

Abstract
We describe a patient with idiopathic pleuroparenchymal fibroelastosis (IPPFE). This rare clinicopathological syndrome is characterized by typical apical alterations op chest imaging, such as pleural thickening and subpleural fibrosis. Thickened visceral pleura and subpleural fibrosis consisting of dense collagen and elastin, are the main histopathological features. Etiology is unknown but a link between recurrent infections (in particular aspergillosis) and autoimmune diseases is suspected. At this time there is no standardized treatment regimen and the prognosis is variable.
Keywords: Pleuroparenchymal fibroelastosis, Interstitial lung disease, Case report
Introduction
In this case report we describe a patient with idiopathic pleuroparenchymal fibroelastosis (IPPFE). We will summarize the patient characteristics and give a brief overview of the cardinal features of this disease.
Case report
A 50-year-old female was referred to our university hospital because of a puzzling case of interstitial lung disease. She presented with a progressive dyspnea on exertion and a non-productive cough since 2 years. There were no other symptoms. She had no relevant history and did not suffer from recurrent respiratory infections. She never smoked nor was she exposed to any potential noxious products or fumes. No major environmental exposures, except a mold problem on a wall in her bedroom were revealed. She worked as a biology teacher and played the flute. There were no symptoms suggesting an underlying systemic disorder.
Upon clinical examination we noted a patient in a good general condition, without signs of chronic hypoxia such as clubbing or cyanosis. Pulmonary auscultation only revealed some fine crackles on the basis of the left lung. Cardiac, abdominal and limb and joint examination was normal. She had no suspect skin lesions. There were no palpable adenopathies.
Pulmonary function testing showed a markedly reduced diffusion capacity (TLco 3.90 mmol/min/Kpa or 45% of the predicted value) and slightly reduced total lung capacity (TLC 4240 ml or 80% of the predicted value).
Extensive biochemical analysis was normal with no signs of (chronic) inflammation. There were no signs of any connective tissue disease. Precipitins and specific IgE's for aspergillus species were negative. There was however an elevated stachybotrys atra precipitin level, probably related to mold exposition in her bedroom.
Bilateral patchy consolidations were noted on a conventional chest radiograph. The alterations were mainly visible in the upper lung zones. A high-resolution chest CT was performed, showing marked bilateral pleural thickening as well as peribronchial cuffing and some (traction) bronchiectasis. Moreover septal thickening was mentioned, most pronounced in the subpleural region (Fig. 1). All these changes were predominantly found in both lung apices and basal regions were relatively spared.
Fig. 1.
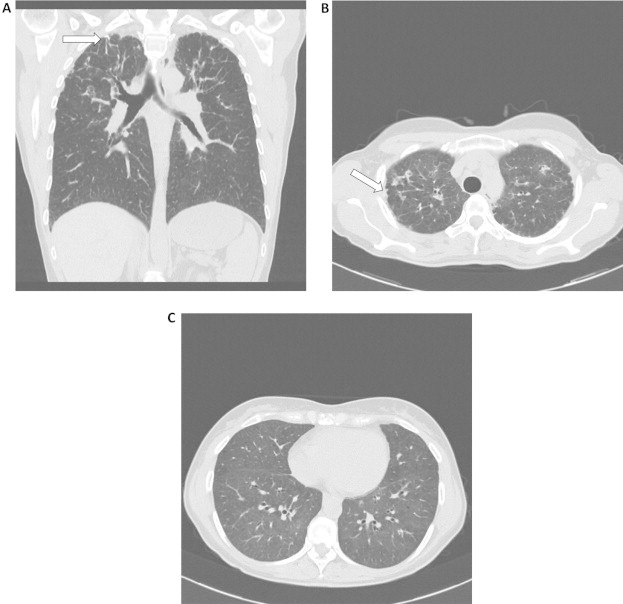
Coronal (A) and axial (B, C) CT scan showing biapical fibrotic pleural and subpleural changes (arrows) together with peri-bronchovascular and linear opacities in the upper part of both lungs and normal lower lung zones.
Bronchoalveolar lavage showed a lymphocytic inflammation (68% lymphocytes) with an increased CD4/CD8 ratio (4.0). Extensive microbiological analysis and transbronchial biopsies were not contributive to diagnosis.
This case was then presented on the interstitial lung diseases multidisciplinary meeting. Based upon these findings, IPPFE was suspected and a video-assisted thoracoscopic surgical (VATS) lung biopsy was proposed to confirm this diagnosis. VATS biopsies were eventually obtained from the right middle and upper lobe. Microscopic examination showed thickened fibrotic pleura. Additional Masson trichrome and elastin staining revealed marked subpleural collagenous and elastinous deposition with an abrupt transition between abnormal and normal lung parenchyma (Fig. 2). Diagnosis of IPPFE was henceforth histopathologically confirmed.
Fig. 2.
