1 CaCl2, 4 Mg-ATP, 0.3 Na-GTP, 8 Na2-phosphocreatine, pH 7.3, adjusted with KOH (295 mOsm · kg−1). For all voltage-clamp experiments (Vhold = −55 mV), errors due to the voltage drop across the series resistance (<20 MΩ) were left uncompensated. ChR2 was activated by a single pulse of light (1 ms for AAV-injected mice, 5 ms for Ai32 mice) delivered by a 473 nm laser using full-field illumination through the objective (5 mW · mm−2) at 30 s intervals. Drugs were bath applied at the indicated concentrations, except reserpine, which was injected intraperitoneally (5 mg · kg−1) 24 h before slicing. All pharmacological reagents were obtained from Tocris Bioscience.
Immunohistochemistry and antibodies.
Deeply anesthetized mice were perfused transcardially with 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.3, and brains were postfixed for 4 h at 4°C. Forty to fifty micrometer sections were cut on a vibratome and processed free-floating. After blocking in 6% normal horse serum (NHS) with 0.2% Triton X-100 for 1 h at room temperature, sections were incubated in primary antibody overnight (goat anti-choline-acetyltransferase; Millipore, AB144P; diluted to 10 μg/ml in 3% NHS with 0.2% Triton X-100), followed by 1–2 h at room temperature in secondary antibody (in 3% NHS). Fluorescence from mCherry and GFP was not immunoenhanced. Sections were mounted in ProLong Antifade reagent with DAPI (Invitrogen), and imaged with a slide scanning microscope (VS110, Olympus). High-resolution images of regions of interest were subsequently acquired with a Leica LS8 confocal microscope (Harvard NeuroDiscovery Center) or an Olympus FV1000 confocal microscope (Harvard Neurobiology Imaging Facility). Individual imaging planes were overlaid and quantified for colocalization in NIH ImageJ and thresholded for display in Photoshop (Adobe). Confocal images represent maximum intensity projections of 15 μm confocal stacks for immunohistochemistry and 6 μm for in situ hybridization.
In situ hybridization.
Double fluorescence in situ hybridization was performed as described previously (Ding et al., 2012). Briefly, brains from adult DatIRES-Cre mice were dissected in PBS and immediately frozen by dipping in liquid nitrogen several times. Brains were cut in 18-μm-thick sections with a cryostat (Leica), postfixed in 4% PFA, acetylated in 1% (v/v) triethanolamine and 0.25% (v/v) acetic anhydride, prehybridized, and hybridized at 65°C using fluorescein-labeled and digoxigenin-labeled antisense probes: Gad2(Gad65) and Cre respectively. Tyramide signal amplification method was performed according to the manufacturer's instructions (PerkinElmer).
Data acquisition and analysis.
Membrane currents and potentials were acquired as described previously (Tritsch et al., 2012), and analyzed in Igor Pro (Wavemetrics). The detection threshold for membrane currents was set at 5 pA. For display and analysis of membrane currents, the first five consecutive traces after threshold was reached for the first time were averaged for each condition. Because optogenetically evoked delayed action potential bursts and inward currents occasionally developed 5–10 min after stimulation onset, recordings were maintained for ≥15 min before determining whether they were present or not. Only currents reaching the threshold were included in the calculation of mean peak current amplitude and time to current peak. For analysis of the decay kinetics for component 1, only recordings that showed clear segregation of components 1 and 2 were used. With the exception of reserpine experiments, for which comparisons were made between treatment groups, all pharmacological analyses were performed by adding drugs to the perfusate and by normalizing light-evoked responses obtained during drug application to baseline responses. For analysis of action potential discharge, 10–50 consecutive acquisitions were overlaid and histograms of spike frequency were plotted using 50 ms bins. Only cells with ≥1 spike per bin were included for analysis. Data are represented as mean ± SEM, and as box plots with superimposed data points in Figures 1F–H and 4E. For statistical analysis, data were compared using the nonparametric Mann–Whitney test (for group comparisons) or the nonparametric Kruskal–Wallis ANOVA followed by Dunn test (for multiple group comparisons; see Fig. 6). p values smaller than 0.05 were considered statistically significant and are specified in the figure and text.
Figure 1.

Characterization of ChatBAC-GFP mice. A, Coronal section of striatum in ChatBAC-GFP mice showing GFP fluorescence (left, green) and ChAT immunolabeling (middle, red). Right, Merged image. DAPI nuclear stain, blue. B, Higher-magnification image within dorsal striatum shows individual CINs double-labeled for GFP (left) and ChAT (middle). All analyzed cells (n = 456 CINs in 2 mice) were positive for both markers. C, Example traces of spontaneous action potential discharge from CINs identified morphologically in wild-type striatal slices (left, black trace) or using GFP fluorescence in slices obtained from ChatBAC-GFP mice (right, green trace). D, Representative voltage response of wild-type (left) and GFP-positive CINs (right) to hyperpolarizing current injections (−100 pA). Note the characteristic membrane potential sag (arrow). E, Overlay of an individual action potential (AP) from a wild-type CIN (black) and GFP-positive cell from a ChatBAC-GFP mouse (green). F–H, No differences were observed in spontaneous firing rate (F), AP half width (G), or input resistance (H) between wild-type CINs (black; n = 38) and GFP-positive cells in ChatBAC-GFP mice (green; n = 33). Circles represent individual data points. Data median and first and third quartiles are shown as box plots.
Figure 4.
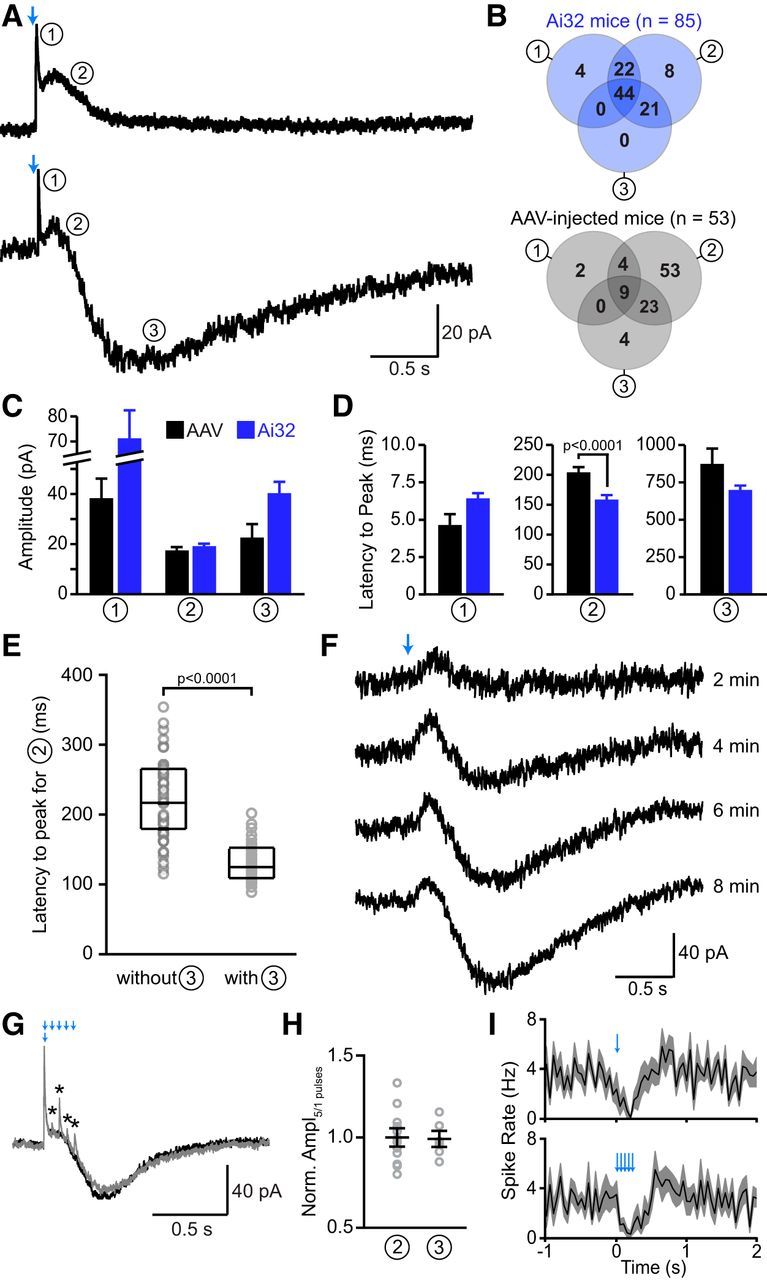
Stimulation of SNc axons evokes several ionic conductances in CINs. A, Two representative whole-cell voltage-clamp recordings from CINs (Vhold = −55 mV) upon optogenetic stimulation of nigrostriatal afferents (blue arrows). Top, Activation of SNc afferents evoked a rapid-onset outward current (circled number 1) followed by a slow outward current (circled number 2). Bottom, In another CIN, light application triggered current components 1 and 2 as well as a delayed inward current (circled number 3). B, Venn diagrams illustrating the prevalence (expressed as a percentage of all recordings in which ≥1 current was evoked by light) of each of the three current components in DatIRES-Cre;Ai32 mice (top, blue) or in DatIRES-Cre virally injected with Cre-dependent ChR2 in the SNc (bottom, gray). Total number of recordings indicated in parentheses. C, D, Average (±SEM) absolute amplitude (C) and latency from light flash onset to current peak (D) of each of the three current components (circled numbers) evoked by stimulation of nigrostriatal afferents in AAV-injected DatIRES-Cre mice (black) or DatIRES-Cre;Ai32 mice (blue). Data were analyzed using the average of the first five sweeps after break-in, or after reaching the threshold of 5 pA (for currents with delayed onsets). Results only include cells with detectable currents. The greater incidence of delayed inward currents in Ai32 mice compared with AAV-infected animals (B) might contribute to the shorter latency of the second current component compared with virally infected mice. E, Box plot with superimposed data points for the latency to peak of the second current component from recordings in which a delayed inward current (component 3) was either absent (left) or present (right). In the presence of the third component, the second component appears significantly faster, suggesting that these two components are partially overlapping. This overlap might explain the reduced latency to peak for the second component in Ai32 mice (D), since the third component was more frequent in this experimental preparation compared with AAV-injected mice (Fig. 4B). F, Individual light-evoked (blue arrow) current responses from a CIN (Vhold = −55 mV) obtained at different times after break-in (indicated at right). Note the development of the delayed inward current (component 3) over time. This was observed in approximately a third of the recordings. G, Representative voltage-clamp recording showing the superimposed average responses of a CIN to alternating light stimuli composed of one (black trace) or five light flashes at 20 Hz (gray trace). Note the additional fast outward currents that are elicited by repetitive stimulation (asterisks). H, Relative amplitude of the second and third current components (circled numbers) upon stimulation with one or five light pulses, as shown in A. For each cell (gray circles), the peak current amplitude evoked by the five-pulse stimulation protocol was divided by the peak current amplitude resulting from the single flash. Black bars indicate mean ± SEM. I, Population perievent spike histograms (mean shown in black, SEM in gray; n = 9 cells) upon stimulation with one (top) and five light pulses (bottom).
