

Abstract
Background
Prion diseases are fatal neurodegenerative disorders whose pathogenesis mechanisms are not fully understood. In this context, the analysis of gene expression alterations occurring in prion-infected animals represents a powerful tool that may contribute to unravel the molecular basis of prion diseases and therefore discover novel potential targets for diagnosis and therapeutics. Here we present the first large-scale transcriptional profiling of brains from BSE-infected cynomolgus macaques, which are an excellent model for human prion disorders.
Results
The study was conducted using the GeneChip® Rhesus Macaque Genome Array and revealed 300 transcripts with expression changes greater than twofold. Among these, the bioinformatics analysis identified 86 genes with known functions, most of which are involved in cellular development, cell death and survival, lipid homeostasis, and acute phase response signaling. RT-qPCR was performed on selected gene transcripts in order to validate the differential expression in infected animals versus controls. The results obtained with the microarray technology were confirmed and a gene signature was identified. In brief, HBB and HBA2 were down-regulated in infected macaques, whereas TTR, APOC1 and SERPINA3 were up-regulated.
Conclusions
Some genes involved in oxygen or lipid transport and in innate immunity were found to be dysregulated in prion infected macaques. These genes are known to be involved in other neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases. Our results may facilitate the identification of potential disease biomarkers for many neurodegenerative diseases.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-434) contains supplementary material, which is available to authorized users.
Keywords: Prion diseases, BSE, Non-human primates, Neurodegeneration, Transcriptome, Microarray, RT-qPCR, Biomarker, Serpina3, Hemoglobin
Background
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are incurable and fatal neurodegenerative disorders that affect both humans and animals; their origin may be sporadic, acquired or genetic [1, 2]. TSEs include Creutzfeldt-Jakob Disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), kuru and fatal familial insomnia (FFI) in humans [2], bovine spongiform encephalopathy (BSE) in cattle [3], scrapie in sheep and goats [4], chronic wasting disease (CWD) in cervids [5], transmissible mink encephalopathy, and feline spongiform encephalopathy (FSE) [6].
A major event that leads to the development of prion diseases is the conversion of the cellular form of the prion protein (PrPC) into an abnormally folded, β-sheet enriched and protease resistant isoform (PrPSc). PrPSc is prone to accumulate and aggregate in the brain of affected individuals [1, 2, 4] leading to neuronal loss, spongiosis and astrogliosis, which are hallmarks of neurodegeneration. The underlying conversion mechanism of PrPC into PrPSc is poorly understood and it is further complicated by the existence of several different strains characterized by distinct tertiary and quaternary structures as well as different clinical patterns [7, 8]. Several hypotheses exist about the contribution of unknown molecules other than PrP to prion propagation [9–11]. To address this issue, several animal studies have investigated the host response to prion infection of different origin and strain. The differential transcription profile after prion infection has been extensively explored (reviewed in [6, 12]); however, most of the studies involved animal models such as mice [13–18], sheep [4, 19–22] and cattle [23–28], all not closely related to humans. Some expression analyses have been conducted in non-human primates focusing mainly on the susceptibility to the infection and the variety of clinical symptoms [29–33], but none has investigated large-scale transcriptome changes due to prion infection. All these investigations suggest that besides the PrP-encoding gene (PRNP in humans), other genes are key players and contribute to the genetic susceptibility to acquired TSEs [6, 34]. The main genes identified so far are related to oxidative stress, mitochondrial apoptotic pathways, endosome/lysosome function, immunity, synapse function, metal ion binding, activated cholesterol biosynthesis, immune and inflammatory response, protease inhibitors, calcium binding proteins, regulation of the actin cytoskeleton, ion transport, cell adhesion, and transcription processes [6]. Dysregulation of these genes seems to cause increased oxidative stress that in turn determines oxidation of proteins, lipids and DNA as well as mitochondrial dysfunction and ER stress [6]. Apart from TSEs, transcriptional changes of these genes are common to other neurodegenerative pathologies [12] and, together with functional proteomics data, may help to identify novel selective biomarkers of prion diseases and neurodegeneration in general.
To accomplish that, we performed a large-scale transcriptional profiling in BSE-infected cynomolgus macaques (Macaca fascicularis). They are known to be an excellent model for studying human acquired prion diseases [32, 33, 35–37], as shown by BSE transmission via the intracranial and oral routes, which lead to a disease pattern comparable to that of human maladies in terms of preclinical incubation time, clinical symptoms and pathophysiology [35]. The objective of this study was to identify genes that are differentially expressed in brain tissue of intracranially infected monkeys compared to non-infected ones using an unbiased genomic approach such as expression microarrays with subsequent data validation by RT-qPCR. Our study aims at revealing biological processes that are relevant to the pathogenesis of human prion diseases using a systematic approach that connects the identified DEGs into potential networks of interacting pathways. This may allow us to discover novel selective markers as potential targets for diagnostic and therapeutic strategies.
Results
PrPSc content in brain tissue
The relative amount of PrPSc in brain homogenate of 6 BSE-infected macaques was examined by Western Blot. Densitometric analysis of the monoglycosylated band revealed that the relative amount of PrPSc strongly differed between the individual macaques. We wondered whether this discrepancy might be due to the preclinical incubation time or rather correspond to the gradual accumulation of PrPSc during the clinical phase of disease as reported for sCJD [38, 39]. As anticipated, we found a significant correlation between PrPSc content and the duration of the symptomatic phase (Figure 1). The correlation analysis includes only the 6 intracranially inoculated macaques. Since these animals were housed in one social group, environmental factors, which may influence the disease course and duration, are identical. Such factors can be different for the orally inoculated animal, which was therefore omitted from the analysis. The infected animals were at an advanced stage of prion disease and the details of their clinical course have been previously described [33]. Briefly, animal A1 showed the shortest duration of disease (17 days) and a short pre-clinical incubation time (931 days) together with the lowest PrPSc content, while animal A5 showed the longest survival period (143 days), compared to an average clinical phase of about 90 days, together with the highest PrPSc content and the second longest pre-clinical phase (1340 days).
Figure 1.
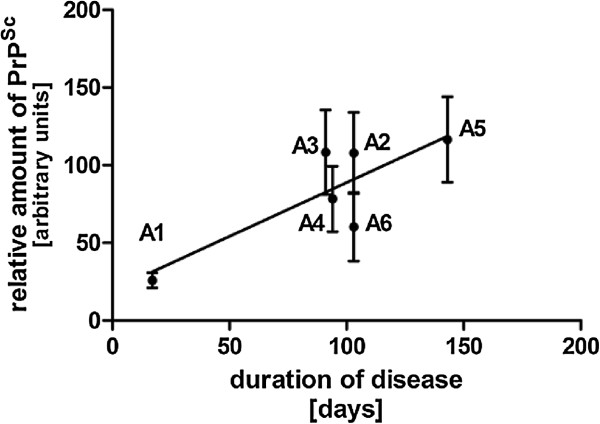
Correlation between PrP Sc content and duration of clinical phase. Western Blot analysis from PK-treated homogenates of brain samples derived from BSE-infected macaques was performed. The monoglycosylated bands of PrPSc were analyzed densitometrically. Relative amounts of PrPSc from brain homogenates were averaged and correlated to the disease duration.
Microarray analysis of brain gene expression in cynomolgus macaques
To investigate differential mRNA expression in BSE-infected macaques we used brain samples from 6 animals that were intracranially challenged [33]. One macaque that was orally infected with 50 mg BSE-homogenate was also included in our study. For comparison purposes, we used 5 brain samples derived from non-infected age- and sex-matched control macaques.
RNA was isolated from the gyrus frontalis superior of all animals and checked for quality by nano-scale electrophoresis, which resulted in an overall RNA Integrity Number (RIN) of about 6. This value is indicative of at least partially degraded RNA within the sample; one possible reason for the reduced RNA integrity may be the procedure utilized to remove the gyrus frontalis superior region from the frozen tissue slide. The biopsy stamp was plugged into a cordless screwdriver that was used to drill a borehole in the frozen tissue block of +/− 1 cm height. This method was chosen to ensure that the material did not thaw; however, the local heat induced by the rotating biopsy stamp may have led to substantial degradation of the RNA. Nonetheless, human brain material exhibiting a comparable RIN value was successfully used for similar studies [40]. All samples were analyzed using the GeneChip® Rhesus Macaque Genome Array (Affymetrix®) that contains 52,024 rhesus probe sets to enable gene expression studies of Macaca mulatta transcriptome interrogating more than 47,000 transcripts. The genomes of M. mulatta and M. fascicularis exhibit a small genetic divergence of approximately 0.4% [41, 42] that presumably allows for the detection of homologue transcripts with high specificity.
Raw data were quality checked and analyzed using Affymetrix® proprietary analysis tools, a hierarchical clustering was performed and a heat map was generated. Then the signals were aligned to the annotation library and a spreadsheet containing gene symbols, p-values and expression fold changes was created. Microarray data were submitted to Gene Expression Omnibus (GEO). The bioinformatics analysis identified 300 probe sets that were up- or down-regulated about twofold (≥ |1.95|). Because among them no candidate appeared using FDR 0.05, we chose as criteria an unadjusted p-value of ≤ 0.005 together with a fold change ≥ |2.0|. Additional file 1 lists the resulting 86 probe sets that were then used to generate the heat map shown in Figure 2.
Figure 2.
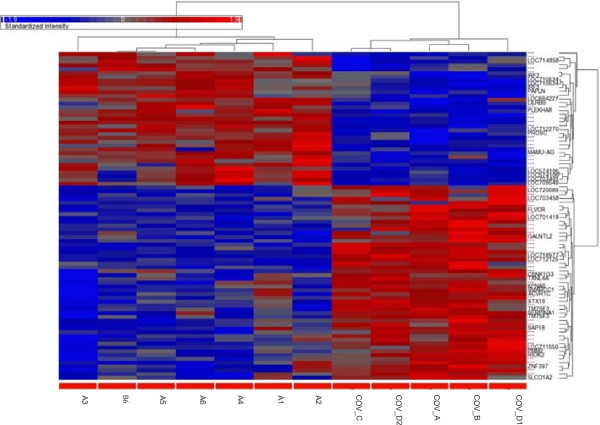
Condition trees of the clustering analysis. The cluster analysis was performed using a hierarchical approach with the average linkage-method (R and Partek® Software, Partek® Inc.): 86 probe sets showed a differential expression with FC ≥ 2. The color represents the level of expression (red: up-regulation, blue: down-regulation) and the sample information is listed across the bottom. The names of the known genes are indicated. More details on all genes are reported in Additional file 1.
Functional classification of differentially expressed genes (DEGs)
We used the Ingenuity Pathways Analysis (IPA®, see section: Availability of supporting data) to annotate genes according to their functional relationships and to determine potential regulatory networks and pathways. Among the 300 differentially expressed (about twofold, (≥ |1.95|) probe sets identified, 105 were associated to mapped IDs; 53 of the latter were identified as network eligible genes, while 86 were identified as function eligible genes. It should be emphasized that the designation of functional class in the present study is neither definitive nor exclusive, as annotation of gene function is incomplete, and multifunctional gene products can be involved in several cellular pathways. First, we identified key biological functions and/or diseases that contain a disproportionately high number of genes from the DEG list compared to the total gene population from the microarray. The analysis was started by identifying the top categories (p < 0.01) of DEGs within three main classes. In the “Diseases and Disorders” class the categories were cancer and developmental disorder, while within the “Molecular and Cellular functions” class most genes were involved in cellular development and cell death/survival. The main categories for the “Physiological System Development and Function” class were tissue morphology as well as nervous system development and function. As a second step, genes were clustered in relation to the main pathways they belong to: the top two canonical pathways in our DEG list were LXR/RXR activation, which is associated with lipid metabolism and transport, and acute phase response signaling.
