

Abstract
To explore epigenetic regulation and the impact of chemokine CXCL14 on colorectal cancer, 7 colorectal cancer cell lines, 107 cases of primary colorectal cancer, and 10 cases of normal colorectal mucosa were evaluated in this study. Methylation specific PCR (MSP), semi-quantitative reverse-transcription PCR (RT-PCR), cell proliferation assay, colony formation, and transwell assay were performed for the evaluation. Complete methylation and loss of CXCL14 expression were found in 5 colorectal cancer cell lines. Partial methylation and weak expression were found in two cell lines. CXCL14 was methylated in 79.4% (85/107) of primary human colorectal cancer. No methylation was found in 10 cases of normal colorectal mucosa. Restoration of CXCL14 expression was induced by the 5-aza-2′-deoxycytidine (DAC) treatment. The cell viability was reduced and colony formation was inhibited by restoration of CXCL14 expression in HCT116 cells, a colorectal cancer cell line. The number of invasive and migration cells was reduced by CXCL14. The expression of MMP-2, Vimentin, and NF-κB was suppressed, and the expression of E-cadherin and IκB-α was induced by CXCL14. In conclusion, CXCL14 is frequently methylated in human colorectal cancer and promoter region hypermethylation silenced CXCL14 expression in colorectal cancer cells. Restoration of CXCL14 expression suppressed colorectal cancer proliferation. CXCL14 inhibits colorectal cancer migration, invasion, and epithelial-to-mesenchymal transition (EMT) by suppressing NF-κB signaling.
Introduction
The incidence of colorectal cancer is the third in men and the second in women for diagnosed cancer in 2008 worldwide and the incidence is increasing relatively rapidly in China (Center et al., 2009; Edwards et al., 2010; Chen et al., 2011; Jemal et al., 2011). Local recurrence or distant metastasis was reported in half of patients after operation. Chemotherapy is the major treatment of metastatic or recurrent colorectal cancer. Cancer is caused by the accumulation of genetic and epigenetic alterations (Riojas et al., 2007; Zhang et al., 2008; Kim et al., 2010; Yin et al., 2012; Jia and Guo, 2013). Chemokines are secreted proteins that belong to a superfamily of small chemotactic cytokines. In humans, the chemokine superfamily is composed of more than 50 small secreted proteins. The recent explosion of research in the field of chemokine function as mediators of tumor progression has led to the possibility that these small, immunomodulatory proteins also play key roles in carcinogenesis. The fact that their receptors are G protein-coupled receptors makes them ideally suited for the development of small molecular inhibitors for potential targeted therapy (Miyazaki et al., 2013). Human CXCL14 gene locates on chromosome 5q31.1. Loss of heterozygosity (LOH) was frequently detected in this region in different cancers (Ogasawara et al., 1996; Nozawa et al., 1998; Beder et al., 2003; Cavalli et al., 2010). Loss or reduced expression of CXCL14 was found in different cancers (Hromas et al., 1999; Starnes et al., 2006; Ito et al., 2010). In this study, we analyzed the epigenetic regulation of CXCL14 expression and the effect of CXCL14 on cell proliferation, invasion, and migration in colorectal cancer.
Materials and Methods
Human tissue samples and cell lines
A total of 107 cases of colorectal cancer were collected immediately after surgical resection at the Chinese PLA General Hospital. Among these, matched cancer and adjacent normal tissue paraffin blocks were available for 30 cases. All definitive pathological diagnoses were classified as tumor stage I (n=15), stage II (n=44), stage III (n=48), or stage IV (n=0), according to NCCN 2009 (National Comprehensive Cancer Network) staging criteria.
Seven colorectal cancer cell lines (HCT116, RKO, HT29, SW620, SW480, LOVO, DLD1) were included in this study. All colorectal cancer cell lines were previously established from primary colorectal cancer (Riojas et al. 2007), and maintained in 90% Gibco RPMI media 1640 (Invitrogen, CA, USA) supplemented with 10% fetal bovine serum. Cells were passaged 1:3 once total confluence (~106 cells) was reached on a 75 cm2 culture flask (NEST Biotechnology, Jiangsu, China).
DAC (5-aza-2′-deoxycytidine) treatment
Colorectal cancer cell lines (HCT116, RKO, HT29, DLD1, SW480, SW620, LOVO) were split to a low density (30% confluence) at 12 h before treatment. Cells were treated with DAC (Sigma, MO, USA) at a concentration of 2 μM. Growth medium, conditioned with DAC at 2 μM, was exchanged every 24 h for a total of 96 h of treatment. At the end of the treatment course, RNA was extracted from the cells as described below.