Figure 6.
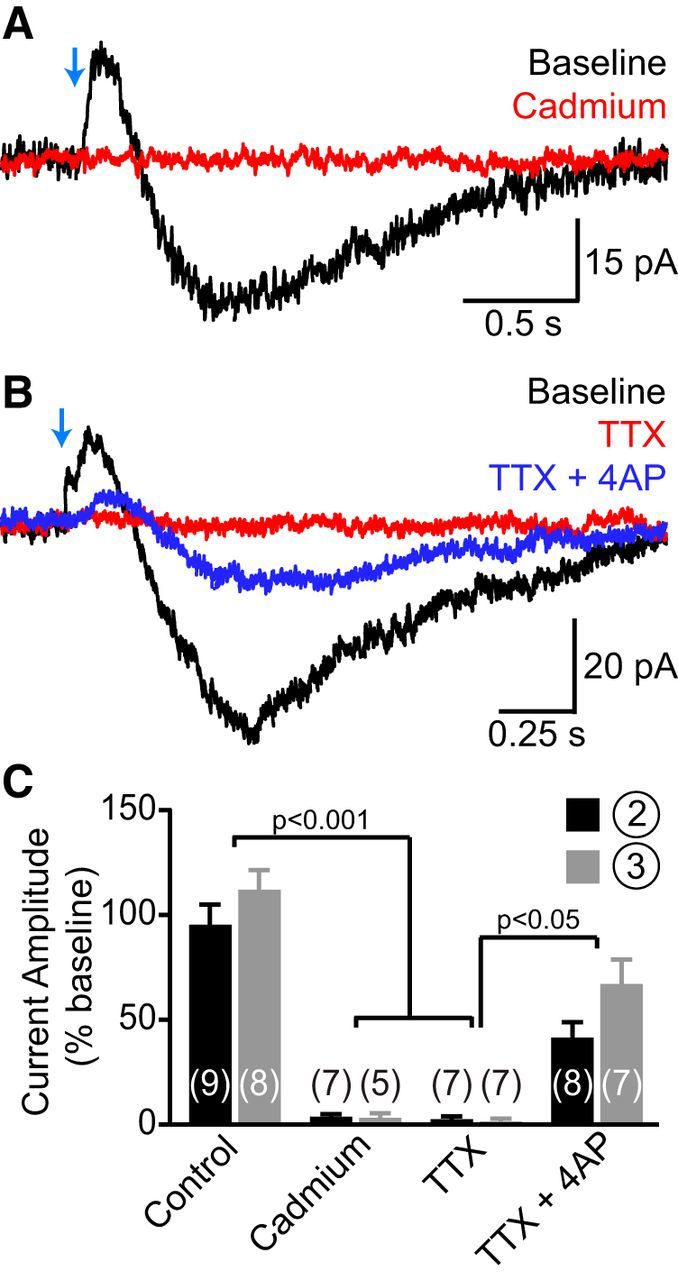
SNc axons directly evoke the second and third current components in CINs. A, Light-evoked current responses from a CIN before (black trace) and after (red trace) bath application of cadmium (30 μm) showing that the second and third components require activation of voltage-gated Ca2+ channels. B, Same as A upon sequential addition of TTX (1 μm, red trace) and TTX plus 4AP (0.5 mm, blue trace) to the perfusate. Note the partial recovery of both second and third current components in TTX plus 4AP (TTX + 4AP), indicating that they are evoked monosynaptically by ChR2-expressing nigrostriatal afferents. C, Histogram of the effect of bath application of control saline, cadmium, TTX, and TTX + 4AP on the peak amplitude of the second (black bars) and third (gray bars) current components (expressed as a percentage of amplitude during a baseline period preceding drug application). Number of recordings indicated in parentheses.
Results
Identification of striatal CINs
Although CINs represent a small percentage of striatal neurons, their distinctive morphological and functional properties have permitted targeting of these cells for electrophysiological recordings in acute brain slices (Bolam et al., 1984; Kawaguchi, 1992, 1993; Bennett and Wilson, 1999). Nevertheless, their identification could be further facilitated by specific expression of fluorophores, such as GFP. Indeed, their soma size is variable, and CINs are not the only interneurons that fire spontaneously in vitro (Tepper et al., 2011). Thus, we obtained BAC transgenic mice that express GFP under transcriptional control of the promoter for the acetylcholine synthetic enzyme ChAT (Tallini et al., 2006). We confirmed the specificity of GFP labeling to CINs in these mice (referred to henceforth as ChatBAC-GFP) by immunolabeling coronal brain sections using antibodies against ChAT (Fig. 1A,B). In the striatum, all GFP-labeled somata were immunopositive for ChAT (n = 456 of 456 cells in two mice), and all ChAT+ cells expressed GFP, indicating that GFP expression is fully penetrant and specific for striatal CINs. Importantly, endogenous GFP fluorescence in these mice is bright enough to unambiguously identify and target CINs for recording in 300-μm-thick striatal slices.
A significant caveat of BAC transgenic mice containing the Chat genomic locus is that they overexpress the vesicular ACh transporter (VAChT), which is encoded by a single exon gene located in an intron of the Chat locus. This results in excessive ACh exocytosis and behavioral abnormalities (Nagy and Aubert, 2012; Kolisnyk et al., 2013). To test whether BAC transgene expression might affect the electrical properties of CINs, we characterized the spontaneous firing rates and membrane properties of GFP+ cells in ChatBAC-GFP mice and compared them to recordings obtained from CINs in slices of dorsal striatum from wild-type mice. The latter were identified in sagittal slices by their large cell body size, and their identity was confirmed electrophysiologically. CINs from wild-type slices spontaneously fired action potentials (mean frequency, 4.5 ± 0.4 Hz; n = 24; Fig. 1C,F) with typical broad waveform (action potential half-width, 1.9 ± 0.1 ms; Fig. 1E,G), exhibited a prominent hyperpolarization-activated current “sag” (Fig. 1D), and displayed passive membrane properties (Rin = 202 ± 13 MΩ; Cin = 77.8 ± 2.3 pF; Fig. 1H) consistent with previous reports (Kawaguchi, 1992, 1993; Bennett and Wilson, 1999; Gittis et al., 2010). We did not detect any difference in GFP+ CINs from ChatBAC-GFP slices in either their response to voltage steps, spontaneous firing, action potential half-width, or membrane resistance (Fig. 1C–H), indicating that neither GFP nor VAChT overexpression significantly alters the electrophysiological properties of CINs. Moreover, optogenetic stimulation of dopaminergic neurons evoked qualitatively similar responses in CINs (see below) in DatIRES-Cre (n = 154) and DatIRES-Cre;ChatBAC-GFP mice (n = 49). Recordings from CINs in both mouse strains were consequently pooled for the remainder of this study. Together, these observations indicate that the basic electrophysiological features of CINs and dopaminergic signaling onto CINs are not influenced by increased cholinergic tone, making ChatBAC-GFP mice suitable for studying the influence that nigrostriatal afferents exert on striatal CINs. Nevertheless, BAC transgenic mice containing several copies of the Chat genomic region—including this line and others (Nagy and Aubert, 2012; Kolisnyk et al., 2013)—should likely not be used for studying the function of neurotransmission from cholinergic neurons.