Identification of biologically relevant networks
To further investigate the global expression response to BSE infection and to define interactions among the identified specific pathways containing the regulated genes, potential networks of interacting DEGs were identified using IPA®. All potential networks with score > 9 (a score ≥ 3 was considered significant, p < 0.001) are listed in Table 1 with information on network genes, score, focus molecules and top functions associated with the focus genes in each network. The highest ranked network identified by IPA® was associated with tissue morphology (specifically the determination of cell quantity), developmental disorder and biological processes controlling cell death and survival (Figure 3a). This network contained genes that are known to be involved in several neurological diseases and nervous system functions, as shown in Figure 3b.
Table 1.
List of 3 Ingenuity networks generated by mapping the focus genes that were differentially expressed between non-infected and BSE-infected samples
| ID | Molecules in network | Score | Focus molecules | Top functions |
|---|---|---|---|---|
| 1 | ACVR1C, AKR1D1, Alp, AMPK, Ap1, APOC1, Calcineurin protein(s), CARTPT, caspase, CD3, CHI3L1, Creb, cytochrome C, DACH1, DLK1, ERK, ERK1/2, F13A1, Focal adhesion kinase, GNRH1, HBA1/HBA2, HBB, HDL, hemoglobin, HEY2, HINT1, HIPK2, Ikk (family), IL1, IRF3, Jnk, KDELR2, LDL, LGALS1, Mapk, MEF2C, Mek, MET, MT2A, N4BP1, NADPH oxidase, NGFR, NR4A2, OTX2, P38 MAPK, p85 (pik3r), Pdgf (complex), PDGF BB, PI3K (complex), PI3K (family), PIK3R3, Pkc(s), PLC gamma, PON3, Pro-inflammatory Cytokine, Ras, SERPINA1, SERPINA3, Shc, SHOC2, SLCO1A2, Sos, STK4, TCF, TCR, TNFSF10, TTR, TWIST1, Vegf, WSB1 | 71 | 35 | Tissue Morphology, Cell Death and Survival, Developmental Disorder |
| 2 | ABR, ACTL6B, ARMC6, ASB6, C10orf137, C6orf211, CAMKV, CHMP2A, CLIC4, CLPP, CSNK1G3, CTBP2, DCLRE1A, DDX19B, DGKE, ECT2, FHL3, FLVCR1, GALNTL5, GLOD4, HEATR6, HSP90AA1, HSPA12A, ITFG1, KLF3, KPNA6, MCTS1, MEIG1, METTL7B, MRPL44, MXD3, MYBPC1, NCLN, NIPBL, NOL4, OSBPL10, PCBP3, PLEKHA8, PMM2, POLR2J, PPAP2C, PRCP, PROSC, RAI2, SAP18, SCAND1, SEPT6, SGTB, SMARCC1, SMC3, SPATA22, SPSB3, SRPK3, SSU72, STAG1, TATDN1, TESPA1, TM7SF3, TNK1, TNNI3K, TP53BP1, TRAPPC2L, TRIP12, TUFM, TXNL4A, UBC, ZNF131, ZNF235, ZNF397, ZNF420 | 54 | 28 | Developmental Disorder, Hereditary Disorder, Hematological Disease |
| 3 | 26 s Proteasome, ADCY, AKR1C1/AKR1C2, Akt, APP, ARL4C, Arntl-Clock, AVP, AVPR1B, CACNA1B, CAMKV, CARTPT, CBLN2, CEACAM6, CLDN10, CLOCK, COX4I2, CTF1, DNAJC12, endocannabinoid, estrogen receptor, FAM46A, FSH, GABRE, GNA15, GPR158, GPX1, GPX2, GSK3A, Histone h3, HMGCR, HNF4A, HSPA12A, Insulin, JPH3, KCNC3, KCNS1, LINGO1, LPAR1, LXN, MGAT2, miR-125b-5p (and other miRNAs w/seed CCCUGAG), Mmp, MST1, NFkB (complex), Npff, OPN1LW, PDX1, PIK3R5, Pka, PKM, PLC, Proinsulin, RAB39A, RAI2, RIOK2, RUFY3, SERPINA3, SMAD5, SMC4, SOX7, SYT17, TCF19, Tnfrsf22/Tnfrsf23, TOR2A, tretinoin, trypsin, TXNL4B, ZBTB44, ZFHX3 | 36 | 21 | Cellular Development, Neurological Disease, Skeletal and Muscular System Development and Function |
Names in lowercase are genes/molecules that are not from the DEG list but are associated with some of them within pathways identified by Ingenuity Pathway Knowledge Base (IPKB).
Figure 3.
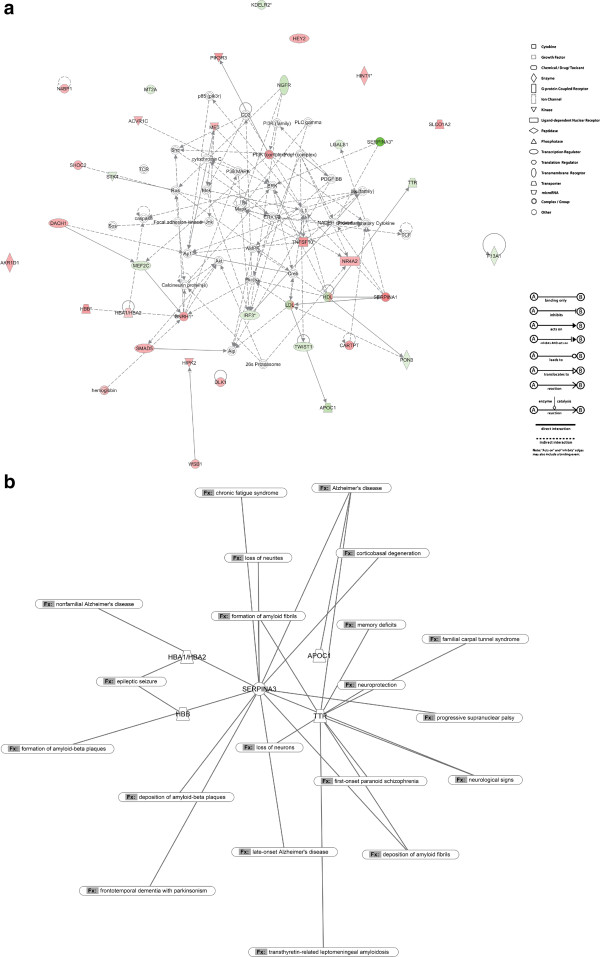
Identification of biologically relevant networks. (a) Top ranking network generated by mapping the focus genes that were differentially expressed in infected animals. Pathway analysis based on the Ingenuity Pathway Knowledge Base (IPKB) is shown. Color shading corresponds to the type of dysregulation: red for up-regulated and green for down-regulated genes according to the microarray fold change calculation method. White open nodes are not from the list of 300 DEGs, but are transcription factors that are associated with the regulation of some of these genes identified by IPKB. The shape of the node indicates the major function of the protein. A line denotes binding of the products of the two genes, while a line with an arrow denotes 'acts on'. A dotted line denotes an indirect interaction. (b) Schematic representation of nervous system-related functions for selected DEGs. The most regulated/interesting DEGs were selected and associated to known nervous system-related functions according to the Ingenuity Pathway Knowledge Base (IPKB) software.
Validation of differentially expressed genes by RT-qPCR
To further confirm the array results using an independent and more sensitive technique, we decided to perform RT-qPCR for a subset of differentially expressed genes. This subset (Additional file 2) was selected in subsequent steps: first, among the 86 probe sets identified during the microarray analysis (Additional file 1) we selected the top 36 with fold change ≥ |2.5| and p ≤ 0.005. Then, after realizing that many were not annotated or did not have a known function, we extended the selection to additional 29 probe sets having fold change ≥ |2.5| but 0.005 ≤ p ≤ 0.05; for the same reasons stated above, we extended the list of candidates one more time using as criteria fold change ≥ |2.5| and p > 0.05 (24 candidates). At this point, having still some cDNA available and only 13 feasible candidate transcripts, we added seven probe sets, corresponding to 5 additional transcripts, selected among the ones with a slightly lower fold change (FC ≥ 2 for at least 1 probe) but possessing an interesting function as revealed by the IPA® analysis or according to the literature. Lastly, HBA2 was added to the list because of its tight relationship with one of the previously selected genes of the hemoglobin complex (HBB), as revealed in the top ranking network from the IPA® analysis (Figure 3a). In summary, we designed RT-qPCR assays for 19 genes (Table 2) and most of them were already known to be involved in neurodegenerative disorders or nervous system regulation, even though very few had been implicated in prion diseases. Among these, we were able to successfully analyze only 11 (reported in Table 3 together with 2 housekeeping genes, ACTB and GAPDH), since the RT-qPCR assays for the remaining 8 genes either showed too low expression (CT > 35) or amplification of trace amounts of residual gDNA. Furthermore, because several gene names have changed since the first annotation was done, updated names from the latest Affymetrix® annotated library are provided in Additional file 2, together with the old ones.
Table 2.
Candidate genes for validation
| Gene | Accession number | Known relation with PrP/nervous system | References |
|---|---|---|---|
| AKR1C1 | NM_001195574.1 | Putative role in myelin formation | [43] |
| HBB | NM_001164428.1 | Putative role in intraneuronal oxygen homeostasis, reduced in Alzheimer's and Parkinson's disease | [44] |
| NCAM1 | XM_001083697.2 | PrP/N-CAM complexes found in prion infected N2a cells | [45] |
| NR4A2 | NM_001266910.2 | Mutations related to dopaminergic dysfunction, including Parkinson schizophrenia and depression | [46, 47] |
| USP16 | NM_001260999.2 | Depletion of USP16 prevented ATMi from restoring transcription after DSB induction | [48] |
| CALB1 | XM_001085269.2 | Plays a protective role in neurodegenerative disorders (depleted in HD) | [49] |
| DACH1 | XM_001082371.2 | Required for normal brain development | [50] |
| LXN | NM_001266988.1 | Marker for the regional specification of the neocortex | [51] |
| PIK3R3 | NM_001266826.1 | Linked to β-amyloid plaque formation in AD brain | [52] |
| SAP18 | NM_001261034.1 | Possibly related to AD | [53] |
| SERPINA3 | NM_001195350.1 | Increased in schizophrenia, SNPs affecting onset and duration of AD | [54, 55] |
| TNFSF10 | NM_001266034.1 | Implicated in pathogenesis of MS (causing demyelination) | [56] |
| HBA2 | NM_001044724.1 | Putative role in intraneuronal oxygen homeostasis, reduced in Alzheimer's and Parkinson's diseases | [44] |
| GNRH1 | NM_001195436.1 | Key regulator of the reproductive neuroendocrine system in vertebrates | [57] |
| IRF3 | NM_001135797 | Putative protective role against prion infection | [58] |
| APOC1 * | AK240617.1 | Binds to ApoE, risk factor for Alzheimer's disease | [59] |
| TM7SF3 | XM_001099269.2 | - | - |
| MYBPC1 | XM_001091952.1 | - | - |
| TTR | NM_001261679 | Amyloid neuropathies, interaction with Aβ | [60] |
List of 19 identified genes selected on the basis of fold change value and known relevance for neurodegeneration. Because of very low signal (LXN, PIK3R3, TNFSF10, GNRH1) or lack of reliable sequence data (CALB1, DACH1, TM7SF3, MYBPC1), only 11 genes (in bold) were successfully analyzed. *Macaca fascicularis transcript.