RNA isolation and semi-quantitative RT-PCR
Total RNA was isolated by Trizol reagent (Life Technologies, MD, USA). Agarose gel (1%) electrophoresis and spectrophotometric analysis (A260:280 nm ratio) were used to evaluate RNA quality and quantity. RNA was stored at −80°C prior to use. First strand cDNA was synthesized according to the manufacturer’s instructions; 5 μg of the total RNA was used to synthesize the first strand cDNA with random six-mer primers using a Superscript III-reverse transcriptase kit (Invitrogen). Following first strand synthesis, the reaction mixture was diluted to 100 μl with water. Subsequently, 2.5 μl of diluted cDNA mixture was used for PCR amplification in a final 25 μl reaction volume. PCR amplification of CXCL14 was carried out using the primers: 5′-TCCGGTCAGCATGAGGCTCC-3′ (forward) and 5′-CACCCTATTCTTCGTAGACC-3′ (reverse). The primer set for the CXCL14 gene was designed to span intronic sequences between adjacent exons in order to control for genomic DNA contamination. A total of 37 cycles of amplification was performed for each of the RT-PCR experiments. As an internal control, GAPDH was amplified with 25 cycles to ensure cDNA quality and quantity for each RT-PCR. Amplified products were analyzed in 1.5% agarose gels.
DNA extraction and bisulfite modification
Biopsy specimens were snap-frozen prior to DNA extraction. Paraffin-embedded tissue samples were sectioned at 10 μm, and four to six tissue sections were deparaffinized in xylene and washed twice with 100% ice-cold ethanol. The deparaffinized tissue sections, snap-frozen fresh tissue, and biopsy samples were digested overnight with proteinase K. After treatment of proteinase K, genomic DNA from colorectal cancer cell lines, 107 cases of primary colorectal cancer, and 10 cases of normal colorectal epithelia were all isolated by phenol-chloroform extraction. Then, DNA was precipitated in ethanol, later dissolved in low TE buffer, and then stored at −20°C. Two μg of genomic DNA was diluted in 50 μl of water. The bisulfite treatment was carried out for 16 h at 50°C. DNA samples were then purified with the Wizard DNA Clean-Up System (Promega, WI, USA), desulfonated with NaOH, and then precipitated with ethanol and resuspended in 20 μl of water.
Methylation-specific PCR (MSP)
MSP primers were designed according to genomic sequences flanking the presumed transcriptional start sites (Guo et al., 2008; Licchesi and Herman 2009). Primer sequences were oligo-synthesized (Invitrogen) to detect bisulfite-induced changes affecting unmethylated (U) and methylated (M) alleles. The MSP of CXCL14 was carried out using primers: 5′-GGGTTTTTTAAAGGTAGAGCGGGAC-3′ (M-sense); 5′-CTACCCTCTCGCTCACACAACACG-3′ (M-anti-sense); 5′-TTGGGTTTTTTAAAGGTAGAGTGGGAT-3′ (U-sense); and 5′-CACTACCCTCTCACTCACACAACACA-3′ (U-antisense). Each MSP reaction included approximately 200 ng of bisulfite-treated DNA, 25 pmol of each primer, 100 pmol dNTPs, 2.5 μl 10× PCR buffer, and 1 unit of Taq Polymerase (Invitrogen) in a final reaction volume of 25 μl. Cycle conditions were: 95°C × 5 min for 1 cycle; 35 cycles × (95°C × 30 s, 60°C × 30 s, 72°C × 30 s); 72°C × 5 min for 1 cycle. Each PCR assay included a positive control, using DNA treated in vitro with Sss I methyltransferase (New England Biolabs, MA, USA), and a negative control, using normal human peripheral lymphocytes. MSP products were analyzed using a 2% agarose gel electrophoresis.
Bisulfite sequencing
DNA from SW620, HCT116, LOVO, RKO, and DLD1 cell lines was sequenced by sodium bisulfite treatment in this study. Bisulfite-treated DNA was amplified using primers flanking the targeted regions, including the MSP amplified region and the transcriptional start site. Sequencing primers were as follows: 5′-AAT-TATAYGATTTAGAAAAGTAGTG-3′ (forward) and 5′-CTATCRCAACRACRCACACCC-3′ (reverse). The size of the PCR product is 180 bp (−81bp to +99 bp). PCR cycle conditions were as follows: 95°C × 5 min for 1 cycle; 35 cycles × (95°C × 30 s, 55°C × 30 s, 72°C × 40 s); 72°C × 5 min for 1 cycle. PCR products were gel purified and cloned into pCR2.1 vectors according to the manufacturer’s protocol (Invitrogen). Colonies were grown on agar plates and randomly selected. Plasmids were then isolated and purified using Wizard mini-prep kits (Promega, Shanghai, China). Integrated PCR fragments were confirmed with EcoRI digestion (New England Biolabs, Beverly, MA, USA) and the cloned PCR fragments were sequenced with the M13 reverse primer via automated sequencing (BGI Sequencing, Beijing, China).