Nigrostriatal afferents modulate CIN firing
To investigate whether phasic activation of nigrostriatal afferents modulates the activity of CINs in the striatum, we virally expressed a conditional allele of ChR2 in the SNc of DatIRES-Cre knock-in mice (see Materials and Methods; Fig. 2A–C). We monitored action potential discharge in CINs recorded in cell-attached or whole-cell current-clamp modes in sagittal slices of dorsal striatum before and after optogenetic activation of SNc axons. In most recordings (n = 59 of 68; 87%), phasic stimulation with a 1 ms flash of 473 nm light significantly altered the firing of CINs over the course of seconds in one of three ways: it evoked either a decrease in spike rate (n = 21 of 59; 36%), a delayed increase in spike rate (n = 6 of 59; 10%), or both (n = 32 of 59; 54%; Fig. 2D–F). The initial inhibitory effect was most pronounced 0.1–0.2 s after light presentation and reduced CIN discharge by 50–100% (mean, 84%). The delayed excitatory response consisted of a transient 1.5–3-fold increase in spike rate that peaked 0.4–0.6 s after optogenetic stimulation and returned to baseline within 1–1.5 s. A single, brief stimulation of SNc afferents therefore significantly modulates the activity of CINs in dorsal striatum over prolonged periods of time.
Figure 2.

Nigrostriatal afferents modulate CIN firing. A, Schematic representation of recording configuration in sagittal brain sections. SNc neurons (red) express ChR2 and project to striatum, where the response of CINs (green) to local optogenetic activation of SNc axons is examined electrophysiologically. B, Sagittal brain section from a DatIRES-Cre;ChatBAC-GFP mouse virally injected with Cre-dependent ChR2-mCherry AAV in SNc. DAPI nuclear stain, blue; ChR2-mCherry, red; GFP, green. C, High-magnification image of the striatum in B showing individual CINs (green) surrounded by ChR2-mCherry expressing nigrostriatal afferents (red). D, E, Representative examples of CIN firing patterns in response to phasic activation (blue arrow) of SNc afferents. CINs responded either with a transient pause in firing (D, pause-only responses) or with a pause in firing that was followed by rebound firing (E, pause/burst responses). This effect could be observed both in cell-attached (left) and whole-cell current-clamp recording configurations (right). F, G, Population perievent spike histograms of the two types of responses exemplified in D and E for DatIRES-Cre mice injected with ChR2-encoding AAV in SNc (F, black traces) or DatIRES-Cre;Ai32 mice, which express ChR2-YFP in all Cre+ neurons (G, blue traces). Solid lines represent mean firing rates, shaded regions represent SEM.
Before investigating the mechanism of CIN modulation by SNc axons, we attempted to devise experimental conditions that reduce the variability of the observed modulatory effects. Variability might result from differences in the extent of viral transduction or levels of ChR2 expression between animals and brain slices. Alternatively, they may stem from heterogeneity in the innervation of CINs by dopamine neurons or in the postsynaptic response of CINs to dopaminergic inputs. To distinguish between some of these possibilities, we repeated our experiments in DatIRES-Cre mice expressing a conditional allele of ChR2 in the Rosa26 locus (Ai32 mice; Madisen et al., 2012). Although light-evoked currents are reduced in these mice compared with virally infected DatIRES-Cre mice, DatIRES-Cre;Ai32 mice offer the advantage of exhibiting more uniform ChR2 expression in all cells containing Cre (Madisen et al., 2012). We first confirmed, using fluorescent RetroBead injections in DatIRES-Cre mice expressing a fluorescent Cre reporter, that all Cre+ neurons projecting into dorsal striatum reside in the ventral midbrain (n = 2 mice; data not shown), thereby excluding confounding contributions from non-nigrostriatal afferents. When ChR2+ axons in slices obtained from DatIRES-Cre;Ai32 mice were stimulated with 5-ms-long light flashes (to minimize variability in stimulation strength), we observed qualitatively similar but less variable modulatory effects on CIN discharge (Fig. 2G): 95% of recorded CINs (n = 41 of 43) exhibited, within a few hundred milliseconds of a stimulus, a transient decrease in firing, which was followed in 31 of these cells (76%) by a prominent burst of action potentials. Thus, the vast majority of CINs respond to phasic nigrostriatal afferent stimulation with a pause followed by a burst. Interestingly, whereas the ChR2-evoked suppression of spiking was immediately detectable, in approximately a third of recordings, bursting tended to develop slowly (within 5–10 min) over the course of the recording, regardless of the recording configuration (cell-attached or whole-cell) or experimental preparation (viral or genetic expression of ChR2). While some of the variability observed in virally infected mice might have originated from nonuniform ChR2 expression between experimental animals, these data suggest that some variability is intrinsic to the synapse under study.
Collectively, these results indicate that SNc axons exert a complex biphasic modulatory influence over the firing behavior of striatal CINs, which bears striking resemblance with the pause–rebound response of putative CINs recorded in vivo in response to behaviorally salient stimuli (Goldberg and Reynolds, 2011; Schulz and Reynolds, 2013).
SNc neuron stimulation evokes several ionic conductances in CINs
Although the cellular and molecular mechanisms underlying the sensory responses of CINs in vivo are unknown, current models favor the involvement of intrinsic membrane conductances (Goldberg and Reynolds, 2011; Schulz and Reynolds, 2013). Several lines of evidence indicate that intrinsic properties are unlikely to play a major role in the phenomena described above. First, the pause was never preceded by an increase in spike frequency (Fig. 2F,G), indicating that it does not result from a postburst afterhyperpolarizing potential. Second, pauses and bursts can occur independently, indicating that the delayed burst is not mediated by rebound spiking. Rebound excitation in CINs is mediated by hyperpolarization-activated cyclic nucleotide-gated (HCN) channels (Wilson, 2005), which are selectively inhibited by ZD7288. Indeed, injection of large hyperpolarizing currents (300 pA for 200 ms) evoked a prominent membrane potential sag characteristic of HCN channel activation during the current step, as well as a burst of action potentials immediately after the step (Fig. 3B,E), both of which were abolished with ZD7288 (50 μm; Fig. 3C,E). By contrast, injection of hyperpolarizing currents similar in amplitude to those observed following SNc axon stimulation (30 pA; Fig. 4) did not evoke an appreciable membrane potential sag during the current step or rebound excitation following the pause (Fig. 3A,D). This suggests that while CINs are capable of evoking a burst following membrane hyperpolarization, the synaptic currents evoked here are too small (at least as measured at the soma) to engage the intrinsic conductances that mediate rebound firing. Finally, when constant negative current was applied to CINs to prevent spontaneous firing, optogenetic stimulation of nigrostriatal afferents evoked a distinct membrane hyperpolarization (−1.3 ± 0.3 mV, n = 9) followed by a prominent slow depolarization (5.3 ± 1.2 mV, n = 8; Fig. 3F). Collectively, these observations suggest that modulation of CIN discharge by SNc neurons arises mainly from several synaptically driven fluctuations in somatic membrane potential.
Figure 3.

The “rebound” firing of CINs is not due to intrinsic membrane properties. A–C, Current-clamp traces from CINs in dorsal striatum upon somatic injection of hyperpolarizing current steps. A, Injection of small current steps (−30 pA, 200 ms) mimicking the amplitude and duration of the second outward current evoked by SNc neuron stimulation did not evoke an obvious rebound burst. B, Injection of larger current steps (−300 pA, 200 ms) evoked a prominent current sag potential (arrow) and rebound burst during and after the step, respectively, both of which were blocked by the HCN channel blocker ZD7288 (50 μm; C). D, E, Population perievent spike histograms of average spike discharge (solid lines) upon somatic injection of −30 pA (D; n = 8 cells) or −300 pA (E; n = 9 cells). Outlines depict SEM. For recordings in the presence of ZD2788 (50 μm), a small depolarizing current (50–80 pA) was continuously applied to the recording pipette to sustain firing. F, Example voltage response of a CIN upon optogenetic stimulation of nigrostriatal afferents. Negative current was injected to prevent the cell from firing, thus revealing subthreshold membrane potential fluctuations. Light stimulation (blue arrow) evokes an initial hyperpolarization and a prominent delayed depolarization, indicating that modulation of CIN discharge by SNc neurons results from synaptically evoked fluctuations in somatic membrane potential.
To reveal the membrane conductances that mediate the sequential hyperpolarization and depolarization of CINs, we performed whole-cell voltage-clamp recordings from CINs in striatal slices obtained from DatIRES-Cre;Ai32 mice. In nearly all recordings (n = 85 of 86; 99%), optogenetic stimulation of SNc axons evoked currents in CINs that were composed of several components exhibiting distinct latencies, amplitudes, and kinetics (Fig. 4A). With low intracellular Cl−, these currents typically included a rapid-onset outward current (component 1; observed in 70% of cells), a slower outward current (component 2; in 95% of cells), and a delayed inward current (component 3; in 65% of cells) when recorded in CINs held at −55 mV. Similar currents were also observed in slices with virally expressed ChR2. Of 58 recordings from CINs, 53 displayed light-evoked currents (91%); 15% of these contained component 1, 89% component 2, and 36% component 3 (Fig. 4B). Each of the three components showed similar magnitudes and time courses when comparing virally injected and Ai32 mice (Fig. 4C,D), except for the latency to peak of the second component (Fig. 4D). This subtle difference is attributed to the greater incidence in DatIRES-Cre;Ai32 mice of recordings displaying a delayed inward current, which truncates the second current component, thereby shortening its apparent latency to peak (Fig. 4B,E). Unless otherwise noted, subsequent analyses combine recordings obtained from both DatIRES-Cre;Ai32 mice and virally infected DatIRES-Cre mice.