Table 3.
Genes analyzed by RT-qPCR
| Gene | Chromosome | Primer sequence | Amplicon length (bp) | Accession number | |
|---|---|---|---|---|---|
| ACTB | 3 | F: | GTTGCGTTACACCCTTTCTTG | 146 | NM_001033084.1 |
| R: | CTGTCACCTTCACCGTTCC | ||||
| GAPDH | 11 | F: | CCTGCACCACCAACTGCTTA | 74 | NM_001195426.1 |
| R: | CATGAGTCCTTCCACGATACCA | ||||
| AKR1C1 | 9 | F: | CCGCCATATTGATTCTGCTCAT | 132 | NM_001195574.1 |
| R: | TGGGAATTGCACCAAAGCTT | ||||
| HBB | 14 | F: | GTCCTCTCCTGATGCTGTTATG | 102 | NM_001164428.1 |
| R: | TTGAGGTTGTCCAGGTGATTC | ||||
| NCAM1 | 14 | F: | GAGCAAGAGGAAGATGACGAG | 150 | XM_001083697.2 |
| R: | GACTTTGAGGTGGATGGTCG | ||||
| NR4A2 | 12 | F: | CCAGTGGAGGGTAAACTCATC | 145 | NM_001266910.2 |
| R: | AGGAGAAGGCAGAAATGTCG | ||||
| USP16 | 3 | F: | GCAGAACTTGTCACAAACACC | 146 | NM_001260999.2 |
| R: | CTAAAGTAAGAGGGCCTGGAG | ||||
| SAP18 | 17 | F: | GGAAATGTACCGTCCAGCGA | 109 | NM_001261034.1 |
| R: | TGCCCTTCTTTCTAGCTTCTGG | ||||
| SERPINA3 | 7 | F: | GCTGGGCATTGAGGAAGTCT | 123 | NM_001195350.1 |
| R: | GTGCCCTCCTCAGACACATC | ||||
| HBA2 | 20 | F: | CGACAAGAGCAACGTCAAGG | 126 | NM_001044724.1 |
| R: | TCGAAGTGGGGGAAGTAGGT | ||||
| IRF3 | 19 | F: | TGGGTTGTGTTTAGCAGAGG | 90 | NM_001135797 |
| R: | GAAAAGTCCCCAACTCCTGAG | ||||
| APOC1* | 19 | F: | TTCTGTCGATGGTCTTGGAAG | 138 | AK240617.1 |
| R: | CACTCTGTTTGATGCGGTTG | ||||
| TTR | 18 | F: | TCACTTGGCATCTCCCCATTC | 114 | NM_001261679 |
| R: | GGTGGAATAGGAGTAGGGGCT | ||||
Primers (F: forward and R: reverse) used for gene amplification, amplicon length and GenBank® accession numbers of the macaque cDNA sequences used for primer design. All primers were designed according to the genome sequence of Macaca mulatta.
*Apolipoprotein C-I (APOC1) primers were designed according to the genome sequence of Macaca fascicularis because the Macaca mulatta mRNA sequence was not annotated (TSA Macaca mulatta Mamu_450725, accession number: JV045807.1). Homology between the two sequences was 99%.
In order to achieve optimal RT-qPCR conditions we performed titration of template and primers as well as optimization of cycling conditions using human cDNA from SH-SY5Y neuroblastoma cells (macaque cDNA was scarce). To assess the specificity of the chosen oligonucleotides prior to performing the quantitative assays, some reactions were carried out using macaque cDNA obtained from control animals to verify the correct amplicon length. Two housekeeping genes, GAPDH and ACTB[61], were used as reference genes to normalize RT-qPCR data. Both genes were monitored across samples derived from infected and control macaques in order to evaluate their expression stability, yielding very similar results (Additional file 3).
At this point we performed the quantitative analysis and in general we observed large intra-assay variability for most genes across different samples, both for infected (Additional file 4) and for control animals (Additional file 5). Interestingly, we found a completely different expression pattern for B6, the only orally-infected sample, compared to the intracranially infected animals, except for a couple of genes (AKR1C1, NCAM1), suggesting that the route of infection might play a role in determining the gene expression changes (Additional file 6). Therefore we decided to rerun the microarray clustering analysis excluding this animal in order to verify its influence on the final results. As shown in Additional file 7, the comparison of the clustering analysis with (panel A) and without (panel B) the orally challenged animal B6 does not show marked differences.
Using SYBR® Green-based RT-qPCR we confirmed the statistically significant up-regulation of TTR (FC = 7.11), SERPINA3 (FC = 18.73) and APOC1 (FC = 6.33) as well as the down-regulation of HBB (FC = 0.19) and HBA2 (FC = 0.22), normalizing the data against GAPDH (Figure 4). Similar results were obtained against ACTB (Additional file 8). For all the other genes the RT-qPCR results confirmed the regulation trend of the microarrays, but without statistical significance (p-value > 0.05).
Figure 4.
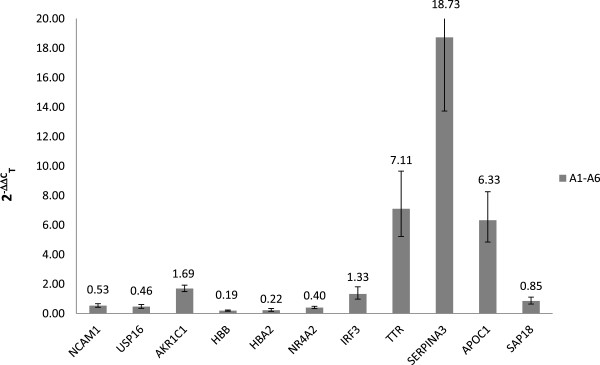
SYBR® Green-based RT-qPCR validation of microarray results. Relative expression levels of 11 genes normalized against GAPDH in BSE-infected cynomolgus macaques.
In order to confirm the SYBR® Green -based results we performed an additional RT-qPCR analysis using FAM-labeled TaqMan® probes, providing more sensitive and specific detection signals for those genes that showed a significant fold change. Using this approach we confirmed the regulation of SERPINA3, APOC1, HBB and HBA2, but not of TTR, which showed comparable trends in FC but lost statistical significance (Figure 5). This may be due to higher variability among triplicates, caused by CT values higher than 35 obtained with the TaqMan® probe chemistry compared to SYBR® Green detection system (Additional file 9).
Figure 5.
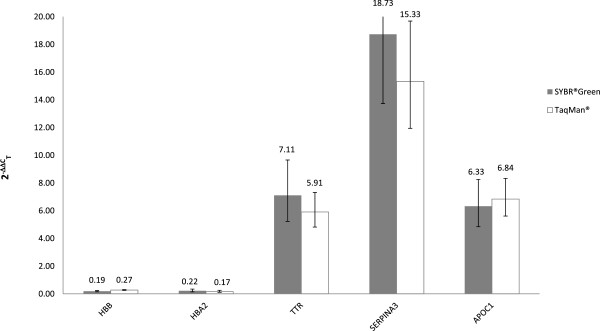
Comparison between SYBR® Green-based and TaqMan® probe-based results. TaqMan® (white) versus SYBR® Green-based (grey) expression levels for each transcript. Both detection systems yielded similar results. Data are normalized against GAPDH. Similar results were obtained with normalization against ACTB (data not shown).
In general, we were able to confirm the results of the array platform obtaining consistent fold change values for all genes analyzed, even though we validated with statistical significance using the specific TaqMan® detection system only four of them: HBB, HBA2, APOC1, SERPINA3 (see Table 4 for details on p-values and FC).
Table 4.
RT-qPCR confirmation of microarray results
| Gene symbol | Microarray fold change | RT-qPCR fold change | |||||
|---|---|---|---|---|---|---|---|
| SYBR® Green | TaqMan® | ||||||
| Min | Max | Mean | FC | P value | FC | P value | |
| AKR1C1 | 2.3 | 2.9 | 2.5 | 1.7 | 0.433 | 2.4 | 0.235 |
| HBB | −2.2 | −2.6 | −2.4 | 0.2 | 0.020 | 0.3 | 0.021 |
| NCAM1 | −1.1 | 2.5 | −0.3 | 0.5 | 0.160 | - | - |
| NR4A2 | 1.1 | −2.1 | −1.6 | 0.4 | 0.248 | - | - |
| USP16 | −1.2 | −5.5 | −2.6 | 0.5 | 0.308 | - | - |
| SAP18 | −1.2 | −2.6 | −1.7 | 0.8 | 0.393 | - | - |
| SERPINA3 | 10.0 | 16.0 | 13.0 | 18.7 | 0.0001 | 15.3 | 0.0005 |
| HBA2 | - | - | - | 0.2 | 0.041 | 0.2 | 0.019 |
| IRF3 | 2.0 | 2.1 | 2.0 | 1.3 | 0.123 | - | - |
| APOC1 | 4.3 | - | 4.3 | 6.3 | 0.047 | 6.8* | 0.028* |
| TTR | 3.1 | - | 3.1 | 7.1 | 0.025 | 5.9 | 0.076 |
Differential expression of selected genes analyzed by microarray and RT-qPCR. For microarray analysis, the lowest (Min), the highest (Max) and the average (Mean) fold change values of all the respective probes are shown. For RT-qPCR analysis, fold change (FC) and statistical significance (p-value) for both SYBR® Green and TaqMan® results are shown. In bold are the genes validated with statistical significance. HBA2 was not present in the array chip.
*Normalization performed vs. ACTB only.
In addition, dealing with animals whose brain material isolation may be susceptible to blood contamination, and as several works in the last few years have shown the presence of active transcription within human red blood cells [62], we decided to analyze the samples also for expression of some erythrocyte markers, such as ALAS2 and RHAG, in order to verify the reliability of the results related to the regulation of both chains of hemoglobin (HBB and HBA2). Although the array data for these genes suggested a negligible and virtually identical presence of blood in both control and infected samples, RT-qPCR analysis revealed a small blood contamination (CT ≥ 34 for ALAS2, CT ≥ 36 for RHAG) within two samples, one control (CovD1) and one infected sample (A4) (Additional file 10 and Additional file 11). In light of these results, we performed an additional gene expression analysis for HBB and HBA2 excluding these two samples. As expected, we obtained slightly different results (FC ~ 0.3 for HBB and 0.2 for HBA2 using TaqMan® probes), but a relevant down-regulation still persisted with statistical significance.
Discussion
The precise mechanisms regulating the molecular processes that lead to neurodegeneration in TSEs remain unknown. Genomic approaches represent unbiased and powerful tools to uncover the molecular basis of these complex mechanisms and they may also contribute to discover new biomarkers for these diseases. Several studies have presented genomic analyses of brain tissues from animal models of TSE; a few of them involved the mRNA profiling of cattle BSE [23, 25–27] or ovine scrapie [4, 19–22, 63] whereas the vast majority was performed on rodent-adapted models of prion disease [13–18, 64]. In several of these prion-infected mice, genomic expression profiles revealed the induction of oxidative and endoplasmic reticulum (ER) stress, activated ER and mitochondrial apoptosis pathways as well as activated cholesterol biosynthesis in the CNS of preclinical mice [64].