Construction of CXCL14 expression vector and transfection assay
Full-length CXCL14 cDNA (NCBI Reference Sequence: NM_004887.4) was cloned by RT-PCR from cDNA derived from placenta into the pCMV6-Entry vector (Origene Technology, MD, USA). The CXCL14 expression vector was verified by DNA sequencing. Transient transfection was performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
MTT assay detection of cell viability
HCT116 cells transfected with empty vector or CXCL14 expression vector or without any transfection (8×103/100 ul/well) were seeded in 96-well plates. Viable proliferating cells were detected by the 3-(4,-dimethy-lthiazol-2-yl)-2,-diphenyl-tetrazoliumbromide (MTT) assay at various time periods (0, 12, 24, 36, 48, 60, and 72 h), using five wells per time period. Cell viability was expressed as optical density (OD), which was detected by an enzyme-linked immunoabsorbent assay reader (Thermo, MA, USA) at 492 nm wavelength.
Colony formation assay
HCT116 cells were grown in six-well culture plates 24 h before transfection. Cells were transfected with empty vector or CXCL14 expression vector according to the manufacturer’s instructions (Invitrogen). After 36 h, the cells were diluted and reseeded 1×104 cells/well in six-well culture plates in triplicates. Growth medium, conditioned with G418 (Invitrogen) at 100 μg/ml, was exchanged every 24 h. After 14 days, the cells were fixed with 75% ethanol for 30 min and stained with 0.2% crystal violet for visualization and counting.
Cell invasion assay
The effect of CXCL14 on cell invasion was detected by the Transwell assay (COSTAR transwell, Corning Incorporated, MA, USA). HCT116 cells were transfected with empty vector or CXCL14 expression vector. 3×105 cells were suspended in 300 μl of serum-free Gibco RPMI Media 1640 and loaded onto the upper compartment of an invasion chamber containing a poly-carbonate membrane with an 8 μm pore size which was coated with a layer of extracellular matrix (ECM; Matrigel™, Becton Dickinson, NJ, USA). After 48 h of incubation, the invasive cells migrated through the ECM layer to the complete medium in the lower compartment. The invasive cells were stained with crystal violet and the number of invaded cells was counted in three independent high powered fields (×100) readings with a light microscope. Statistical analysis was applied among the groups.
Analysis of cell migration
The effect of CXCL14 on cell migration was detected by using the Transwell assay in the absence of the ECM layer. HCT116 cells were transfected with empty vector or CXCL14 expression vector. After 48 h, CXCL14 expressed cells and empty vector transfected cells were harvested and suspended in the serum free Gibco RPMI media 1640. Cell suspensions were then placed into the upper well at a concentration of 1×104 cells/100 μl separately, while the complete medium with 15% fetal bovine serum was placed into the lower well (500 μl). The chamber was incubated for 4 h. Non-migrated cells on the upper surface were scraped gently and washed out with PBS three times. HCT116 cells migrated to the lower surface of the membrane were stained with crystal violet and counted in three independent high powered fields (×100) with light microscope. Statistical analysis was applied among groups.
Protein preparation and western blotting
HCT116 cells were transfected with CXCL14 expression vector or empty control vector. 48 h later, transfected cells were harvested and lysed in ice-cold Tris buffer (20 mmol/l Tris; pH 7.5) containing 137 mmol/l of NaCl, 2 mmol/l of EDTA, 1% Triton X, 10% glycerol, 50 mmol/l of NaF, 1 mmol/l of DTT, and a protease inhibitor cocktail (Roche Applied Science, IN, USA). 40 μg of cell lysate was loaded into each lane. The protein lysates were then separated by SDS-PAGE and electroblotted onto polyvinylidene fluoride membranes (Hybond-P, Amersham, PA, USA). After blocking with 5% nonfat milk and 0.1% Tween-20 in Tris buffered saline, the membranes were incubated with rabbit antibodies of anti-CXCL14 (Abcam, MA, USA), anti-MMP-2 (Bioworld Technology, MN, USA), anti-vimentin (Bioworld Technology) or rabbit antibody of anti-beta-actin (Bioworld Technology). Beta-actin antibody was used as a loading control. The blots were visualized using enhanced chemiluminescence (Pierce Bioscience, IL, USA).