The results described above were obtained using single light flashes, which presumably activate all ChR2-positive SNc afferents in the field of view synchronously. However, in vivo recordings indicate that midbrain dopamine neurons phasically fire 1–5 action potentials over a few hundred milliseconds in response to salient stimuli (Schultz et al., 1997; Morris et al., 2004). We therefore compared recordings in which SNc afferents were stimulated with single pulses or short trains (five flashes at 20 Hz) of light in an alternating and interleaved manner (Fig. 4G). Train stimulation did not alter the occurrence of any of the three components (first component: 7 of 12 cells; second component: 12 of 12 cells; third component: 6 of 12 cells for both single-pulse and train stimulation), and did not change the amplitude of the second and third components (Fig. 4H). In cells with a prominent first component, however, we occasionally observed additional synaptic events riding on top of the second component that were time-locked to the light stimuli (Fig. 4G). These additional outward currents might also contribute to the subtle widening of the pause in spiking following train stimulation (Fig. 1I).
Together, our results indicate that phasic stimulation of nigrostriatal afferents recruits several, temporally offset ionic conductances. The latter likely mediate the biphasic modulation of CIN firing: based on their prevalence, sign, and time course, the first two current components may participate in the membrane hyperpolarization that slows CIN pacemaking shortly after light stimulation, whereas the third component likely underlies the delayed burst. Indeed, like the late burst, the inward current occasionally developed over 5–10 min of light stimulation (Fig. 4F).
First component
Under our recording conditions, the first component averaged 46.1 ± 11.3 pA in amplitude, displayed fast kinetics (10–90% rise time, 1.3 ± 0.1 ms; decay τ, 12.2 ± 0.7 ms; n = 26) and was entirely blocked by SR95531, a selective GABAA receptor antagonist (Fig. 5A,B), indicating that it represents a GABAergic IPSC. This current exhibited a synaptic latency of 3.3 ± 0.5 ms from flash onset in virally infected DatIRES-Cre mice (n = 5) and 4.0 ± 0.2 ms in DatIRES-Cre;Ai32 mice (n = 32), consistent with monosynaptic transmission. Moreover, the IPSC was not affected by bath application of the ionotropic glutamate receptor antagonists NBQX and 3-[(±)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid (CPP; n = 8; data not shown), confirming that it does not result from activation of a local interneuron. In 27% of recordings, inhibiting GABAA receptors revealed an EPSC mediated by activation of ionotropic glutamate receptors (Fig. 5B), which might originate from glutamate corelease by dopaminergic axons (Stuber et al., 2010; Tecuapetla et al., 2010; Tritsch et al., 2012). Because of its small conductance relative to that of IPSCs (EPSC: 0.6 ± 0.1 pS, n = 16; IPSC: 7.0 ± 0.9 pS, n = 37, p < 0.01), the net initial effect of synaptic stimulation of SNc axons on membrane potential is a small IPSP (Fig. 3C) and a suppression of action potential firing (Fig. 2D–G). Therefore, the synaptic origin and functional contribution of EPSCs were not investigated further.
Figure 5.
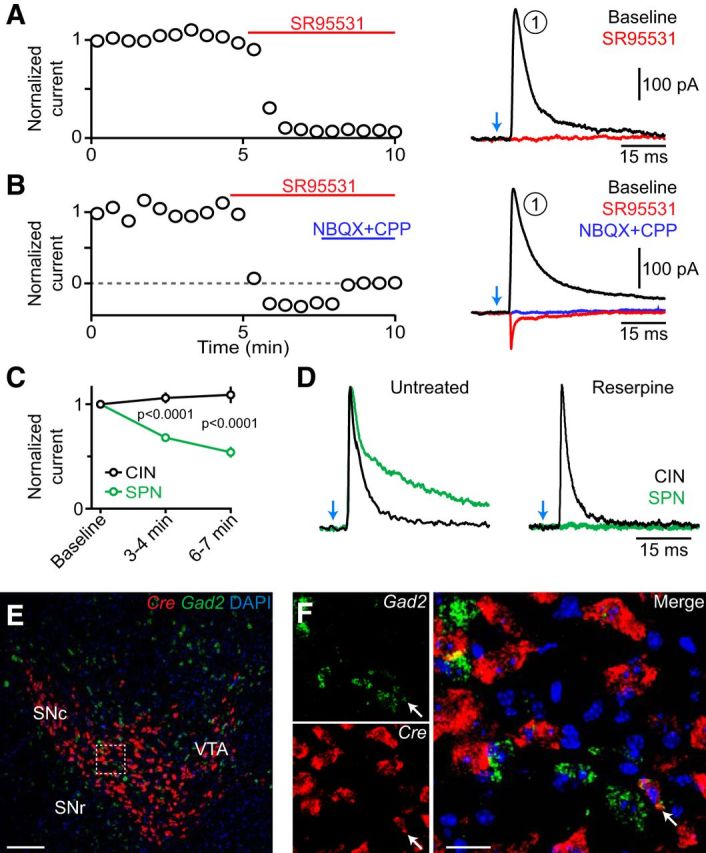
A reserpine-insensitive GABAergic current underlies the first component. A, Left, Peak amplitude of light-evoked IPSCs (current component 1; normalized to the first response) is sensitive to pharmacological inhibition of GABAA receptors with SR95531 (10 μm, bath application denoted by red bar). Right, Example traces obtained before and after application of SR95531. B, Same as A, for a recording in which inhibition of GABAA receptors with SR95531 (10 μm, red) revealed a glutamatergic EPSC that was antagonized by coapplication of NBQX and CPP (both at 10 μm, blue). C, Mean (±SEM) amplitude of GABAergic IPSCs (normalized to baseline) evoked by optogenetic stimulation of nigrostriatal afferents (stimulation interval, 30 s) in CINs (black) and SPNs (green) over time. Note that IPSCs in CINs do not “run down.” D, Overlay of representative peak-normalized IPSCs evoked by stimulation of nigrostriatal afferents (blue arrow) in CINs (black traces) or SPNs (green traces) from control slices (left) or slices obtained from reserpine-treated mice (right). Note the differences in decay kinetics and sensitivity to reserpine between GABAergic IPSCs recorded in CINs and SPNs. E, F, Double fluorescent in situ hybridization of Cre (red) and Gad2 (green) on coronal midbrain sections from DatIRES-Cre mice. The boxed area in E indicates the area shown at higher magnification in F. Arrow indicates example of a double-positive cell (occurrence: 0.7% of all Cre+ cells). Nuclear DAPI staining in blue. Scale bars: E, 200 μm; F, 50 μm.
The monosynaptic IPSC observed in CINs is reminiscent of the GABAA receptor-mediated currents recorded in SPNs following the stimulation of midbrain dopamine neurons (Tritsch et al., 2012). We therefore reasoned that IPSCs in CINs might similarly result from GABA corelease from dopaminergic axons. However, IPSCs evoked by optogenetic activation of nigrostriatal fibers in CINs did not run down in amplitude with time (Fig. 5C), and they displayed significantly faster decay kinetics compared with dopaminergic IPSCs in SPNs (τ = 56 ± 3.8 ms; p < 0.001 vs IPSCs in CINs; Fig. 5D; Tritsch et al., 2012, 2014; Ishikawa et al., 2013). To directly test whether IPSCs in CINs are evoked by GABA release from dopaminergic neurons, we recorded light-evoked IPSCs in CINs and SPNs in slices obtained from control DatIRES-Cre;Ai32 mice (n = 6 mice) and in age-matched and genotype-matched littermates treated with reserpine (n = 6 mice). Reserpine is a specific and irreversible antagonist of the vesicular monoamine transporter (VMAT), which has been shown to block GABA release from dopaminergic terminals (Tritsch et al., 2012). To our surprise, whereas reserpine effectively eliminated light-evoked IPSCs in SPNs (n = 7 of 7 cells), it did not prevent IPSCs recorded in CINs within the same slices [IPSC amplitude: reserpine, 81 ± 21 pA; control, 116 ± 25 pA; IPSC incidence: reserpine, 15 of 27 CINs (56%); control, 12 of 20 CINs (60%); p > 0.4 for both; Fig. 5D]. Thus, ChR2-evoked IPSCs in CINs differ fundamentally from those recorded in SPNs, suggesting that they originate from distinct populations of GABAergic synapses.