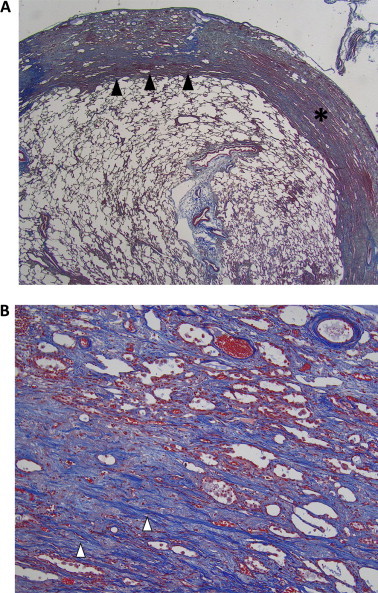
Pathology specimen obtained by video-assisted thoracoscopic surgery. Masson trichrome stain shows a thickened visceral pleura (asterisk) and subpleural deposition of collagen en elastin (white arrowheads) with an abrupt transition between normal and abnormal parenchyma (black arrowheads). A:×12.5 magnification and B:×100 magnification.
Our patient was treated with steroids that were gradually tapered. Currently she still is being treated with methylprednisolone 4 mg once daily. During this treatment the diffusion capacity improved moderately (TLco 4.46 mmol/min/Kpa or 53% of the predicted value after 6 months of treatment). Whether exposition to stachybotrys contributed to disease development remains unclear. Nevertheless sanitation of her room was recommended.
Discussion
IPPFE is a rare clinicopathological syndrome first described in 2004 [1] with distinctive radiological and histopathological findings [2,3]. The recent published updated classification of idiopathic interstitial pneumonias (IIP) classifies IPPFE in the group of rare IIP [4].
Presenting symptoms are dyspnea on exertion, a dry cough and recurrent respiratory tract infections. Spontaneous pneumothorax and pneumomediastinum have been reported.
Imaging shows upper and middle lobe pleural thickening and subpleural fibrosis, in absence of lower lobe involvement. Honeycombing, traction bronchiectasis and reticular abnormalities are noted.
Histophathological findings are thickened visceral pleura and subpleural fibrosis consisting of dense collagen and elastin (hence fibroelastosis). Transition from pathological to normal parenchyma is abrupt. Fibroblast foci and lymphocytic inflammation is variably observed.
Etiology is unknown but recurrent infections (in particular by aspergillus species), autoimmune diseases and genetic predisposition seem to be linked. Several case reports of patients who developed IPPFE after they underwent bone marrow transplantation have been published [5]. Pleuroparenchymal fibroelastosis is also reported in lung transplant patients suffering from restrictive allograft syndrome [6].
There is no consensus about treatment, although corticosteroids are routinely used. Reddy et al [3] suggested that patients with infection or autoimmunity require a specific approach. Patients with a family history of IPPFE deserve a close follow-up due to a more aggressive disease course. Due to the extreme rarity of this syndrome, an experienced multidisciplinary team is essential in (timely) identifying this disease as well as establishing an adequate approach and follow-up. Therefore it is our belief that (early) referral of IIP patients, especially when posing diagnostic difficulties, to a center with an expert multidisciplinary panel is indicated.
In summary, we present a patient with IPPFE, a rare clinicopathological syndrome. This case demonstrates the importance of multidisciplinary approach in interstitial lung diseases.
Acknowledgment
Wim Wuyts is a Senior Clinical Investigator of the Research Foundation – Flanders (1.8.325.12N) and holder of the Crystal Chair in Interstitial lung diseases.
References
- 1.Frankel S., Cool D., Lynch D., Brown K. Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest. 2004;126:2007–2013. doi: 10.1378/chest.126.6.2007. [DOI] [PubMed] [Google Scholar]
- 2.Piciucchi S., Tomassetti S., Casoni G., Sverzellati N., Carloni A., Dubini A. High resolution CT and histological findings in idiopathic pleuroparenchymal fibroelastosis. Respir Res. 2011;12:111. doi: 10.1186/1465-9921-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reddy T., Tominaga M., Hansell D., von der Thusen J., Rassl D., Parfrey H. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J. 2012;40:377–385. doi: 10.1183/09031936.00165111. [DOI] [PubMed] [Google Scholar]
- 4.Travis W.D., Costabel U., Hansell D.M., King T., Lynch D., Nicholson A. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Von der Thüsen J.H., Hansell D.M., Tominaga M., Veys P., Ashworth M., Owens C. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol. 2011;24(12):1633–1639. doi: 10.1038/modpathol.2011.114. [DOI] [PubMed] [Google Scholar]
- 6.Ofek E., Sato M., Saito T., Wagnetz U., Roberts H., Chaparro C. Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Mod Pathol. 2013 Mar;26(3):350–356. doi: 10.1038/modpathol.2012.171. [DOI] [PubMed] [Google Scholar]