Statistical analysis
Statistical analysis was carried out using χ2 test and p<0.05 was considered statistically significant.
Results
CXCL14 was silenced by promoter region hypermethylation in colorectal cancer cell lines
CXCL14 expression was detected by semi-quantitative RT-PCR in colorectal cancer cell lines. Low level expression of CXCL14 was found in SW620 and LOVO cells and loss of expression was found in HCT116, SW480, RKO, HT29, and DLD1 cells (Figure 1A). Complete methylation was detected in HCT116, RKO, HT29, DLD1, and SW480 cell lines, and partial methylation was revealed in SW620 and LOVO cell lines (Figure 1B). It suggests that loss of CXCL14 expression was correlated with promoter region hypermethylation. Restoration of CXCL14 expression was found in HCT116, SW480, RKO, HT29, and DLD1 cells, and increased expression was found in SW620 and LOVO cells after DAC treatment. To validate MSP results and the density of methylation, bisulfite sequencing of CXCL14 promoter region was performed in colorectal cancer cell lines. Consistent with the MSP results, dense methylation in the promoter region was found in HCT116, RKO, and DLD1 cells, and partial methylation was detected in LOVO and SW620 cells (Figure 1C).
Figure 1.

CXCL14 was silenced by hypermethylation of promoter region in colorectal cancer cell lines. A. Expression of CXCL14 was analyzed by semi-quantitative RT-PCR in colorectal cancer cell lines (HCT116, RKO, HT29, DLD1, SW480, SW620, LOVO) before and after DAC. B. MSP results of the CXCL14 gene in colorectal cancer cell lines (HCT116, RKO, HT29, DLD1, SW480, SW620, LOVO). C. BSSQ of the CXCL14 in the promoter region of colorectal cancer cell lines (SW620, HCT116, LOVO, RKO, DLD1). CXCL14 was methylated in HCT116, RKO, HT29, DLD1, and SW480 cell lines, partially methylated in SW620 and LOVO cell lines. The region amplified by MSP is labeled with a double-headed arrow. Filled circles represent methylated CpG sites and open circles denote unmethylated CpG sites. Notes: −, before DAC treatment; +, after DAC treatment; BSSQ, bisulfite sequencing; DAC, 5-aza-2′-deoxycytidine; H2O, double distilled water; IVD, in vitro methylated DNA; M, methylated alleles; MSP, methylation-specific PCR; NL, normal blood lymphocyte DNA; TSS, transcriptional start site; U, unmethylated alleles.
CXCL14 was frequently methylated in primary colorectal cancer
To see if CXCL14 methylation happens to primary colorectal cancer, CXCL14 methylation in 107 cases of colorectal cancer tissue and 10 cases of normal colorectal mucosa was detected by MSP. CXCL14 was methylated in 79.4% (85 of 107) of primary colorectal cancer, but no methylation was found in normal mucosa (Figure 2). As shown in Table 1, no association was found between CXCL14 methylation and age, gender, TNM stage, tumor size, tumor differentiation, and lymph node metastasis.
Figure 2.
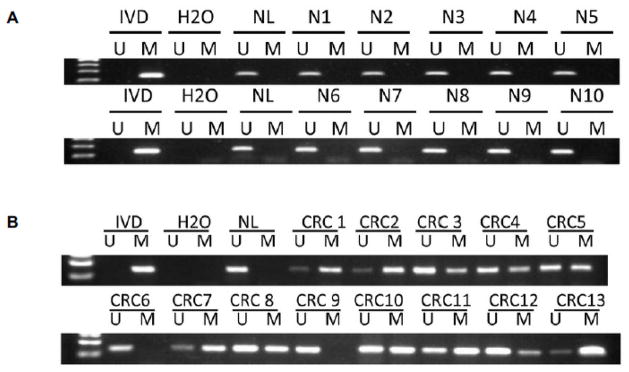
CXCL14 was frequently methylated in colorectal cancer. A. MSP result of CXCL14 in normal colorectal epithelia. B. MSP result of CXCL14 in colorectal cancer tissues. Notes: IVD, in vitro methylated DNA; M, methylated alleles; U, unmethylated alleles; NL, normal blood lymphocyte DNA; NC, normal colorectal epithelia; CRC, colorectal cancer tissue.
Table 1.