The ventral midbrain contains a population of GABAergic projection neurons that innervate CINs but not SPNs in the striatum (Brown et al., 2012). We previously reported that ∼3% of Cre+ cells in the SNc and ventral tegmental area (VTA) of DatIRES-Cre mice that extend axons into the striatum are immunonegative for tyrosine hydroxylase and VMAT2 (Tritsch et al., 2012), indicating that Cre is expressed in a subpopulation of “nondopaminergic” neurons in this mouse line. We therefore hypothesized that the reserpine-insensitive IPSCs we specifically observed in CINs might originate in these Cre+ nondopaminergic cells. To test this, we performed double fluorescence in situ hybridization against Cre and Gad2, which encodes the GABA synthetic enzyme GAD65. Because midbrain dopamine neurons do not express Gad2 (Tritsch et al., 2014), colocalization of mRNA for Cre and Gad2 would indicate that Cre also distributes to “classical” GABAergic neurons in the SNc and VTA of DatIRES-Cre mice. Indeed, we observed a few Cre+ SNc/VTA cells (17 of 2494; 0.7%) that also expressed Gad2 (Fig. 5E,F), indicating that some of the GABAergic projection neurons that specifically target striatal CINs (Brown et al., 2012) might be optogenetically stimulated in these mice, giving rise to the first GABAergic current component in CINs.
Second and third components
When the voltage of CINs was held at −55 mV, the second and third current components respectively averaged 18.5 ± 0.9 pA (range: 5–47 pA) and −37 ± 3.9 pA (range: −5 to −148 pA) in amplitude. Both components were eliminated by the voltage-gated Na+ and Ca2+ channel blockers tetrodotoxin (TTX) and cadmium, respectively, but were maintained in TTX and 4-aminopyridine (4AP; Fig. 6). These results therefore indicate that the second and third current components result from direct, action potential-evoked neurotransmission from nigrostriatal afferents (Petreanu et al., 2009; Cruikshank et al., 2010), and that their underlying conductances are not carried by voltage-gated Na+ channels or 4AP-sensitive K+ channels. Component 2 was sensitive to intracellular dialysis of cesium and displayed a current–voltage relation consistent with a K+ conductance (data not shown), indicating that this outward current is mediated K+ efflux. By contrast, the third component was not associated with clear changes in membrane conductance and exhibited a range of current–voltage relations across cells (data not shown), suggesting that the optogenetically evoked excitation of CINs might involve multiple conductances.
Although our extracellular and current-clamp recordings do not support the involvement of intrinsic, hyperpolarization-activated membrane conductances in generating the third component (Fig. 3), we nonetheless investigated whether HCN channels in distal dendrites might underlie the slow inward current. Consistent with our earlier findings, the HCN-channel blocker ZD7288 (50 μm) had no effect on the amplitude of the third component in voltage-clamp recordings (111.2 ± 21.5% of control, n = 4), suggesting that the second and third components represent two independent synaptic currents.
The slow kinetics of both second and third components (latency from flash onset to current peak: second component, 177.5 ± 6.9 ms, n = 103; third component, 729 ± 28.7 ms, n = 58) presumably reflect the activation of metabotropic receptor signaling cascades. In agreement with this, antagonists of ionotropic glutamate (using NBQX and CPP) and GABAA (SR95531) receptors did not affect either current (Fig. 7C). CINs express several metabotropic receptors for transmitters released by nigrostriatal axons, including D1-family and D2-family receptors, GABAB receptors, and group I–III metabotropic glutamate receptors (Yan et al., 1997; Pisani et al., 1999, 2002; Bell et al., 2002). Bath application of CGP55845, (S)-MCPG, LY341495, (RS)-α-cyclopropyl-4-phosphophenylglycine (CPPG), and (RS)-α-methylserin-O-phosphate (MSOP) did not appreciably reduce the amplitude of the second or third components compared with baseline (Fig. 7C), indicating that they are not evoked by activation of GABAB or metabotropic glutamate receptors. By contrast, a mixture of pharmacological blockers of D1-type and D2-type dopamine receptors (SCH23390 plus SKF82566 plus sulpiride plus L-741,626) eliminated the second component but not the third, indicating that the slow outward current is mediated by dopamine receptor activation. To determine which receptor family mediates the second component, we repeated this experiment using inhibitors specific for D1-like or D2-like receptors. While preventing the activation of D1-type receptors was ineffective at reducing the amplitude of the slow outward current (Fig. 7A,C), the D2 receptor family-specific antagonist sulpiride consistently eliminated the second component (Fig. 7B,C). A similar effect was observed upon bath application of a structurally unrelated inhibitor of D2-family receptors, L-741,626 (Fig. 7C). Moreover, the prevalence of the second component was significantly reduced in slices obtained from reserpine-treated DatIRES-Cre;Ai32 mice (only one current was observed in 27 CIN recordings from six mice) compared with littermate controls (current observed in 20 of 20 recordings from six mice; Fig. 7D). Together, these findings indicate that synaptic release of dopamine from SNc axons recruits a hyperpolarizing K+ current in striatal CINs requiring the activation of D2-family receptors.
Figure 7.
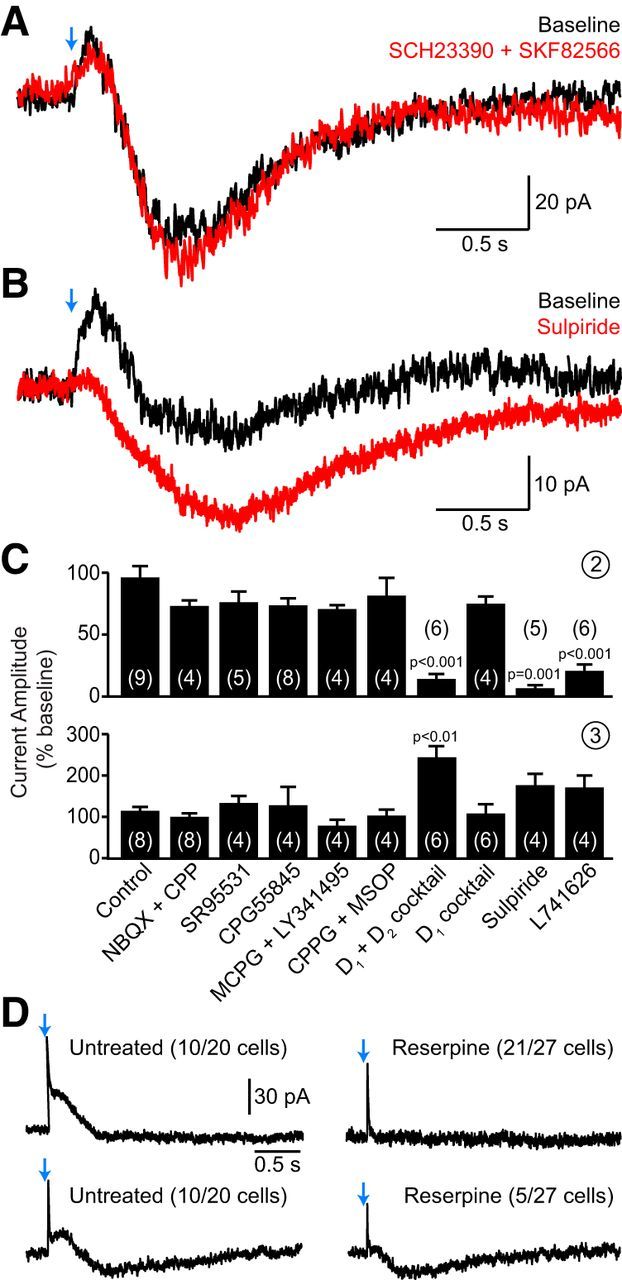
Synaptic activation of D2 receptors underlies the second current component. A, Representative voltage-clamp recording of CIN currents evoked by ChR2 stimulation (blue arrow) before (black trace) and after (red trace) addition of a mixture of D1-family receptor inhibitors (0.5 μm SCH23390 plus 0.5 μm SKF82566) to the perfusate. B, Same as A after bath application of the D2-like receptor antagonist sulpiride (10 μm). C, Group effects of pharmacological antagonists on the amplitude (normalized to baseline) of light-evoked second (top) and third (bottom) current components in CINs. Final drug concentrations (in μm) as follows: 10 NBQX, 10 R-CPP, 10 SR95531, 5 CGP55845, 500 (S)-MCPG, 50 LY341495, 300 CPPG, 200 MSOP, 1 SCH23390, 1 SKF82566, 1 sulpiride, 1 L-741,626. D1 plus D2 mixture, SCH23390 plus SKF82566 plus sulpiride plus L-741,626. D1 mixture, SCH23390 plus SKF82566. Number of recordings indicated in parentheses. The increased amplitude of the delayed inward current in the presence of D2 receptor antagonists suggests that the second and third current components overlap in time. D, Representative voltage-clamp CIN recordings from control DatIRES-Cre;Ai32 mice (left) and reserpine-treated littermates (right). Two examples are shown in each condition, representing the two current response types observed (incidence indicated in parentheses) following optogenetic stimulation of SNc axons (blue arrows). In untreated mice, light application evoked either the first two current components (top) or all three (bottom). In reserpine-treated mice, CINs exhibited either the first component alone (top) or the first and third component (bottom). The second current component was never observed in treated mice.