We report here the first large-scale transcriptome analysis of the superior frontal gyrus of BSE-infected macaques. This region was selected based on its histopathological and functional relevance in the majority of neurodegenerative disorders [65] and because it corresponds to Brodmann areas 10 and 11, known to be involved in strategic processes in memory recall, various executive functions as well as in planning, reasoning, and decision making [66], all processes known to be disturbed by neurodegeneration. In general, RT-qPCR results confirmed the regulation trend seen in the microarray platform for all the 11 genes analyzed with very similar values using either GAPDH or ACTB for normalization. For five of them (HBB, HBA2, TTR, SERPINA3, APOC1) we obtained statistical significance with one or both qPCR detection systems utilized in this study (SYBR® Green and TaqMan® probes) and some of them were involved in the top two canonical pathways identified during the functional classification reported in the Results section: APOC1 and TTR are part of the LXR/RXR activation pathway, which is associated with lipid metabolism and transport, whereas SERPINA3 and TTR are involved in the acute phase response signaling pathway. All the other genes seemed to fall in the grey zone of both platforms and therefore their FC values could not be considered reliable.
When validating the array results by RT-qPCR, the first evidence obtained was a marked variability among the samples of the same group, either control or infected animals. Unlike other animal models, nonhuman primates are usually not inbred. Therefore, differences in the genomic background of the animals in our study may have contributed to the variability in the time of disease onset [33] and in gene expression within the same group. Paradoxically, for some genes that resulted strongly regulated (APOC1, HBB, HBA2) the variability resulted even more accentuated within the control group if compared to that of the infected group. The experimental and control animals were housed in different animal facilities and this may have generated slight differences in diet and/or housing conditions that may have contributed to the above-mentioned effect.
We also reported a peculiar dysregulation pattern of the orally infected sample (B6) for several genes, showing a completely opposite trend compared to intracranially infected animals. Although no data are available for PrPSc deposition in brain or other tissues of this animal, the significantly longer incubation period (1950 days compared to an average of 1100 days for the other animals) could suggest a correlation between the mRNA expression profile and the route of infection [67]. Nonetheless, this different pattern may be due to the age difference at the time of euthanasia: 7.1 +/− 0.7 years for the intracranially infected macaques versus 9.9 years for the orally infected animal.
Concerning hemoglobin (Hb), a few years ago its expression was unexpectedly discovered in mesencephalic dopaminergic neurons of different mouse strains, as well as in rats and humans affected by Parkinson's disease (PD) and multiple sclerosis (MS) [68–70].
Hb expression is known to decrease in neurons of PD, Alzheimer’s disease (AD), argyrophilic grain disease (AGD) and dementia with Lewy bodies (DLB) brains [44] as well as in the CNS of scrapie-infected mice [13, 14]. Also, it has been shown that Hb binds to Aβ enhancing its aggregation and co-localizes in amyloid plaques in AD brains [71]. If we consider a possible similar interaction with β-rich PrPSc isoforms in prion diseases, we can hypothesize that in our animal model down-regulated Hb fails to promote aggregation of the prion protein, thus leading to a higher presence of toxic species like oligomers [72]. Moreover, in PD it has been hypothesized that Hb may act as oxygen storage molecule in oligodendrocytes [68]. Oxygen would be later released to neighboring neurons in hypoxia conditions to maintain the aerobic metabolism [68, 69]. When down-regulated, Hb would not be available for this function and cells would be damaged by the defective oxygen homeostasis. Our results indicated a strong down-regulation (about 70-80% lower expression than normal) of both HBB and HBA2 in symptomatic advanced-stage BSE-infected macaques. The data were analyzed with a very stringent procedure after excluding any major effect of potential blood contamination, thus confirming the robustness of the results.
Taken together, all these data indicate a possible general role for hemoglobin in neurodegenerative disorders, possibly related to an alteration of O2 homeostasis and oxidative metabolism [68]. One point that needs further investigation is whether this alteration (down-regulation) occurs as an early/late consequence of the disease, or may act as a susceptibility factor that influences the onset of the pathology. Furthermore, future studies may investigate the localization of the observed down-regulation in terms of cell population: it could involve neurons as well as astrocytes or microglia.
Another crucial molecule, APOC1, was significantly up-regulated in BSE-infected brains samples compared to controls. Apolipoprotein C-I, whose gene APOC1 is part of the APOE/C-I/C-IV/C-II gene cluster, (apoC-I) is a small 6.6 kDa component of lipoproteins (mainly HDL) that is known to inhibit receptor-mediated lipoprotein clearance, especially particles containing apoE [73]. Increasing evidence indicates a role for this gene in neurodegenerative disorders, especially in AD and MS [74–76]. A disruption in lipid metabolism and signaling is one of the early alterations apparent in many neurodegenerative diseases, including prion diseases [77, 78]; indeed, cholesterol metabolites are investigated by a number of studies aimed at the identification of early biomarkers for neurodegenerative disorders [79–81]. Several genes involved in cholesterol metabolism and lipid biosynthesis have been found to be up-regulated in preclinical scrapie-infected mice [64]. Since APOC1 is able to activate cholesterol esterification via lecithin-cholesterol acyltransferase [75], its up-regulation could lead to an increase in cholesterol biosynthesis, consistent with the concomitant presence of prion disease. In fact, in vitro studies have shown that depletion of cellular cholesterol reduces the conversion of PrPC to PrPSc[82] and evidence exists also in AD, where altered cholesterol metabolism has been found [83]. Hypercholesterolemia has also been shown to influence amyloid precursor protein processing [84]. One explanation for altered cholesterol homeostasis affecting prion disease development could lie in the fact that PrP is localized in cholesterol-rich lipid rafts [85].
SERPINA3, a serpin peptidase inhibitor involved in acute phase response pathways, is another gene that we found highly regulated in our animal model. It is extensively reported to be regulated in other neurodegenerative disease models and in particular it is well known to interact with APP to promote amyloid plaque formation —a hallmark of AD [86]. Indeed, increased levels of SERPINA3 have been found in the brain and peripheral blood of AD patients [87], mainly due to persistent and almost chronic inflammation [88]. In prion disease studies, SERPINA3 was found increased in brains of scrapie-infected mice [77], in mice infected with RML prior to clinical onset [89] as well as in urine and cerebrospinal fluid of CJD patients [90]. Being an acute phase protein, its up-regulation is explained by the onset of an inflammatory condition, particularly as a response of the innate immune system [91]. Interestingly, two β-sheets of SERPINA3 exhibit a polymorphism mimicking changes in the serpin structure that normally occur during the formation of its stable complex with the target proteinase. In this conformation, SERPINA3 can bind Aβ, thus imposing a β-strand conformation that upon dissociation leads to a faster formation of fibrils [86]. Therefore, an intriguing hypothesis may be envisioned in which PrP conversion into β-sheet conformation can be assisted by SERPINA3, which would accelerate the formation of toxic species like PrP oligomers.
Transthyretin, a protein in the same pathway of acute phase response as SERPINA3, was found to be up-regulated at the transcription level in our BSE-infected macaques according to the SYBR® Green assay. Even though we were not able to confirm the statistical significance using the TaqMan® assay, this gene seems to be of interest. Indeed, TTR, carrier of the thyroid hormone thyroxine (T4) in serum and CSF, is associated with systemic amyloidosis in humans [92], but also with an anti-amyloidogenic effect preventing Aβ deposition in neuronal cell cultures [93]. Moreover, increased mRNA and protein levels have been shown in neurons from the AD mouse model ‘APP23’ and in human AD brain with a neuroprotective role [94, 95]. Even in prion models TTR levels have been found strongly increased in the cortex of scrapie-infected mice [15]. Our study now provides indication that up-regulation of TTR may also be found in BSE-infected macaques, further reinforcing the hypothesis of a common mechanism in AD and TSEs. Taken together, these data may suggest innate immune system activation and inflammatory response in these diseases [96], leading to a sustained up-regulation of both SERPINA3 and TTR genes simultaneously: SERPINA3 as inflammation effect, TTR as attempt to neutralize the infectious agent preventing its deposition. However, analysis of the microarray data did not reveal relevant deregulation of other genes typically involved in neuroinflammation and/or immune response, such as cytokines and other mediators. Even though some authors have reported alteration of these pathways [97], in our array IL6, TNFα, GFAP and CD68 showed a fold change < |2|, suggesting that inflammatory responses may not be particularly severe in this model.
One last point that remains to be addressed is the expression of the prion protein gene itself (PRNP in humans) upon infection. Because of shortage of cDNA, we were not able to validate its levels in our samples. Nevertheless, our microarray data did not identify any changes between control and infected samples, at least at the mRNA level. This is in agreement with findings reported for BSE-infected cattle [3], but differs from the situation in sporadic CJD patients who show reduced mRNA expression [97]. Whether this disagreement is related to the host or the infectious agent needs to be explored.
Conclusions
To our knowledge, this is the first genome-wide expression study in the gyrus frontalis superior region of cynomolgus macaques inoculated with BSE. Using microarray and RT-qPCR technologies we identified a gene signature able to distinguish infected macaques from control animals. These results could be extremely helpful in understanding the progression of the disease, allowing for the identification of some key players which, if not being the cause of the onset, could be some of the target genes affected by the disease. Therefore, after deeper investigations to validate these targets at the protein level and confirm their specificity for prion diseases, they may be exploited as potential biomarkers to set up pre-clinical diagnostic tests.
In particular, our findings support the hypothesis of a potential shared mechanism underlying the onset and the development of all neurodegenerative disorders, as the majority of our DEGs are known to be involved in other diseases such as AD or PD. This is in concordance with very recent data supporting the idea of a unifying role of prions in these diseases in general and maybe a prion-like behavior for most neurodegenerative disorders [98]. Furthermore, some of the DEG transcripts we found are present also in blood (hemoglobin, transthyretin, serpin peptidase inhibitor) and among them hemoglobin exhibited decreased expression throughout the entire course of the infection, including preclinical time points, in mouse models. Therefore, there is the intriguing possibility to employ these "readily available" biomarkers for diagnostic purposes, especially if additional studies will confirm the expression level of the proteins encoded by these DEGs in brain and/or blood tissue.
In general, our results suggest that, in order to identify potential biomarkers and drug targets for prion diseases and other neurodegenerative disorders, a combination of various pathways has to be targeted, including oxygen homeostasis, lipid metabolism and inflammation response.
In summary, large-scale transcriptome analyses of human TSEs are rare [97, 99] and primate models are a valid approach to better understand the mechanisms of these fatal diseases. Even with all the limitations discussed above, our BSE-infected macaques are, to our knowledge, the closest available model for human vCJD and these results, obtained with an unbiased methodology as the gene expression microarray technology, are contributing to shed some light on the molecular basis of TSEs as well as neurodegeneration as a whole.
Methods
Ethics statement
Ethics approval for the study was issued by the Lower Saxony Ministry for consumer protection and food safety (509.42502/08/07.98). Animal experimentation was performed in accordance with section 8 of the German Animal Protection Law in compliance with EC Directive 86/609.
Samples
Samples were derived from six BSE-infected macaques, Macaca fascicularis (A1 to A6) that were intracranially inoculated with a single dose of 50 mg brain homogenate (10% wt/vol) [33, 37]. One cynomolgus macaque (B6) was orally inoculated with the same material; inoculation was performed per os, as single dose.
Brain material from five age- and sex-matched non-infected cynomolgus macaques (CovA, CovB, CovC, CovD1, CovD2) was obtained from Covance Laboratory Münster GmbH and processed in an equivalent manner.