Clinicopathologic Features and Methylation Status in 107 CRC Patients.
| Clinical Parameters | Number | CXCL14 Methylation Status | p-value# | |
|---|---|---|---|---|
| Methylated 85 (79.4%) | Unmethylated 22 (20.6%) | |||
| Gender | ||||
| Male | 71 | 56 (78.9%) | 15 (21.1%) | |
| Female | 36 | 28 (77.8%) | 8 (22.2%) | p=0.90 |
| Age | ||||
| ≥60 | 53 | 45 (84.9%) | 8 (15.1%) | |
| <60 | 54 | 40 (74.1%) | 14 (25.9%) | p=0.17 |
| TNM stage | ||||
| I | 15 | 10 (66.7%) | 5 (33.3%) | |
| II | 44 | 38 (86.4%) | 6 (13.6%) | p>0.05 |
| III | 48 | 37 (77.1%) | 9 (22.9%) | |
| IV | 0 | |||
| Tumor size (cm) | ||||
| ≥5 | 60 | 50 (83.3%) | 10 (16.7%) | |
| <5 | 47 | 35 (74.5%) | 12 (25.5%) | p=0.26 |
| Differentiation | ||||
| Poor | 22 | 17 (77.3%) | 5 (22.7%) | |
| Moderate | 78 | 63 (80.8%) | 15 (19.2%) | p>0.05 |
| Well | 7 | 5 (71.4%) | 2 (8.6%) | |
| Lymphatic metastasis | ||||
| Negative | 58 | 47 (81.0%) | 11 (19.0%) | |
| Positive | 49 | 38 (77.6%) | 11 (22.4%) | p=0.66 |
Note:
p-value is obtained from χ2 test. p<0.05 is statistically significant.
Restoration of CXCL14 expression inhibits the viability of HCT116 cells
To analyze the impact of CXCL14 on colorectal cancer cell viability, MTT assay was employed. The number of viable proliferating cells was reduced significantly by re-expression of CXCL14 in HCT116 cells (Figure 3A, p<0.05). As shown in Figure 3B, colony formation was inhibited by restoration of CXCL14 expression in HCT116 cells.
Figure 3.

Restoration of CXCL14 expression inhibited HCT116 cell proliferation. A. The proliferation curve. The CXCL14 re-expressed group was significantly lower compared with two control groups (*p<0.05). B. The clone number was fewer in the CXCL14-expressed group compared with the empty vector group in HCT116 cells (p<0.05). No clone was formed in the absence of vectors after G418 screening. Average of tumor clones was presented by a bar chart. Each experiment was repeated in triplicates.
Colorectal cancer cell invasion and migration were inhibited by CXCL14
To evaluate the effect of CXCL14 on colorectal cancer invasion, the transwell assay was employed in this study. The invasive cell number of each high powered field was 159±11 in the CXCL14 expressed HCT116 cell group and 945±8 in the empty vector group under the microscope (Figure 4A). The number of invasive cells was reduced significantly in CXCL14-expressed HCT116 cell group (p<0.001). Cell migration was detected by transwell assay in the absence of ECM coating. The number of migrated cells of each high powered field was 254±8 in CXCL14 re-expressed HCT116 cells and 1103±5 in empty vector group (Figure 4B). The number of migrated cells is significantly reduced in CXCL14 forced expression group (p<0.001). These findings indicate that CXCL14 suppresses cellular invasion and migration in colorectal cancer.
Figure 4.

The invasion and migration of HCT116 cells were inhibited by CXCL14. A. The number of invasive cells was significantly reduced in the CXCL14 expressed group compared with the empty vector groups (p<0.001). B. The number of migrated cells was significantly reduced in the CXCL14 expressed group compared with the empty vector groups (p<0.001).
Restoration of CXCL14 suppressed matrix metalloproteinase-2 (MMP-2), Vimentin, and NF-κB expression, and increased E-cadherin and IκB-α expression
To further explore the mechanisms of CXCL14 on colorectal cancer invasion and migration, western blot was employed. The expression of MMP-2, Vimentin, and NF-κB was reduced and the expression of E-cadherin and IκB-α was increased considerably after restoration of CXCL14 expression in HCT116 cells. These results suggest that the expression of the key components related to cancer invasion and migration were inhibited by CXCL14.