Our data show that the third component is not triggered by receptors for dopamine, glutamate, or GABA. However, synaptic signaling by nigrostriatal afferents is not limited to these transmitters. Dopaminergic axons have the ability to take up and release other monoamines, such as serotonin and noradrenaline (Giros et al., 1991; Zhou et al., 2005), both of which evoke a slow depolarizing current in CINs (Pisani et al., 2003; Bonsi et al., 2007). However, inhibition of α2-adrenergic (1 μm yohimbine, n = 3) and β-adrenergic receptors [10 μm (S)-propanolol, n = 3] as well as serotonin receptor types 2 (1 μm MDL11939, n = 3), 6 (10 μm SB258585, n = 2), and 7 (5 μm SB269970, n = 2), did not block the third component. The antipsychotic asenapine maleate (10–100 nm; n = 3), which broadly inhibits several monoaminergic receptors, was similarly ineffective (data not shown). In addition, optogenetically evoked inward currents could still be observed in slices obtained from DatIRES-Cre;Ai32 mice treated with reserpine, although their incidence was reduced compared with untreated littermates (Fig. 7D). The slow depolarizing current is therefore unlikely to be directly evoked by monoamines.
Dopamine neurons contain several other neuropeptides and transmitters that may stimulate metabotropic receptors on CINs, including cholecystokinin (CCK), neurotensin (NTS), substance P, and purine nucleosides (Tzschentke, 2001; Bentivoglio and Morelli, 2005). To identify whether release of any of these molecules underlies the third component, we screened pharmacological antagonists of candidate receptors. However, inhibiting either CCK1/2 receptors (using 1 μm L365260; n = 3), NTS receptors (0.5 μm SR142948; n = 3), neurokinin receptors 1–3 (1 μm each of SR140333, GR159897, and SB222200; n = 3), or puninergic type 1 (0.1 μm CGS15943; n = 3) and 2 (100 μm suramin; n = 3) receptors did not eliminate the burst or inward current. Blocking muscarinic acetylcholine receptors (10 μm scopolamine; n = 2), nitric oxide synthesis [100 μm Nω-nitro-l-arginine methyl ester; n = 3] or brain neurotrophic factor signaling through TrkA and TrkB receptors (200 nm K252a; n = 3) was similarly ineffective (data not shown). These findings therefore suggest that the delayed excitation of CINs does not arise from any of the candidate receptor families tested and may represent an unidentified neurotransmitter released from striatonigral neurons.
D2 receptor activation underlies the inhibition of CINs
Our data show that phasic stimulation of nigrostriatal afferents evokes two outward currents in CINs: one GABAergic and the other dopaminergic (Figs. 5, 7). These currents may collectively or independently mediate the hyperpolarization that suppresses CIN firing. To distinguish the contributions of the first and second current components on membrane potential, we monitored the discharge of CINs in slices obtained from control or reserpinized DatIRES-Cre;Ai32 littermates. As expected, optogenetic stimulation of nigrostrial afferents reliably paused the discharge of all recorded CINs in control slices (n = 14; Fig. 8A,B). By contrast, light stimulation did not appreciably decrease the firing of CINs in reserpine-treated slices (n = 12). Because dopamine depletion with reserpine abolishes the second outward current but not the first (Figs. 5D, 7D), these results suggest that the light-evoked pause in CINs requires dopaminergic signaling. This conclusion was tested using cell-attached recordings from CINs in untreated DatIRES-Cre;Ai32 mice by comparing the discharge pattern of CINs before and after bath application of SR95531 or sulpiride. While blocking GABAA receptors did not significantly affect the light-evoked suppression of firing (n = 9 CINs; Fig. 8C,D), antagonism of D2-type receptors eliminated it (n = 8; Fig. 8E,F). Furthermore, the fraction of CIN recordings exhibiting a clear pause in firing (90% in AAV-injected DatIRES-Cre mice, 95% in DatIRES-Cre;Ai32 mice) closely matches the proportion of voltage-clamp recordings displaying a second component (89 and 95%, respectively) but not the proportions showing a first component (15 and 70%, respectively; Fig. 4B). Collectively, these results show that D2-family receptors mediate the pause in firing evoked by stimulation of SNc axons.
Figure 8.

The pause in CIN firing evoked by SNc neuron stimulation is mediated by D2 receptors. A, Representative cell-attached recording from CINs obtained from a control DatIRES-Cre;Ai32 mouse (top, black trace) or a reserpine-treated littermate (bottom, red trace) upon optogenetic stimulation of nigrostriatal afferents (blue arrows). B, Population perievent spike histogram (normalized to baseline) for all CIN recordings obtained in control (n = 14, black) and reserpine-treated slices (n = 12, red) following light stimulation of SNc axons (blue arrow). Solid lines represent the mean, shaded regions the SEM. C, E, Example cell-attached recordings from CINs in slices of dorsal striatum expressing ChR2 in SNc axons before (baseline, black traces) or after bath perfusion of the GABAA receptor blocker SR95531 (10 μm; C, red trace) or the D2 receptor antagonist sulpiride (1 μm; E, red trace). D, F, Same as B for extracellular CIN recordings before (black) and after (red) bath application of SR95531 (n = 9, D) or sulpiride (n = 8, F). Note that the light-evoked suppression of firing is sensitive to D2 receptor block, but not GABAA receptor antagonism.
Discussion
We examined the influence that SNc neurons exert on a class of interneurons that critically modulate striatal function. We determined that stimulation of SNc axons directly recruits several temporally offset ionic conductances in CINs that sequentially suppress and increase their discharge over the course of seconds. Our results indicate that synaptically released dopamine primarily inhibits CINs through D2 receptors, whereas another unidentified transmitter initiates delayed excitation in these cells. Thus, in addition to their direct effects on SPNs, midbrain dopamine neurons modulate striatal output by dynamically controlling CINs. Moreover, our data suggest that phasic dopaminergic activity may directly participate in the characteristic pause–rebound sensory response that CINs exhibit in vivo.
SNc neurons synaptically inhibit CINs
Early clinical observations revealed that ACh levels rise as dopamine levels fall in Parkinson's disease (Barbeau, 1962; Stoof et al., 1992; Aosaki et al., 2010). Electrophysiological experiments in primates noted that pauses in putative CINs coincide with bursts of action potentials in dopaminergic neurons (Morris et al., 2004; Joshua et al., 2009) and depend on the activation of dopamine receptors (Aosaki et al., 1994). The data presented here demonstrate that midbrain dopaminergic projections can directly control the excitability of CINs in dorsal striatum. A single activation of SNc axons inhibits CIN spiking in a D2 receptor-dependent manner. The pause in firing coincides with a slow, D2 receptor-dependent outward K+ current whose amplitude is sufficient to silence CINs. This current likely mediates the pause in firing, although additional modulation of cell-intrinsic conductances underlying regenerative firing may also contribute. In agreement with widespread expression of D2 receptors in dorsal striatum CINs (Yan et al., 1997), the pause and slow outward current were observed in nearly all CINs and were sustained for the recording duration (20–40 min), indicating that inhibition by dopamine neurons constitutes a general and robust property of CINs.
Stimulation of nigrostriatal afferents also evoked monosynaptic GABAergic IPSCs in some CINs, which are distinct from the VMAT-dependent GABAergic IPSCs recorded in SPNs. These observations imply that stimulation of nigrostriatal axons engages separate sets of GABAergic synapses onto SPNs and CINs. One hypothesis is that dopamine neurons contain two distinct groups of GABAergic vesicles—one of which requires VMAT function, while the other does not—and that these vesicles distribute to different presynaptic terminals depending on the identity of the postsynaptic neuron. Alternatively, our in situ data indicate that ChR2 expression in DatIRES-Cre mice might extend to a population of GABAergic neurons, the activation of which may account for the reserpine-insensitive IPSCs selectively observed in CINs. In support of this hypothesis, nigrostriatal GABAergic neurons specifically innervate CINs but not neighboring SPNs (Tzschentke, 2001; Brown et al., 2012). Conversely, the specificity of IPSCs evoked by GABA coreleased from dopaminergic terminals to SPNs could reflect a lack of postsynaptic GABAA receptors at dopaminergic synapses in CINs or differences in dopaminergic presynaptic terminals targeting CINs and SPNs, respectively.
Regardless of their synaptic origin, GABAergic IPSCs in CINs did not appreciably suppress spiking under our recording conditions. However, when activated repeatedly, their effectiveness in pausing CINs increases (Fig. 4G,I), in agreement with previous observations (Brown et al., 2012).
Nigrostriatal afferents promote a delayed excitation of CINs
A striking effect of nigrostriatal afferent stimulation is the sustained increase in CIN firing that follows the pause. HCN channels have been shown to mediate rebound excitation following CIN hyperpolarization (Wilson, 2005). However, bursts were only observed in a subset (∼70%) of recordings with pauses in firing, and injection of hyperpolarizing current steps mimicking the slow outward currents that develop in CINs after SNc axon stimulation failed to induce bursts of comparable amplitude and duration (Fig. 3). Furthermore, since D2 receptor antagonists block the outward current but not the slow inward current (Fig. 7) and the latter is insensitive to pharmacological inhibition of HCN channels, intrinsic membrane conductances are unlikely to underlie the delayed excitation of CINs. Instead, our data suggest that the second and third components arise independently from the synaptic release of several transmitters.