Tissue and RNA extraction
At autopsy of seven BSE-infected cynomolgus macaques at advanced stage of disease and five non-infected control animals, one hemisphere of the brain was sliced dorso-ventrally and snap-frozen on dry-ice plates. The gyrus frontalis superior region was macroscopically identified on the frozen tissue and removed via a biopsy stamp. Total RNA (RNA > 200 bases) was isolated by manually homogenizing the material with micro pestles (Kisker Biotech GmbH) in TRIzol (Invitrogen). RNA isolation was performed according to the supplier’s instructions. Following RNA isolation, a DNase I digestion was performed using 1 unit of enzyme per μg RNA (Fermentas) for 30 min at 37°C, and heat inactivated for 5 min at 95°C followed by precipitation with Sodium Acetate/Ethanol. RNA was checked for quantity and purity on a Spectrophotometer 2000 (PEQLAB) and for integrity of the 18S and 28S ribosomal bands by capillary electrophoresis using the 2100 Bioanalyzer (Agilent Technologies).
Immunoblot analysis
PK-treated (50 μg/mL for 1 hour at 37°C) and untreated brain homogenates corresponding to 0.7 mg or 0.3 mg brain tissue, respectively, were separated on 12% Bis/Tris Acrylamide gels (NuPAGE, Invitrogen) and transferred to nitrocellulose membranes (Protran, Schleicher & Schüll, Germany). Detection of macaque PrPSc was performed using the monoclonal anti-PrP antibody 11C6 and a Peroxidase conjugated anti-mouse IgG-antibody (Sigma-Aldrich, Germany). Signal was visualized using a chemiluminescent substrate (Super Signal West Pico, Pierce) and high sensitivity films (Amersham). Densitometric analysis of PrPSc was performed using the Image J program 1.37v.
Microarray analysis using the GeneChip® Rhesus Macaque genome array
Samples were labeled using the GeneChip® 3’IVT Express Kit (Affymetrix®). Reverse transcription of RNA was performed using 500 ng of total RNA to synthesize first-strand cDNA. This cDNA was then converted into a double-stranded DNA template for transcription. In vitro transcription included a linear RNA amplification (aRNA) and the incorporation of a biotin-conjugated nucleotide. The aRNA was then purified to remove unincorporated NTPs, salts, enzymes, and inorganic phosphate. The labeled aRNA of each animal was fragmented (50–100 bp) and hybridized to a GeneChip® Rhesus Macaque Genome Array (Cat N° 900656; Affymetrix®). The degree of fragmentation and the length distribution of the aRNA were checked by capillary electrophoresis using the Agilent 2100 Bioanalyzer (Agilent Technologies).
The hybridization was performed for 16 h at 1 × g and 45°C in the GeneChip® Hybridization Oven 640 (Affymetrix®). Washing and staining of the arrays were performed on the Gene Chip® Fluidics Station 450 (Affymetrix®) according to the manufacturer's recommendations. The antibody signal amplification and washing and staining protocol were used to stain the arrays with streptavidin R-phycoerythrin (SAPE; Invitrogen). To amplify staining, SAPE solution was added twice with a biotinylated anti-streptavidin antibody (Vector Laboratories, Burlingame, CA, USA) staining step in-between. Arrays were scanned using the GeneChip® Scanner 3000 7G (Affymetrix®).
Microarray data analysis
Intensity data from the CEL. files were imported to the Partek® software including a quality control based on internal controls. All chips passed the quality control and were analyzed using the Limma package [100] of Bioconductor [101, 102] and the Partek® software. The microarray data discussed in this paper were generated conforming to the MIAME guidelines and are deposited in the NCBI’s Gene Expression Omnibus (GEO) database [103]. They are accessible through GEO series accession number GSE52436 (see section: Availability of supporting data).
The microarray data analysis consisted of the following steps: 1. quantile method normalization, 2. global clustering and PCA-analysis, 3. fitting the data to a linear model, 4. detection of differential gene expression and 5. over-representation analysis of differentially expressed genes. Quantile-normalization was applied to the log2-transformed intensity values as a method for between-array normalization to ensure that the intensities had similar distributions across arrays.
For cluster analysis, we used a hierarchical approach with the average linkage-method. Distances were measured as 1 - Pearson's Correlation Coefficient. The PCA was performed using the princomp-function in the Partek® software. To estimate the average group values for each gene and assess differential gene expression, a simple linear model was fitted to the data, and group-value averages and standard deviations for each gene were obtained. To find genes with significant expression changes between groups, empirical Bayes statistics were applied to the data by moderating the standard errors of the estimated values [100].
P-values were obtained from the moderated t-statistic and corrected for multiple testing with the Benjamini–Hochberg method [104]. The p-value adjustment guarantees a smaller number of false positive findings by controlling the false discovery rate (FDR). For each gene, the null hypothesis, that there is no differential expression between degradation levels, was rejected when its FDR was lower than 0.05. Because no candidates appeared using FDR 0.05, we made the selection using another p-value (unadjusted p-value ≤ 0.005) and a fold change ≥ |2|.
Reverse transcription and RT-qPCR
Validation by quantitative reverse transcription real-time PCR (RT-qPCR) was performed using gene-specific primer pairs. cDNA synthesis was accomplished using 100 ng RNA, 10 ng random hexamer primer, 2 mM dNTPs, 0.5 U RNase inhibitor and 5 U reverse transcriptase (Bioline) in 1× reaction buffer. For each sample a negative control was carried along by omission of the reverse transcriptase (−RT control).
The cDNA was diluted 1:10 prior to RT-qPCR. Ten ng RNA equivalent was added to the reaction mix including 2× iQ™ SYBR® Green Supermix (Bio-Rad Laboratories, Inc.), 400 nM of the corresponding forward and reverse primer (Sigma), and quantified in technical triplicates on an iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.). All primers used for RT-qPCR are listed in Table 3.
After initial denaturation for 3 min at 95°C, 45 cycles were performed at 95°C for 15 sec and 58°C for 1 min. Differential gene expression of candidates was normalized to GAPDH and ACTB expression. –RT controls were included in the plates for each primer pair and sample. The relative expression ratio was calculated using the ΔΔCT method [105, 106]. Significance was calculated with the unpaired student t-test (p < 0.05). Melting curve analysis and gel electrophoresis of amplification products were performed for each primer pair to verify that artificial products or primer dimers were not responsible for the signals obtained. Some results were further confirmed using TaqMan® MGB probes and iQ™ Multiplex Powermix (Bio-Rad Laboratories, Inc.). The primer sequences, the reaction setup and the cycling conditions were the same as described above.
The probe sequences used for the detection of specific targets were:
GAPDH: 5’-FAM CTGGCCAAGGTCATCCATGA-3’;
ACTB: 5’-FAM-ACAAGATGAGATTGGCATGGC-3’;
HBB: 5’-FAM-AAGTGCTTGGTGCCTTTAGTGATGG-3’;
HBA2: 5’-FAM-TGGCGAGTATGGTGCGGAGG-3’;
SERPINA3: 5’-FAM-TTCCTGGCCCCTGTGATCCC-3’;
TTR: 5’-FAM-ATCGTTGGCTGTGAATACCACCTCTG-3’;
APOC1: 5’-FAM-TGGAGGACAAGGCTTGGGAAGTG-3’.
Availability of supporting data
The microarray data set supporting the results of this article is available in the Gene Expression Omnibus (GEO) repository, [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wnmjowqqhrcpzod&acc=GSE52436].
The DEGs were analyzed for their functions, pathways and networks using Ingenuity Pathways Analysis-IPA® [http://www.ingenuity.com/products/ipa/try-ipa-for-free].
Electronic supplementary material
Additional file 1: List of 86 differentially expressed probe sets with p values ≤ 0.005 and FC ≥ │2│. Probe ID, Gene Symbol, Gene Name and RefSeq Transcript IDs annotation as of release 29 of the Affymetrix® Rhesus Annotation library (01/July/09). P-values and fold changes are reported for all 86 probe sets. (XLSX 18 KB)
Additional file 2: List of 97 differentially expressed probe sets selected as RT-qPCR candidates. Probe IDs and Previous Gene Symbol annotation as of release 29 of the Affymetrix® Rhesus Annotation library (01/July/09). Current Gene Symbol annotation as of the latest Affymetrix® Rhesus Annotation library (release 32 - 09/June/11). Gene Name and RefSeq Transcript IDs as of Ensembl release 72 (June 2013). Annotation using alignment with the human genome has been performed (as stated in the gene name column) for the most highly regulated probe sets with unknown macaque annotation. P-values and fold changes are reported for all genes. (XLSX 57 KB)
Additional file 3: Evaluation of reference gene expression stability across non-infected and BSE-infected samples. For each sample, average values of absolute CTs (+/−SD) of triplicate wells for GAPDH (grey) and ACTB (white) are shown. (PDF 47 KB)
Additional file 4: ΔC T values for all genes showing variability among BSE-infected samples. ΔCT values (+/−SD) normalized against GAPDH. Very similar results were obtained with normalization against ACTB (data not shown). (PDF 58 KB)
Additional file 5: ΔC T values for all genes showing variability among non-infected samples. ΔCT values (+/−SD) normalized against GAPDH. Very similar results were obtained with normalization against ACTB (data not shown). (PDF 59 KB)
Additional file 6: ΔΔC T values of selected genes in the infected samples. ΔΔCT values (+/−SD) for HBB, NR4A2, NCAM1, USP16 and AKR1C1 normalized against GAPDH in the orally-infected animal B6 (white) compared to intracranially infected samples A1-A6 (grey). Only 5 genes were analyzed for animal B6 due to shortage of cDNA. (PDF 27 KB)
Additional file 7: Cluster analysis. Cluster analysis was performed using a hierarchical approach with the average linkage-method for all animals (panel A) or excluding the orally infected one, B6 (panel B). (PDF 68 KB)
Additional file 8: SYBR® Green-based RT-qPCR validation of microarray results. Relative expression levels of 11 genes in BSE-infected cynomolgus macaques normalized against ACTB as reference gene. (PDF 47 KB)
Additional file 9: Comparison between SYBR® Green -based and TaqMan® probe-based results for TTR. Average values of absolute CTs (+/− SD) of triplicate wells for TTR obtained with SYBR® Green (grey) and TaqMan® probe (white) detection methods in BSE-infected samples are shown. (PDF 29 KB)
Additional file 10: RT-qPCR analysis of blood specific marker RHAG. CT values for the erythrocyte marker RHAG were monitored across BSE-infected (solid fill) and non-infected (dotted fill) samples. Human blood cDNA was used as positive control (gradient fill). Note that for almost all the samples CT values were ≥ 35 therefore indicating a very low expression level. Primer sequence (3’-5’): RHAG: F = AGGCAAGCTCAACATGGTTC, R = GGGTGAATTGCCATATCCGC. (PDF 56 KB)
Additional file 11: RT-qPCR analysis of blood specific marker ALAS2. CT values for the erythrocyte marker ALAS2 were monitored across BSE-infected (solid fill) and non-infected (dotted fill) samples. Human blood cDNA was used as positive control (gradient fill). Note that for almost all the samples CT values were ≥ 35 therefore indicating a very low expression level. Primer sequence (3’-5’): ALAS2: F = TCCCTTCATGCTGTCGGAAC, R = GAGCTAGGCAGATCTGTTTTGAA. (PDF 56 KB)
Acknowledgements
The authors wish to thank Dr. Lisa Gasperini for the RT-qPCR initial technical support, Dr. Paolo Vatta for suggestions on primer design and useful discussions, and Prof. Stefano Gustincich for useful discussions. This work was supported by the European Regional Development Fund, Cross-Border Cooperation Italy–Slovenia, Programme 2007–2013, Strategic Project TRANS2CARE to GL and by the European Community’s Seventh Framework Programme (FP7/2007–2013) under grant agreement n° 222887—the PRIORITY project to GL. Samples were derived from an EU study supported by grants QLK1-CT-2002-01096 and BMH4-CT-98-6029.