Discussion
CXCL14, also known as BRAK (breast and kidney expressed chemokine), was initially identified from the kidney and breast tissue. CXCL14 mRNA and protein are ubiquitously expressed in normal tissues, but are absent in tumor cell lines and in primary tumors (Hromas et al., 1999). CXCL14 is down-regulated by DNA methylation in prostate and lung cancer (Song et al., 2010; Tessema et al., 2010). It was reported that CXCL14 was expressed in normal colonic tissue and reduced in colonic cancer cell lines, but other reports made it confusing (Hromas et al., 1999; Frederick et al., 2000; Zeng et al., 2013). In vivo, CXCL14 mRNA expression was increased in inflammatory cells in the microenvironment of a variety of solid malignancies (Frederick et al., 2000). To explore the expression and the regulation of CXCL14 in colorectal cancer, 7 colorectal cancer cell lines were screened by semi-quantitative RT-PCR and MSP. Loss of CXCL14 expression was frequently observed in these cell lines, and reduced or loss of CXCL14 expression was correlated with promoter region hypermethylation. The CXCL14 expression was further validated to be regulated by promoter region methylation with DAC treatment. Frequent methylation was found in primary colorectal cancer. These results suggest that CXCL14 methylation may serve as colorectal cancer detection marker and CXCL14 may be involved in colorectal carcinogenesis.
Absence of CXCL14 expression in many malignant tissues is in agreement with the deficiency of effective antitumor immune responses in cancer patients. CXCL14 may act as chemo-attractant for monocytes and dendritic cells (DC). DC could bind CXCL14 with high affinity and be activated by CXCL14 (Shellenberger et al., 2004; Shurin et al., 2005). Down-regulation of CXCL14 expression in tumor tissues might represent a new mechanism of tumor evasion, which allows tumor cells to escape recognition by antigen-presenting cells. CXCL14 was reported to suppress breast cancer proliferation both in vitro and in vivo (Gu et al., 2012). To explore the effect of CXCL14 on colorectal cancer proliferation, colony formation and MTT assay were performed in this study. The proliferation of HCT116 was inhibited by CXCL14 significantly. CXCL14 was reported to suppress breast cancer metastasizing to lung in MDA-MB-231 xenograft mice. In human breast cancer, CXCL14 expression was associated with lymph node metastasis in reverse, and CXCL14 expression was associated with overall survival (Gu et al., 2012). But the effect of CXCL14 on colorectal cancer metastasis remains unclear.
In this study, we analyzed the impact of CXCL14 on colorectal cancer invasion and migration by transwell assay. The cellular invasion and migration were suppressed by CXCL14 significantly. The function of CXCL14 on tumor invasion and migration is controversial in breast and pancreatic cancer, and CXCL14 was involved in NF-κB signaling (Wente et al., 2008; Park et al., 2013). But CXCL14 on NF-κB signaling was not reported in colorectal cancer. NF-κB activation was reported to promote tumor growth, invasion, and metastasis in many tumors, including colorectal cancer. Matrix metalloproteinases (MMPs), produced by tumor cells, are key players in the degradation of the extracellular matrix and basement membranes and are thus important in tumor invasion. MMP2 is a prognostic factor in many solid tumors and its expression is regulated by NF-κB signaling. Restoration of CXCL14 suppressed MMP-2 expression in HCT116 cell. It suggests that CXCL14 suppresses colorectal cancer invasion and migration by inhibiting NF-κB signaling. Epithelial-to-mesenchymal transition (EMT) was originally known to be one of the key physiological phenomena in the process of embryo development or wound healing. EMT was promoted by NF-κB signaling (Huber et al., 2004; Sakamoto and Maeda, 2010). In cancer metastasis, induction and maintenance of EMT may be accelerated in cancer cells. It was reported that the activity of NF-κB signaling was influenced by CXCL14 and further altered by MMP2 and MMP9. Our study demonstrated that restoration of CXCL14 suppressed Vimentin and NF-κB expression, and increased E-cadherin and IκB-α expression. These results indicate that inhibition of EMT by CXCL14 was related to NF-κB signaling in colorectal cancer, and suggest that CXCL14 inhibits colorectal cancer metastasis by suppressing NF-κB signaling.
Conclusion
CXCL14 is frequently methylated in colorectal cancer and methylation of CXCL14 may serve as a potential colorectal cancer detection marker. CXCL14 expression was regulated by promoter region hypermethylation. Restoration of CXCL14 expression suppressed colorectal cancer proliferation. CXCL14 inhibits colorectal cancer metastasis by suppressing NF-κB signaling.
Figure 5.

Restoration of CXCL14 down-regulates the expression of MMP-2, Vimentin, NF-κB, and up-regulates the expression of E-cadherin and IκB-α. Expression of MMP-2, Vimentin, and NF-κB was decreased in HCT116 cells transfected with CXCL14 compared with the HCT116 cells just transfected with empty vector. But E-cadherin and IκB-α were down-regulated by restoration of CXCL14. Re-expression of CXCL14 was found in CXCL14 transfected HCT116 cells.
Footnotes
Disclosure
J.G.H. is a consultant to MDxHealth. The other authors report no conflicts of interest.