We were unable to identify a transmitter receptor that underlies the inward current. Because most of the 24 receptor antagonists tested were applied individually, we cannot exclude the possibility that several redundant receptor families participate in the delayed response. Nevertheless, the ineffectiveness of the nonselective antagonists asenapine maleate and reserpine argue against the involvement of monoaminergic receptors. Coupled with the insensitivity to blockade of GABA and glutamate receptors, this raises the possibility that nigrostriatal neurons release an additional unidentified transmitter.
The late increase in CIN firing is more variable than the pause in two regards. First, ∼30% of CINs do not display a delayed burst or slow inward current. The source of this heterogeneity is unknown but may arise from intrinsic heterogeneity of midbrain dopamine neurons (Lammel et al., 2014). Second, the magnitudes of the burst and late inward current vary greatly between, as well as within, recordings. Interestingly, the sensory responses of CINs in vivo also display considerable variability, particularly the rebound excitation (Schulz and Reynolds, 2013). In approximately a third of our recordings, the delayed response was not immediately apparent, but slowly emerged with subsequent stimuli. These observations suggest that the expression of the delayed excitation in CINs is plastic and dependent on neuromodulation within the striatum.
While this manuscript was under review, a study addressing the modulation of CIN firing by dopamine neurons in different regions of the striatum was published (Chuhma et al., 2014). Consistent with our results, the study shows that synaptic activation of D2 receptors on CINs in dorsal striatum gates a K+ conductance that pauses firing in a GABAA receptor-independent manner. The additional detection of the first GABAergic component in our study may be attributed to a more positive holding potential in voltage-clamp recordings, which facilitates identification of this small current. Although Chuhma et al. (2014) report occasional bursts of activity following the pause, they did not further investigate the underlying mechanisms. This difference in focus may have originated in the variable and time-dependent nature of the burst, which was only detected in ∼30% of CINs using viral labeling of dopamine neurons.
Implications for the characteristic pause–rebound responses of CINs in vivo
Striatal CINs in vivo exhibit a distinctive response to salient sensory events that consists of a prominent pause in firing followed by a variable burst of action potentials, often called rebound excitation. Our findings indicate that activation of dopaminergic neurons is sufficient to evoke the pause as well as the burst through the synaptic actions of dopamine and an unknown mechanism, respectively. This hypothesis is consistent with prior work in vitro showing that pharmacological activation of D2 receptors decreases CIN firing, as well as reports in vivo indicating that sensory pauses in CINs coincide with phasic activation of dopamine neurons (Morris et al., 2004) and depend on dopaminergic signaling (Aosaki et al., 1994). In addition, sensory responses in CINs mirror the activity of dopamine neurons during behavioral conditioning, such that they respond more frequently and vigorously to unpredicted rewards and to sensory cues that reliably predict reward (Ravel et al., 2003; Apicella et al., 2009).
Some studies have failed to detect significant modulation of CIN pauses to cues that differentially stimulate dopamine neurons, and have described prominent CIN pauses to rewarding, neutral, and aversive events under conditions that do not readily engage dopamine neurons (Morris et al., 2004; Joshua et al., 2008), arguing against a direct effect of dopamine. However, because suppression of firing is truncated at zero, the ability to detect modulatory influences is limited. The D2 receptor-mediated suppression of CIN firing may be saturated at or downstream of the level of dopamine receptors, such that changes in dopamine release may not be reflected in the strength or duration of the pause. Moreover, the heterogeneity of midbrain dopamine neurons (Lammel et al., 2014) makes it possible that under-sampled populations of cells account for the sensory responses of striatal CINs.
Finally, several distinct mechanisms may underlie qualitatively similar pause–rebound CIN responses in vivo. GABAergic neurons in the VTA phasically increase their firing to aversive events (Cohen et al., 2012), and high-frequency stimulation of these cells evokes a pause followed by hyperpolarization-activated burst in striatal CINs (Brown et al., 2012). Striatal GABAergic interneurons can similarly depress CIN firing (Sullivan et al., 2008; English et al., 2012). Recent evidence also suggests that synchronous activation of CINs triggers neurotransmitter release from dopaminergic terminals in the striatum (Cachope et al., 2012; Threlfell et al., 2012), possibly evoking a “burst–pause–burst” response in CINs without changes in the discharge of dopamine neurons.
Our data add to a large body of work describing complex interactions between the dopaminergic and cholinergic systems, and shows that dopamine neurons are not limited to a tonic, permissive role in CIN excitability. Instead, our results imply that phasic dopamine release is transiently paired with a global decrease in cholinergic signaling in the striatum. A greater understanding of the effects of nigrostriatal afferents on striatal circuits will prove essential to elucidate their contribution to movement selection and motor learning in health and disease.
Footnotes
This work was supported by grants from the National Institutes of Health to C.G. (RO1NSO64583) and B.L.S. (NS046579). C.S. was supported by a fellowship from the postdoctoral program of the German Academic Exchange Service (Deutscher Akademischer Austausch Dienst). N.X.T. was supported by a fellowship from the Nancy Lurie Marks Family Foundation. We thank Nicole Mulder for genotyping, and members of the Sabatini laboratory for helpful discussions.
The authors declare no competing financial interest.
References
- Aosaki T, Graybiel AM, Kimura M. Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science. 1994;265:412–415. doi: 10.1126/science.8023166. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Kiuchi K, Kawaguchi Y. Dopamine D1-like receptor activation excites rat striatal large aspiny neurons in vitro. J Neurosci. 1998;18:5180–5190. doi: 10.1523/JNEUROSCI.18-14-05180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Miura M, Suzuki T, Nishimura K, Masuda M. Acetylcholine-dopamine balance hypothesis in the striatum: an update. Geriatr Gerontol Int. 2010;10(Suppl 1):S148–S157. doi: 10.1111/j.1447-0594.2010.00588.x. [DOI] [PubMed] [Google Scholar]
- Apicella P, Deffains M, Ravel S, Legallet E. Tonically active neurons in the striatum differentiate between delivery and omission of expected reward in a probabilistic task context. Eur J Neurosci. 2009;30:515–526. doi: 10.1111/j.1460-9568.2009.06872.x. [DOI] [PubMed] [Google Scholar]
- Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, Tomac AC. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–390. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- Barbeau A. The pathogenesis of Parkinson's disease: a new hypothesis. Can Med Assoc J. 1962;87:802–807. [PMC free article] [PubMed] [Google Scholar]
- Bell MI, Richardson PJ, Lee K. Functional and molecular characterization of metabotropic glutamate receptors expressed in rat striatal cholinergic interneurones. J Neurochem. 2002;81:142–149. doi: 10.1046/j.1471-4159.2002.00815.x. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ. Spontaneous activity of neostriatal cholinergic interneurons in vitro. J Neurosci. 1999;19:5586–5596. doi: 10.1523/JNEUROSCI.19-13-05586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentivoglio M, Morelli M. The organization and circuits of mesencephalic dopaminergic neurons and the distribution of dopamine receptors in the brain. In: Dunnett SB, et al., editors. Handbook of chemical neuroanatomy, vol. 21, dopamine. San Diego: Elsevier; 2005. pp. 1–107. [Google Scholar]
- Bolam JP. Synapses of identified neurons in the neostriatum. Ciba Found Symp. 1984;107:30–47. doi: 10.1002/9780470720882.ch3. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Wainer BH, Smith AD. Characterization of cholinergic neurons in the rat neostriatum. A combination of choline acetyltransferase immunocytochemistry, Golgi-impregnation and electron microscopy. Neuroscience. 1984;12:711–718. doi: 10.1016/0306-4522(84)90165-9. [DOI] [PubMed] [Google Scholar]
- Bonsi P, Cuomo D, Ding J, Sciamanna G, Ulrich S, Tscherter A, Bernardi G, Surmeier DJ, Pisani A. Endogenous serotonin excites striatal cholinergic interneurons via the activation of 5-HT 2C, 5-HT6, and 5-HT7 serotonin receptors: implications for extrapyramidal side effects of serotonin reuptake inhibitors. Neuropsychopharmacology. 2007;32:1840–1854. doi: 10.1038/sj.npp.1301294. [DOI] [PubMed] [Google Scholar]
- Brown MT, Tan KR, O'Connor EC, Nikonenko I, Muller D, Lüscher C. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature. 2012;492:452–456. doi: 10.1038/nature11657. [DOI] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, Cheer JF. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41. doi: 10.1016/j.celrep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Grande C, Usiello A, Gubellini P, Erbs E, Martin AB, Pisani A, Tognazzi N, Bernardi G, Moratalla R, Borrelli E, Calabresi P. Receptor subtypes involved in the presynaptic and postsynaptic actions of dopamine on striatal interneurons. J Neurosci. 2003;23:6245–6254. doi: 10.1523/JNEUROSCI.23-15-06245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Mingote S, Moore H, Rayport S. Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron. 2014;81:901–912. doi: 10.1016/j.neuron.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JY, Haesler S, Vong L, Lowell BB, Uchida N. Neuron-type-specific signals for reward and punishment in the ventral tegmental area. Nature. 2012;482:85–88. doi: 10.1038/nature10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron. 2010;65:230–245. doi: 10.1016/j.neuron.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoer P, Abercrombie ED. Physiological release of striatal acetylcholine in vivo: modulation by D1 and D2 dopamine receptor subtypes. J Pharmacol Exp Ther. 1996;277:775–783. [PubMed] [Google Scholar]