Abbreviations
- RT-qPCR
Reverse transcriptase quantitative polymerase chain reaction
- HBB
Hemoglobin, beta
- HBA2
Hemoglobin, alpha 2
- TTR
Transthyretin
- APOC1
Apolipoprotein C-I
- SERPINA3
serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 3
- TSE
Transmissible spongiform encephalopathy
- CJD
Creutzfeldt-Jakob disease
- GSS
Gerstmann-Sträussler-Scheinker syndrome
- FFI
Fatal familial insomnia
- BSE
Bovine spongiform encephalopathy
- CWD
Chronic wasting disease
- FSE
Feline spongiform encephalopathy
- PrPC
Cellular prion protein
- PrPSc
Scrapie prion protein
- PRNP
Prion protein
- ER
Endoplasmic reticulum
- DEG
Differentially expressed gene
- RIN
RNA integrity number
- FDR
False discovery rate
- IPA
Ingenuity pathways analysis
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- ACTB
Actin, beta
- AKR1C1
Aldo-keto reductase family 1, member C1
- NCAM1
Neural cell adhesion molecule 1
- USP16
Ubiquitin specific peptidase 16
- NR4A2
Nuclear receptor subfamily 4, group A, member 2
- ALAS2
Aminolevulinate, delta-, synthase 2
- RHAG
Rh-associated glycoprotein
- FC
Fold change
- Hb
Hemoglobin
- SN
Substantia nigra
- PD
Parkinson's disease
- MS
Multiple sclerosis
- AD
Alzheimer’s disease
- AGD
Argyrophilic grain disease
- DLB
Dementia with Lewy bodies
- Hpt
Haptoglobin
- CNS
Central nervous system
- HDAC
Histone deacetylase
- APP
Amyloid beta precursor protein
- PS1
Presenilin 1
- RML
Rocky mountain laboratory
- Aβ
Amyloid beta
- HDL
High density lipoprotein
- LDL
Low density lipoprotein
- APOE
Apolipoprotein E
- LOAD
Late onset Alzheimer’s disease
- IRF3
Interferon regulatory factor 3
- vCJD
Variant Creutzfeldt-Jakob disease
- DNase I
Deoxyribonuclease I
- dNTP
2'-deoxynucleoside 5'-triphosphate
- MGB
Minor groove binder
- PCA
Principal component analysis
- PK
Proteinase K.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MB conceived and designed the RT-qPCR validation studies, carried out the initial optimization experiments, supervised all the experiments as well as the data interpretation, and wrote the manuscript. SV carried out the functional classification of the DEGs, performed most of the RT-qPCR experiments with the related statistical analysis, and provided the initial draft of the manuscript. ACS performed RNA isolation and cDNA preparation of BSE-infected and non-infected brain material, and contributed to the drafting of the manuscript. JM performed the immunoblot analysis. DM provided tissues and contributed to the drafting of the manuscript. GSR and LO performed microarrays and primary data analysis. GL conceived and designed the whole project, contributed to the drafting of the manuscript and gave final approval of the version to be published. All authors read and approved the final manuscript.
Contributor Information
Maura Barbisin, Email: maura.barbisin@sissa.it.
Silvia Vanni, Email: silvia.vanni@sissa.it.
Ann-Christin Schmädicke, Email: schmaedicke@med.ovgu.de.
Judith Montag, Email: montag.judith@mh-hannover.de.
Dirk Motzkus, Email: dmotzkus@dpz.eu.
Lennart Opitz, Email: lopitz@fgcz.ethz.ch.
Gabriela Salinas-Riester, Email: gsalina@gwdg.de.
Giuseppe Legname, Email: legname@sissa.it.
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colby DW, Prusiner SB. Prions. Cold Spring Harbor Perspect Biol. 2011;3(1):a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang Y, Xiang W, Hawkins SA, Kretzschmar HA, Windl O. Transcriptional changes in the brains of cattle orally infected with the bovine spongiform encephalopathy agent precede detection of infectivity. J Virol. 2009;83(1):9464–9473. doi: 10.1128/JVI.00352-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filali H, Vidal E, Bolea R, Marquez M, Marco P, Vargas A, Pumarola M, Martin-Burriel I, Badiola JJ. Gene and protein patterns of potential prion-related markers in the central nervous system of clinical and preclinical infected sheep. Vet Res. 2013;44(3):14. doi: 10.1186/1297-9716-44-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basu U, Almeida LM, Dudas S, Graham CE, Czub S, Moore SS, Guan LL. Gene expression alterations in rocky mountain elk infected with chronic wasting disease. Prion. 2012;6(3):282–301. doi: 10.4161/pri.19915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basu U, Guan LL, Moore SS. Functional genomics approach for identification of molecular processes underlying neurodegenerative disorders in prion diseases. Curr Genomics. 2012;13(5):369–378. doi: 10.2174/138920212801619223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Legname G, Nguyen HO, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A. 2006;103(50):19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318(5852):930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 9.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A. 2012;109(22):8546–8551. doi: 10.1073/pnas.1204498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A. 2012;109(28):E1938–E1946. doi: 10.1073/pnas.1206999109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller MB, Wang DW, Wang F, Noble GP, Ma J, Woods VL, Jr, Li S, Supattapone S. Cofactor molecules induce structural transformation during infectious prion formation. Structure. 2013;21(11):2061–2068. doi: 10.1016/j.str.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benetti F, Gustincich S, Legname G. Gene expression profiling and therapeutic interventions in neurodegenerative diseases: a comprehensive study on potentiality and limits. Expert Opin Drug Discov. 2012;7(3):245–259. doi: 10.1517/17460441.2012.659661. [DOI] [PubMed] [Google Scholar]
- 13.Booth S, Bowman C, Baumgartner R, Sorensen G, Robertson C, Coulthart M, Phillipson C, Somorjai RL. Identification of central nervous system genes involved in the host response to the scrapie agent during preclinical and clinical infection. J Gen Virol. 2004;85(Pt 11):3459–3471. doi: 10.1099/vir.0.80110-0. [DOI] [PubMed] [Google Scholar]
- 14.Kim HO, Snyder GP, Blazey TM, Race RE, Chesebro B, Skinner PJ. Prion disease induced alterations in gene expression in spleen and brain prior to clinical symptoms. Adv Appl Bioinform Chem. 2008;1:29–50. doi: 10.2147/aabc.s3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riemer C, Neidhold S, Burwinkel M, Schwarz A, Schultz J, Kratzschmar J, Monning U, Baier M. Gene expression profiling of scrapie-infected brain tissue. Biochem Biophys Res Commun. 2004;323(2):556–564. doi: 10.1016/j.bbrc.2004.08.124. [DOI] [PubMed] [Google Scholar]
- 16.Xiang W, Windl O, Wunsch G, Dugas M, Kohlmann A, Dierkes N, Westner IM, Kretzschmar HA. Identification of differentially expressed genes in scrapie-infected mouse brains by using global gene expression technology. J Virol. 2004;78(201):11051–11060. doi: 10.1128/JVI.78.20.11051-11060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorensen G, Medina S, Parchaliuk D, Phillipson C, Robertson C, Booth SA. Comprehensive transcriptional profiling of prion infection in mouse models reveals networks of responsive genes. BMC Genomics. 2008;9:114. doi: 10.1186/1471-2164-9-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skinner PJ, Abbassi H, Chesebro B, Race RE, Reilly C, Haase AT. Gene expression alterations in brains of mice infected with three strains of scrapie. BMC Genomics. 2006;7:114. doi: 10.1186/1471-2164-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filali H, Martin-Burriel I, Harders F, Varona L, Lyahyai J, Zaragoza P, Pumarola M, Badiola JJ, Bossers A, Bolea R. Gene expression profiling and association with prion-related lesions in the medulla oblongata of symptomatic natural scrapie animals. PLoS One. 2011;6(5):e19909. doi: 10.1371/journal.pone.0019909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hedman C, Lyahyai J, Filali H, Marin B, Serrano C, Monleon E, Moreno B, Zaragoza P, Badiola JJ, Martin-Burriel I, Bolea R. Differential gene expression and apoptosis markers in presymptomatic scrapie affected sheep. Vet Microbiol. 2012;159(1-2):23–32. doi: 10.1016/j.vetmic.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 21.Filali H, Martin-Burriel I, Harders F, Varona L, Serrano C, Acin C, Badiola JJ, Bossers A, Bolea R. Medulla oblongata transcriptome changes during presymptomatic natural scrapie and their association with prion-related lesions. BMC Genomics. 2012;13:399. doi: 10.1186/1471-2164-13-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filali H, Martin-Burriel I, Harders F, Varona L, Hedman C, Mediano DR, Monzon M, Bossers A, Badiola JJ, Bolea R. Gene expression profiling of mesenteric lymph nodes from sheep with natural scrapie. BMC Genomics. 2014;15:59. doi: 10.1186/1471-2164-15-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almeida LM, Basu U, Khaniya B, Taniguchi M, Williams JL, Moore SS, Guan LL. Gene expression in the medulla following oral infection of cattle with bovine spongiform encephalopathy. J Toxic Environ Health A. 2011;74(2-4):110–126. doi: 10.1080/15287394.2011.529061. [DOI] [PubMed] [Google Scholar]
- 24.Khaniya B, Almeida L, Basu U, Taniguchi M, Williams JL, Barreda DR, Moore SS, Guan LL. Microarray analysis of differentially expressed genes from Peyer's patches of cattle orally challenged with bovine spongiform encephalopathy. J Toxic Environ Health A. 2009;72(17-18):1008–1013. doi: 10.1080/15287390903084199. [DOI] [PubMed] [Google Scholar]
- 25.Almeida LM, Basu U, Williams JL, Moore SS, Guan LL. Microarray analysis in caudal medulla of cattle orally challenged with bovine spongiform encephalopathy. Genet Mol Res. 2011;10(4):3948–3962. doi: 10.4238/2011.October.25.5. [DOI] [PubMed] [Google Scholar]
- 26.Tang Y, Xiang W, Terry L, Kretzschmar HA, Windl O. Transcriptional analysis implicates endoplasmic reticulum stress in bovine spongiform encephalopathy. PLoS One. 2010;5(12):e14207. doi: 10.1371/journal.pone.0014207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basu U, Almeida L, Olson NE, Meng Y, Williams JL, Moore SS, Guan LL. Transcriptome analysis of the medulla tissue from cattle in response to bovine spongiform encephalopathy using digital gene expression tag profiling. J Toxic Environ Health A. 2011;74(2-4):127–137. doi: 10.1080/15287394.2011.529062. [DOI] [PubMed] [Google Scholar]
- 28.Panelli S, Strozzi F, Capoferri R, Barbieri I, Martinelli N, Capucci L, Lombardi G, Williams JL. Analysis of gene expression in white blood cells of cattle orally challenged with bovine amyloidotic spongiform encephalopathy. J Toxic Environ Health A. 2011;74(2-4):96–102. doi: 10.1080/15287394.2011.529059. [DOI] [PubMed] [Google Scholar]