Contributor Information
Baoping Cao, Department of Gastroenterology and Hepatology, Chinese PLA General Hospital, Beijing 100853, China
Yunsheng Yang, Department of Gastroenterology and Hepatology, Chinese PLA General Hospital, Beijing 100853, China
Yuanming Pan, Department of Gastroenterology and Hepatology, Chinese PLA General Hospital, Beijing 100853, China
Yan Jia, Department of Gastroenterology and Hepatology, Chinese PLA General Hospital, Beijing 100853, China
Malcolm V. Brock, The Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, Maryland 21231, USA
James G. Herman, The Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, Maryland 21231, USA
Mingzhou Guo, Department of Gastroenterology and Hepatology, Chinese PLA General Hospital, Beijing 100853, China
References
- Beder LB, Gunduz M, Ouchida M, Fukushima K, Gunduz E, Ito S, Sakai A, Nagai N, Nishizaki K, Shimizu K. Genome-wide analyses on loss of heterozygosity in head and neck squamous cell carcinomas. Lab Invest. 2003;83(1):99–105. doi: 10.1097/01.lab.0000047489.26246.e1. [DOI] [PubMed] [Google Scholar]
- Cavalli LR, Riggins RB, Wang A, Clarke R, Haddad BR. Frequent loss of heterozygosity at the interferon regulatory factor-1 gene locus in breast cancer. Breast Cancer Res Treat. 2010;121(1):227–231. doi: 10.1007/s10549-009-0509-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin. 2009;59(6):366–378. doi: 10.3322/caac.20038. [DOI] [PubMed] [Google Scholar]
- Chen WQ, Zhang SW, Zou XN, Zhao P. Cancer incidence and mortality in china, 2006. Chin J Cancer Res. 2011;23(1):3–9. doi: 10.1007/s11670-011-0003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, Jemal A, Schymura MJ, Lansdorp-Vogelaar I, Seeff LC, van Ballegooijen M, Goede SL, Ries LA. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544–573. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick MJ, Henderson Y, Xu X, Deavers MT, Sahin AA, Wu H, Lewis DE, El-Naggar AK, Clayman GL. In vivo expression of the novel CXC chemokine BRAK in normal and cancerous human tissue. Am J Pathol. 2000;156(6):1937–1950. doi: 10.1016/S0002-9440(10)65067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu XL, Ou ZL, Lin FJ, Yang XL, Luo JM, Shen ZZ, Shao ZM. Expression of CXCL14 and its anticancer role in breast cancer. Breast Cancer Res Treat. 2012;135(3):725–735. doi: 10.1007/s10549-012-2206-2. [DOI] [PubMed] [Google Scholar]
- Guo M, Ren J, Brock MV, Herman JG, Carraway HE. Promoter methylation of HIN-1 in the progression to esophageal squamous cancer. Epigenetics. 2008;3(6):336–341. doi: 10.4161/epi.3.6.7158. [DOI] [PubMed] [Google Scholar]
- Hromas R, Broxmeyer HE, Kim C, Nakshatri H, Christopherson K, 2nd, Azam M, Hou YH. Cloning of BRAK, a novel divergent CXC chemokine preferentially expressed in normal versus malignant cells. Biochem Biophys Res Commun. 1999;255(3):703–706. doi: 10.1006/bbrc.1999.0257. [DOI] [PubMed] [Google Scholar]
- Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114(4):569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Ozawa S, Ikoma T, Yajima N, Kiyono T, Hata R. Expression of a chemokine BRAK/CXCL14 in oral floor carcinoma cells reduces the settlement rate of the cells and suppresses their proliferation in vivo. Biomed Res. 2010;31(3):199–206. doi: 10.2220/biomedres.31.199. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Jia Y, Guo M. Epigenetic changes in colorectal cancer. Chin J Cancer. 2011, 2013;32(1):21–30. doi: 10.5732/cjc.011.10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Lee J, et al. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev. 2010;29(1):181–206. doi: 10.1007/s10555-010-9207-6. [DOI] [PubMed] [Google Scholar]