- Dimova R, Vuillet J, Nieoullon A, Kerkerian-Le Goff L. Ultrastructural features of the choline acetyltransferase-containing neurons and relationships with nigral dopaminergic and cortical afferent pathways in the rat striatum. Neuroscience. 1993;53:1059–1071. doi: 10.1016/0306-4522(93)90489-3. [DOI] [PubMed] [Google Scholar]
- Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294–307. doi: 10.1016/j.neuron.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JB, Oh WJ, Sabatini BL, Gu C. Semaphorin 3E-plexin-D1 signaling controls pathway-specific synapse formation in the striatum. Nat Neurosci. 2012;15:215–223. doi: 10.1038/nn.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsáki G, Deisseroth K, Tepper JM, Koos T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci. 2012;15:123–130. doi: 10.1038/nn.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, el Mestikawy S, Bertrand L, Caron MG. Cloning and functional characterization of a cocaine-sensitive dopamine transporter. FEBS Lett. 1991;295:149–154. doi: 10.1016/0014-5793(91)81406-X. [DOI] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30:2223–2234. doi: 10.1523/JNEUROSCI.4870-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Reynolds JN. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience. 2011;198:27–43. doi: 10.1016/j.neuroscience.2011.08.067. [DOI] [PubMed] [Google Scholar]
- Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström MA. Mechanisms for selection of basic motor programs—roles for the striatum and pallidum. Trends Neurosci. 2005;28:364–370. doi: 10.1016/j.tins.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Otaka M, Huang YH, Neumann PA, Winters BD, Grace AA, Schlüter OM, Dong Y. Dopamine triggers heterosynaptic plasticity. J Neurosci. 2013;33:6759–6765. doi: 10.1523/JNEUROSCI.4694-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshua M, Adler A, Mitelman R, Vaadia E, Bergman H. Midbrain dopaminergic neurons and striatal cholinergic interneurons encode the difference between reward and aversive events at different epochs of probabilistic classical conditioning trials. J Neurosci. 2008;28:11673–11684. doi: 10.1523/JNEUROSCI.3839-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshua M, Adler A, Prut Y, Vaadia E, Wickens JR, Bergman H. Synchronization of midbrain dopaminergic neurons is enhanced by rewarding events. Neuron. 2009;62:695–704. doi: 10.1016/j.neuron.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Hikida T, Watanabe D, Ichinose H, Nagatsu T, Kreitman RJ, Pastan I, Nakanishi S. Synaptic integration mediated by striatal cholinergic interneurons in basal ganglia function. Science. 2000;289:633–637. doi: 10.1126/science.289.5479.633. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Large aspiny cells in the matrix of the rat neostriatum in vitro: physiological identification, relation to the compartments and excitatory postsynaptic currents. J Neurophysiol. 1992;67:1669–1682. doi: 10.1152/jn.1992.67.6.1669. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolisnyk B, Guzman MS, Raulic S, Fan J, Magalhães AC, Feng G, Gros R, Prado VF, Prado MA. ChAT-ChR2-EYFP mice have enhanced motor endurance but show deficits in attention and several additional cognitive domains. J Neurosci. 2013;33:10427–10438. doi: 10.1523/JNEUROSCI.0395-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology. 2014;76:351–359. doi: 10.1016/j.neuropharm.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AJ, 3rd, Gu X, Zanella S, Kidney J, Gu H, Mao Y, Hooks BM, Boyden ES, Buzsáki G, Ramirez JM, Jones AR, Svoboda K, Han X, et al. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci. 2012;15:793–802. doi: 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, Surmeier DJ. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci. 2004;24:10289–10301. doi: 10.1523/JNEUROSCI.2155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–143. doi: 10.1016/j.neuron.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Nagy PM, Aubert I. Overexpression of the vesicular acetylcholine transporter increased acetylcholine release in the hippocampus. Neuroscience. 2012;218:1–11. doi: 10.1016/j.neuroscience.2012.05.047. [DOI] [PubMed] [Google Scholar]
- Pakhotin P, Bracci E. Cholinergic interneurons control the excitatory input to the striatum. J Neurosci. 2007;27:391–400. doi: 10.1523/JNEUROSCI.3709-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–1145. doi: 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickel VM, Chan J. Spiny neurons lacking choline acetyltransferase immunoreactivity are major targets of cholinergic and catecholaminergic terminals in rat striatum. J Neurosci Res. 1990;25:263–280. doi: 10.1002/jnr.490250302. [DOI] [PubMed] [Google Scholar]
- Pisani A, Calabresi P, Centonze D, Marfia GA, Bernardi G. Electrophysiological recordings and calcium measurements in striatal large aspiny interneurons in response to combined O2/glucose deprivation. J Neurophysiol. 1999;81:2508–2516. doi: 10.1152/jn.1999.81.5.2508. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Centonze D, Calabresi P, Bernardi G. Activation of D2-like dopamine receptors reduces synaptic inputs to striatal cholinergic interneurons. J Neurosci. 2000;20:RC69. doi: 10.1523/JNEUROSCI.20-07-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Catania MV, Giuffrida R, Morari M, Marti M, Centonze D, Bernardi G, Kingston AE, Calabresi P. Metabotropic glutamate 2 receptors modulate synaptic inputs and calcium signals in striatal cholinergic interneurons. J Neurosci. 2002;22:6176–6185. doi: 10.1523/JNEUROSCI.22-14-06176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Centonze D, Martorana A, Fusco F, Sancesario G, De Persis C, Bernardi G, Calabresi P. Activation of β1-adrenoceptors excites striatal cholinergic interneurons through a cAMP-dependent, protein kinase-independent pathway. J Neurosci. 2003;23:5272–5282. doi: 10.1523/JNEUROSCI.23-12-05272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Ravel S, Legallet E, Apicella P. Responses of tonically active neurons in the monkey striatum discriminate between motivationally opposing stimuli. J Neurosci. 2003;23:8489–8497. doi: 10.1523/JNEUROSCI.23-24-08489.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Schulz JM, Reynolds JN. Pause and rebound: sensory control of cholinergic signaling in the striatum. Trends Neurosci. 2013;36:41–50. doi: 10.1016/j.tins.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, Surmeier DJ. Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci. 2007;10:1458–1466. doi: 10.1038/nn1972. [DOI] [PubMed] [Google Scholar]
- Stoof JC, Drukarch B, de Boer P, Westerink BH, Groenewegen HJ. Regulation of the activity of striatal cholinergic neurons by dopamine. Neuroscience. 1992;47:755–770. doi: 10.1016/0306-4522(92)90027-Y. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30:8229–8233. doi: 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MA, Chen H, Morikawa H. Recurrent inhibitory network among striatal cholinergic interneurons. J Neurosci. 2008;28:8682–8690. doi: 10.1523/JNEUROSCI.2411-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallini YN, Shui B, Greene KS, Deng KY, Doran R, Fisher PJ, Zipfel W, Kotlikoff MI. BAC transgenic mice express enhanced green fluorescent protein in central and peripheral cholinergic neurons. Physiol Genomics. 2006;27:391–397. doi: 10.1152/physiolgenomics.00092.2006. [DOI] [PubMed] [Google Scholar]
- Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, Berlin J, Deisseroth K, Rice ME, Tepper JM, Koos T. Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J Neurosci. 2010;30:7105–7110. doi: 10.1523/JNEUROSCI.0265-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Tecuapetla F, Koós T, Ibáñez-Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2011;4:150. doi: 10.3389/fnana.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F, Ghiglieri V, Giampà C, Fusco FR, Picconi B, Calabresi P. The distinct role of medium spiny neurons and cholinergic interneurons in the D(2)/A(2)A receptor interaction in the striatum: implications for Parkinson's disease. J Neurosci. 2011;31:1850–1862. doi: 10.1523/JNEUROSCI.4082-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through noncanonical release of GABA. Nature. 2012;490:262–266. doi: 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Oh WJ, Gu C, Sabatini BL. Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. eLife. 2014;3:e01936. doi: 10.7554/eLife.01936. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschentke TM. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol. 2001;63:241–320. doi: 10.1016/S0301-0082(00)00033-2. [DOI] [PubMed] [Google Scholar]
- Wilson CJ. The mechanism of intrinsic amplification of hyperpolarizations and spontaneous bursting in striatal cholinergic interneurons. Neuron. 2005;45:575–585. doi: 10.1016/j.neuron.2004.12.053. [DOI] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf NJ, Butcher LL. Cholinergic neurons in the caudate-putamen complex proper are intrinsically organized: a combined Evans blue and acetylcholinesterase analysis. Brain Res Bull. 1981;7:487–507. doi: 10.1016/0361-9230(81)90004-6. [DOI] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. D5 dopamine receptors enhance Zn2+-sensitive GABA(A) currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron. 1997;19:1115–1126. doi: 10.1016/S0896-6273(00)80402-X. [DOI] [PubMed] [Google Scholar]
- Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Salas R, Zhang L, De Biasi M, Dani JA. Corelease of dopamine and serotonin from striatal dopamine terminals. Neuron. 2005;46:65–74. doi: 10.1016/j.neuron.2005.02.010. [DOI] [PubMed] [Google Scholar]