- 29.Race B, Meade-White KD, Miller MW, Barbian KD, Rubenstein R, LaFauci G, Cervenakova L, Favara C, Gardner D, Long D, Parnell M, Striebel J, Priola SA, Ward A, Williams ES, Race R, Chesebro B. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg Infect Dis. 2009;15(9):1366–1376. doi: 10.3201/eid1509.090253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenwood AD, Vincendeau M, Schmadicke AC, Montag J, Seifarth W, Motzkus D. Bovine spongiform encephalopathy infection alters endogenous retrovirus expression in distinct brain regions of cynomolgus macaques (Macaca fascicularis) Mol Neurodegener. 2011;6(1):44. doi: 10.1186/1750-1326-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montag J, Hitt R, Opitz L, Schulz-Schaeffer WJ, Hunsmann G, Motzkus D. Upregulation of miRNA hsa-miR-342-3p in experimental and idiopathic prion disease. Mol Neurodegener. 2009;4:36. doi: 10.1186/1750-1326-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herzog C, Riviere J, Lescoutra-Etchegaray N, Charbonnier A, Leblanc V, Sales N, Deslys JP, Lasmezas CI. PrPTSE distribution in a primate model of variant, sporadic, and iatrogenic Creutzfeldt-Jakob disease. J Virol. 2005;79(22):14339–14345. doi: 10.1128/JVI.79.22.14339-14345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Montag J, Schulz-Schaeffer W, Schrod A, Hunsmann G, Motzkus D. Asynchronous onset of clinical disease in BSE-infected Macaques. Emerg Infect Dis. 2013;19(7):1125–1127. doi: 10.3201/eid1907.120438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamguney G, Giles K, Glidden DV, Lessard P, Wille H, Tremblay P, Groth DF, Yehiely F, Korth C, Moore RC, Tatzelt J, Rubinstein E, Boucheix C, Yang X, Stanley P, Lisanti MP, Dwek RA, Rudd PM, Moskovitz J, Epstein CJ, Cruz TD, Kuziel WA, Maeda N, Sap J, Ashe KH, Carlson GA, Tesseur I, Wyss-Coray T, Mucke L, Weisgraber KH, et al. Genes contributing to prion pathogenesis. J Gen Virol. 2008;89(Pt 7):1777–1788. doi: 10.1099/vir.0.2008/001255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lasmézas CI, Comoy E, Hawkins S, Herzog C, Mouthon F, Konold T, Auvré F, Correia E, Lescoutra-Etchegaray N, Salès N, Wells G, Brown P, Deslys J-P. Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet. 2005;365(9461):781–783. doi: 10.1016/S0140-6736(05)71001-1. [DOI] [PubMed] [Google Scholar]
- 36.Holznagel E, Yutzy B, Schulz-Schaeffer W, Kruip C, Hahmann U, Bierke P, Torres JM, Kim YS, Thomzig A, Beekes M, Hunsmann G, Loewer J. Foodborne transmission of bovine spongiform encephalopathy to nonhuman primates. Emerg Infect Dis. 2013;19(5):712–720. doi: 10.3201/eid1905.120274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yutzy B, Holznagel E, Coulibaly C, Stuke A, Hahmann U, Deslys JP, Hunsmann G, Lower J. Time-course studies of 14-3-3 protein isoforms in cerebrospinal fluid and brain of primates after oral or intracerebral infection with bovine spongiform encephalopathy agent. J Gen Virol. 2007;88(Pt 22):3469–3478. doi: 10.1099/vir.0.83128-0. [DOI] [PubMed] [Google Scholar]
- 38.Cali I, Castellani R, Alshekhlee A, Cohen Y, Blevins J, Yuan J, Langeveld JP, Parchi P, Safar JG, Zou WQ, Gambetti P. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain. 2009;132(Pt 10):2643–2658. doi: 10.1093/brain/awp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39(6):767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 40.Antonell A, Llado A, Altirriba J, Botta-Orfila T, Balasa M, Fernandez M, Ferrer I, Sanchez-Valle R, Molinuevo JL. A preliminary study of the whole-genome expression profile of sporadic and monogenic early-onset Alzheimer's disease. Neurobiol Aging. 2013;34(7):1772–1778. doi: 10.1016/j.neurobiolaging.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 41.Osada N, Uno Y, Mineta K, Kameoka Y, Takahashi I, Terao K. Ancient genome-wide admixture extends beyond the current hybrid zone between Macaca fascicularis and M. mulatta. Mol Ecol. 2010;19(14):2884–2895. doi: 10.1111/j.1365-294X.2010.04687.x. [DOI] [PubMed] [Google Scholar]
- 42.Osada N, Hashimoto K, Kameoka Y, Hirata M, Tanuma R, Uno Y, Inoue I, Hida M, Suzuki Y, Sugano S, Terao K, Kusuda J, Takahashi I. Large-scale analysis of Macaca fascicularis transcripts and inference of genetic divergence between M. fascicularis and M. mulatta. BMC Genomics. 2008;9:90. doi: 10.1186/1471-2164-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Dufort I, Rheault P, Luu-The V. Characterization of a human 20alpha-hydroxysteroid dehydrogenase. J Mol Endocrinol. 2000;25(2):221–228. doi: 10.1677/jme.0.0250221. [DOI] [PubMed] [Google Scholar]
- 44.Ferrer I, Gomez A, Carmona M, Huesa G, Porta S, Riera-Codina M, Biagioli M, Gustincich S, Aso E. Neuronal hemoglobin is reduced in Alzheimer's disease, argyrophilic grain disease, Parkinson's disease, and dementia with Lewy bodies. J Alzheimers Dis. 2011;23(3):537–550. doi: 10.3233/JAD-2010-101485. [DOI] [PubMed] [Google Scholar]
- 45.Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol. 2001;314(5):1209–1225. doi: 10.1006/jmbi.2000.5183. [DOI] [PubMed] [Google Scholar]
- 46.Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, Vassilatis DK. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2003;33(1):85–89. doi: 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- 47.Xing G, Zhang L, Russell S, Post R. Reduction of dopamine-related transcription factors Nurr1 and NGFI-B in the prefrontal cortex in schizophrenia and bipolar disorders. Schizophr Res. 2006;84(1):36–56. doi: 10.1016/j.schres.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 48.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141(6):970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soontornniyomkij V, Risbrough VB, Young JW, Soontornniyomkij B, Jeste DV, Achim CL. Hippocampal calbindin-1 immunoreactivity correlate of recognition memory performance in aged mice. Neurosci Lett. 2012;516(1):161–165. doi: 10.1016/j.neulet.2012.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Backman M, Machon O, Van Den Bout CJ, Krauss S. Targeted disruption of mouse Dach1 results in postnatal lethality. Dev Dyn. 2003;226(1):139–144. doi: 10.1002/dvdy.10210. [DOI] [PubMed] [Google Scholar]
- 51.Liu Q, Yu L, Gao J, Fu Q, Zhang J, Zhang P, Chen J, Zhao S. Cloning, tissue expression pattern and genomic organization of latexin, a human homologue of rat carboxypeptidase A inhibitor. Mol Biol Rep. 2000;27(4):241–246. doi: 10.1023/A:1010971219806. [DOI] [PubMed] [Google Scholar]
- 52.Silver M, Janousova E, Hua X, Thompson PM, Montana G, Alzheimer's Disease Neuroimaging I Identification of gene pathways implicated in Alzheimer's disease using longitudinal imaging phenotypes with sparse regression. NeuroImage. 2012;63(3):1681–1694. doi: 10.1016/j.neuroimage.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guttula SV, Allam A, Gumpeny RS. Analyzing microarray data of Alzheimer's using cluster analysis to identify the biomarker genes. Int J Alzheimers Dis. 2012;2012:649456. doi: 10.1155/2012/649456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry. 2007;7:46. doi: 10.1186/1471-244X-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamboh MI, Minster RL, Kenney M, Ozturk A, Desai PP, Kammerer CM, DeKosky ST. Alpha-1-antichymotrypsin (ACT or SERPINA3) polymorphism may affect age-at-onset and disease duration of Alzheimer's disease. Neurobiol Aging. 2006;27(10):1435–1439. doi: 10.1016/j.neurobiolaging.2005.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mc Guire C, Beyaert R, van Loo G. Death receptor signalling in central nervous system inflammation and demyelination. Trends Neurosci. 2011;34(12):619–628. doi: 10.1016/j.tins.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 57.Stevenson TJ, Hahn TP, MacDougall-Shackleton SA, Ball GF. Gonadotropin-releasing hormone plasticity: a comparative perspective. Front Neuroendocrinol. 2012;33(3):287–300. doi: 10.1016/j.yfrne.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishibashi D, Atarashi R, Fuse T, Nakagaki T, Yamaguchi N, Satoh K, Honda K, Nishida N. Protective role of interferon regulatory factor 3-mediated signaling against prion infection. J Virol. 2012;86(9):4947–4955. doi: 10.1128/JVI.06326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lucatelli JF, Barros AC, Silva VK, Machado Fda S, Constantin PC, Dias AA, Hutz MH, de Andrade FM. Genetic influences on Alzheimer's disease: evidence of interactions between the genes APOE, APOC1 and ACE in a sample population from the South of Brazil. Neurochem Res. 2011;36(8):1533–1539. doi: 10.1007/s11064-011-0481-7. [DOI] [PubMed] [Google Scholar]
- 60.Li X, Buxbaum JN. Transthyretin and the brain re-visited: is neuronal synthesis of transthyretin protective in Alzheimer's disease? Mol Neurodegener. 2011;6:79. doi: 10.1186/1750-1326-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noriega NC, Kohama SG, Urbanski HF. Microarray analysis of relative gene expression stability for selection of internal reference genes in the rhesus macaque brain. BMC Mol Biol. 2010;11:47. doi: 10.1186/1471-2199-11-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabanova S, Kleinbongard P, Volkmer J, Andree B, Kelm M, Jax TW. Gene expression analysis of human red blood cells. Int J Med Sci. 2009;6(4):156–159. doi: 10.7150/ijms.6.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cosseddu GM, Andreoletti O, Maestrale C, Robert B, Ligios C, Piumi F, Agrimi U, Vaiman D. Gene expression profiling on sheep brain reveals differential transcripts in scrapie-affected/not-affected animals. Brain Res. 2007;1142:217–222. doi: 10.1016/j.brainres.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 64.Brown AR, Rebus S, McKimmie CS, Robertson K, Williams A, Fazakerley JK. Gene expression profiling of the preclinical scrapie-infected hippocampus. Biochem Biophys Res Commun. 2005;334(1):86–95. doi: 10.1016/j.bbrc.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 65.Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA. Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiol Genomics. 2008;33(2):240–256. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koechlin E, Hyafil A. Anterior prefrontal function and the limits of human decision-making. Science. 2007;318(5850):594–598. doi: 10.1126/science.1142995. [DOI] [PubMed] [Google Scholar]