- Licchesi JD, Herman JG. Methylation-specific PCR. Methods Mol Biol. 2009;507:305–323. doi: 10.1007/978-1-59745-522-0_22. [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Takabe K, Yeudall WA. Chemokines, chemokine receptors and the gastrointestinal system. World J Gastroenterol. 2013;19(19):2847–2863. doi: 10.3748/wjg.v19.i19.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa H, Oda E, Ueda S, Tamura G, Maesawa C, Muto T, Taniguchi T, Tanaka N. Functionally inactivating point mutation in the tumor-suppressor IRF-1 gene identified in human gastric cancer. Int J Cancer. 1998;77(4):522–527. doi: 10.1002/(sici)1097-0215(19980812)77:4<522::aid-ijc8>3.0.co;2-w. 1998. [DOI] [PubMed] [Google Scholar]
- Ogasawara S, Tamura G, Maesawa C, Suzuki Y, Ishida K, Satoh N, Uesugi N, Saito K, Satodate R. Common deleted region on the long arm of chromosome 5 in esophageal carcinoma. Gastroenterology. 1996;110(1):52–57. doi: 10.1053/gast.1996.v110.pm8536888. [DOI] [PubMed] [Google Scholar]
- Park CR, You DJ, Kim DK, Moon MJ, Lee C, Oh SH, Ahn C, Seong JY, Hwang JI. CXCL14 enhances proliferation and migration of NCI-H460 human lung cancer cells overexpressing the glycoproteins containing heparan sulfate or sialic acid. J Cell Biochem. 2013;114(5):1084–1096. doi: 10.1002/jcb.24449. [DOI] [PubMed] [Google Scholar]
- Riojas MA, Guo M, Glöckner SC, Machida EO, Baylin SB, Ahuja N. Methylation-induced silencing of ASC/TMS1, a pro-apoptotic gene, is a late-stage event in colorectal cancer. Cancer Biol Ther. 2007;6(11):1710–1716. doi: 10.4161/cbt.6.11.4829. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Maeda S. Targeting NF-kappaB for colorectal cancer. Expert Opin Ther Targets. 2010;14(6):593–601. doi: 10.1517/14728221003769903. [DOI] [PubMed] [Google Scholar]
- Shellenberger TD, Wang M, Gujrati M, Jayakumar A, Strieter RM, Burdick MD, Ioannides CG, Efferson CL, El-Naggar AK, Roberts D, Clayman GL, Frederick MJ. BRAK/CXCL14 is a potent inhibitor of angiogenesis and a chemotactic factor for immature dendritic cells. Cancer Res. 2004;64(22):8262–8270. doi: 10.1158/0008-5472.CAN-04-2056. [DOI] [PubMed] [Google Scholar]
- Shurin GV, Ferris RL, Tourkova IL, Perez L, Lokshin A, Balkir L, Collins B, Chatta GS, Shurin MR. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol. 2005;174(9):5490–5498. doi: 10.4049/jimmunol.174.9.5490. [DOI] [PubMed] [Google Scholar]
- Song EY, Shurin MR, Tourkova IL, Gutkin DW, Shurin GV. Epigenetic mechanisms of promigratory chemokine CXCL14 regulation in human prostate cancer cells. Cancer Res. 2010;70(11):4394–4401. doi: 10.1158/0008-5472.CAN-10-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starnes T, Rasila KK, Robertson MJ, Brahmi Z, Dahl R, Christopherson K, Hromas R. The chemokine CXCL14 (BRAK) stimulates activated NK cell migration: implications for the down-regulation of CXCL14 in malignancy. Exp Hematol. 2006;34(8):1101–1105. doi: 10.1016/j.exphem.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Tessema M, Klinge DM, Yingling CM, Do K, Van Neste L, Belinsky SA. Re-expression of CXCL14, a common target for epigenetic silencing in lung cancer, induces tumor necrosis. Oncogene. 2010;29(37):5159–5170. doi: 10.1038/onc.2010.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente MN, Mayer C, Gaida MM, Michalski CW, Giese T, Bergmann F, Giese NA, Büchler MW, Friess H. CXCL14 expression and potential function in pancreatic cancer. Cancer Lett. 2008;259(2):209–217. doi: 10.1016/j.canlet.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Yin D, Jia Y, Brock MV, Herman JG, Han C, Su X, Liu Y, Guo M. SOX17 methylation inhibits its antagonism of Wnt signaling pathway in lung cancer. Discov Med. 2012;14(74):33–40. [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Yang X, Cheng L, Liu R, Lei Y, Dong D, Li F, Lau QC, Deng L, Nice EC, Xie K, Huang C. Chemokine CXCL14 is associated with prognosis in patients with colorectal carcinoma after curative resection. J Transl Med. 2013;11:6. doi: 10.1186/1479-5876-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Glockner SC, Guo M, Machida EO, Wang DH, Easwaran H, Van Neste L, Herman JG, Schuebel KE, Watkins DN, Ahuja N, Baylin SB. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res. 2008;68(8):2764–2772. doi: 10.1158/0008-5472.CAN-07-6349. [DOI] [PMC free article] [PubMed] [Google Scholar]