- 67.Langevin C, Andreoletti O, Le Dur A, Laude H, Beringue V. Marked influence of the route of infection on prion strain apparent phenotype in a scrapie transgenic mouse model. Neurobiol Dis. 2011;41(1):219–225. doi: 10.1016/j.nbd.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 68.Biagioli M, Pinto M, Cesselli D, Zaninello M, Lazarevic D, Roncaglia P, Simone R, Vlachouli C, Plessy C, Bertin N, Beltrami A, Kobayashi K, Gallo V, Santoro C, Ferrer I, Rivella S, Beltrami CA, Carninci P, Raviola E, Gustincich S. Unexpected expression of alpha- and beta-globin in mesencephalic dopaminergic neurons and glial cells. Proc Natl Acad Sci U S A. 2009;106(36):15454–15459. doi: 10.1073/pnas.0813216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Richter F, Meurers BH, Zhu C, Medvedeva VP, Chesselet MF. Neurons express hemoglobin alpha- and beta-chains in rat and human brains. J Comp Neurol. 2009;515(5):538–547. doi: 10.1002/cne.22062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Broadwater L, Pandit A, Clements R, Azzam S, Vadnal J, Sulak M, Yong VW, Freeman EJ, Gregory RB, McDonough J. Analysis of the mitochondrial proteome in multiple sclerosis cortex. Biochim Biophys Acta. 2011;1812(5):630–641. doi: 10.1016/j.bbadis.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci. 2010;11(5):361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu CW, Liao PC, Yu L, Wang ST, Chen ST, Wu CM, Kuo YM. Hemoglobin promotes Abeta oligomer formation and localizes in neurons and amyloid deposits. Neurobiol Dis. 2004;17(3):367–377. doi: 10.1016/j.nbd.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 73.Cudaback E, Li X, Yang Y, Yoo T, Montine KS, Craft S, Montine TJ, Keene CD. Apolipoprotein C-I is an APOE genotype-dependent suppressor of glial activation. J Neuroinflammation. 2012;9:192. doi: 10.1186/1742-2094-9-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ki CS, Na DL, Kim DK, Kim HJ, Kim JW. Genetic association of an apolipoprotein C-I (APOC1) gene polymorphism with late-onset Alzheimer's disease. Neurosci Lett. 2002;319(2):75–78. doi: 10.1016/S0304-3940(01)02559-9. [DOI] [PubMed] [Google Scholar]
- 75.Petit-Turcotte C, Stohl SM, Beffert U, Cohn JS, Aumont N, Tremblay M, Dea D, Yang L, Poirier J, Shachter NS. Apolipoprotein C-I expression in the brain in Alzheimer's disease. Neurobiol Dis. 2001;8(6):953–963. doi: 10.1006/nbdi.2001.0441. [DOI] [PubMed] [Google Scholar]
- 76.Huang R, Hughes M, Mobley S, Lanham I, Poduslo SE. APOE genotypes in African American female multiple sclerosis patients. Neurosci Lett. 2007;414(1):51–56. doi: 10.1016/j.neulet.2006.12.049. [DOI] [PubMed] [Google Scholar]
- 77.Xiang W, Hummel M, Mitteregger G, Pace C, Windl O, Mansmann U, Kretzschmar HA. Transcriptome analysis reveals altered cholesterol metabolism during the neurodegeneration in mouse scrapie model. J Neurochem. 2007;102(3):834–847. doi: 10.1111/j.1471-4159.2007.04566.x. [DOI] [PubMed] [Google Scholar]
- 78.Katsel P, Li C, Haroutunian V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer's disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer's disease? Neurochem Res. 2007;32(4-5):845–856. doi: 10.1007/s11064-007-9297-x. [DOI] [PubMed] [Google Scholar]
- 79.Bach C, Gilch S, Rost R, Greenwood AD, Horsch M, Hajj GN, Brodesser S, Facius A, Schadler S, Sandhoff K, Beckers J, Leib-Mosch C, Schatzl HM, Vorberg I. Prion-induced activation of cholesterogenic gene expression by Srebp2 in neuronal cells. J Biol Chem. 2009;284(45):31260–31269. doi: 10.1074/jbc.M109.004382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gilch S, Bach C, Lutzny G, Vorberg I, Schatzl HM. Inhibition of cholesterol recycling impairs cellular PrP(Sc) propagation. Cell Mol Life Sci. 2009;66(24):3979–3991. doi: 10.1007/s00018-009-0158-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leoni V, Shafaati M, Salomon A, Kivipelto M, Bjorkhem I, Wahlund LO. Are the CSF levels of 24S-hydroxycholesterol a sensitive biomarker for mild cognitive impairment? Neurosci Lett. 2006;397(1-2):83–87. doi: 10.1016/j.neulet.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 82.Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol. 1995;129(1):121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101(7):2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7(4):321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 85.Patel NV, Forman BM. Linking lipids, Alzheimer's and LXRs? Nucl Recept Signal. 2004;2:e001. doi: 10.1621/nrs.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Janciauskiene S, Wright HT. Inflammation, antichymotrypsin, and lipid metabolism: autogenic etiology of Alzheimer's disease. Bioessays. 1998;20(12):1039–1046. doi: 10.1002/(SICI)1521-1878(199812)20:12<1039::AID-BIES10>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 87.Matsubara E, Hirai S, Amari M, Shoji M, Yamaguchi H, Okamoto K, Ishiguro K, Harigaya Y, Wakabayashi K. Alpha 1-antichymotrypsin as a possible biochemical marker for Alzheimer-type dementia. Ann Neurol. 1990;28(4):561–567. doi: 10.1002/ana.410280414. [DOI] [PubMed] [Google Scholar]
- 88.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hwang D, Lee IY, Yoo H, Gehlenborg N, Cho JH, Petritis B, Baxter D, Pitstick R, Young R, Spicer D, Price ND, Hohmann JG, Dearmond SJ, Carlson GA, Hood LE. A systems approach to prion disease. Mol Syst Biol. 2009;5:252. doi: 10.1038/msb.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miele G, Seeger H, Marino D, Eberhard R, Heikenwalder M, Stoeck K, Basagni M, Knight R, Green A, Chianini F, Wuthrich RP, Hock C, Zerr I, Aguzzi A. Urinary alpha1-antichymotrypsin: a biomarker of prion infection. PLoS One. 2008;3(12):e3870. doi: 10.1371/journal.pone.0003870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cray C, Zaias J, Altman NH. Acute phase response in animals: a review. Comp Med. 2009;59(6):517–526. [PMC free article] [PubMed] [Google Scholar]
- 92.Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014;11(11):50–57. doi: 10.1007/s11897-013-0182-4. [DOI] [PubMed] [Google Scholar]
- 93.Cascella R, Conti S, Mannini B, Li X, Buxbaum JN, Tiribilli B, Chiti F, Cecchi C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim Biophys Acta. 2013;1832(12):2302–2314. doi: 10.1016/j.bbadis.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 94.Li X, Masliah E, Reixach N, Buxbaum JN. Neuronal production of transthyretin in human and murine Alzheimer's disease: is it protective? J Neurosci. 2011;31(35):12483–12490. doi: 10.1523/JNEUROSCI.2417-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci U S A. 2008;105(7):2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McGeer PL, McGeer EG. History of innate immunity in neurodegenerative disorders. Front Pharmacol. 2011;2:77. doi: 10.3389/fphar.2011.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Llorens F, Ansoleaga B, Garcia-Esparcia P, Zafar S, Grau-Rivera O, Lopez-Gonzalez I, Blanco R, Carmona M, Yague J, Nos C, Del Rio JA, Gelpi E, Zerr I, Ferrer I. PrP mRNA and protein expression in brain and PrP(c) in CSF in Creutzfeldt-Jakob disease MM1 and VV2. Prion. 2013;7(5):383–393. doi: 10.4161/pri.26416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336(6088):1511–1513. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xiang W, Windl O, Westner IM, Neumann M, Zerr I, Lederer RM, Kretzschmar HA. Cerebral gene expression profiles in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 2005;58(2):242–257. doi: 10.1002/ana.20551. [DOI] [PubMed] [Google Scholar]
- 100.Smyth GK: Linear models and empirical bayes methods for assessing differential expression in microarray experiments.Stat Appl Genet Mol Biol 2004.,3(1): Article3 [DOI] [PubMed]
- 101.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 103.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57(1):289–300. [Google Scholar]
- 105.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 106.Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270(1):41–49. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: List of 86 differentially expressed probe sets with p values ≤ 0.005 and FC ≥ │2│. Probe ID, Gene Symbol, Gene Name and RefSeq Transcript IDs annotation as of release 29 of the Affymetrix® Rhesus Annotation library (01/July/09). P-values and fold changes are reported for all 86 probe sets. (XLSX 18 KB)
Additional file 2: List of 97 differentially expressed probe sets selected as RT-qPCR candidates. Probe IDs and Previous Gene Symbol annotation as of release 29 of the Affymetrix® Rhesus Annotation library (01/July/09). Current Gene Symbol annotation as of the latest Affymetrix® Rhesus Annotation library (release 32 - 09/June/11). Gene Name and RefSeq Transcript IDs as of Ensembl release 72 (June 2013). Annotation using alignment with the human genome has been performed (as stated in the gene name column) for the most highly regulated probe sets with unknown macaque annotation. P-values and fold changes are reported for all genes. (XLSX 57 KB)
Additional file 3: Evaluation of reference gene expression stability across non-infected and BSE-infected samples. For each sample, average values of absolute CTs (+/−SD) of triplicate wells for GAPDH (grey) and ACTB (white) are shown. (PDF 47 KB)
Additional file 4: ΔC T values for all genes showing variability among BSE-infected samples. ΔCT values (+/−SD) normalized against GAPDH. Very similar results were obtained with normalization against ACTB (data not shown). (PDF 58 KB)
Additional file 5: ΔC T values for all genes showing variability among non-infected samples. ΔCT values (+/−SD) normalized against GAPDH. Very similar results were obtained with normalization against ACTB (data not shown). (PDF 59 KB)
Additional file 6: ΔΔC T values of selected genes in the infected samples. ΔΔCT values (+/−SD) for HBB, NR4A2, NCAM1, USP16 and AKR1C1 normalized against GAPDH in the orally-infected animal B6 (white) compared to intracranially infected samples A1-A6 (grey). Only 5 genes were analyzed for animal B6 due to shortage of cDNA. (PDF 27 KB)
Additional file 7: Cluster analysis. Cluster analysis was performed using a hierarchical approach with the average linkage-method for all animals (panel A) or excluding the orally infected one, B6 (panel B). (PDF 68 KB)
Additional file 8: SYBR® Green-based RT-qPCR validation of microarray results. Relative expression levels of 11 genes in BSE-infected cynomolgus macaques normalized against ACTB as reference gene. (PDF 47 KB)
Additional file 9: Comparison between SYBR® Green -based and TaqMan® probe-based results for TTR. Average values of absolute CTs (+/− SD) of triplicate wells for TTR obtained with SYBR® Green (grey) and TaqMan® probe (white) detection methods in BSE-infected samples are shown. (PDF 29 KB)
Additional file 10: RT-qPCR analysis of blood specific marker RHAG. CT values for the erythrocyte marker RHAG were monitored across BSE-infected (solid fill) and non-infected (dotted fill) samples. Human blood cDNA was used as positive control (gradient fill). Note that for almost all the samples CT values were ≥ 35 therefore indicating a very low expression level. Primer sequence (3’-5’): RHAG: F = AGGCAAGCTCAACATGGTTC, R = GGGTGAATTGCCATATCCGC. (PDF 56 KB)
Additional file 11: RT-qPCR analysis of blood specific marker ALAS2. CT values for the erythrocyte marker ALAS2 were monitored across BSE-infected (solid fill) and non-infected (dotted fill) samples. Human blood cDNA was used as positive control (gradient fill). Note that for almost all the samples CT values were ≥ 35 therefore indicating a very low expression level. Primer sequence (3’-5’): ALAS2: F = TCCCTTCATGCTGTCGGAAC, R = GAGCTAGGCAGATCTGTTTTGAA. (PDF 56 KB)