

Abstract
PknG from Mycobacterium tuberculosis is a Ser/Thr protein kinase that regulates key metabolic processes within the bacterial cell as well as signaling pathways from the infected host cell. This multidomain protein has a conserved canonical kinase domain with N- and C-terminal flanking regions of unclear functional roles. The N-terminus harbors a rubredoxin-like domain (Rbx), a bacterial protein module characterized by an iron ion coordinated by four cysteine residues. Disruption of the Rbx-metal binding site by simultaneous mutations of all the key cysteine residues significantly impairs PknG activity. This encouraged us to evaluate the effect of a nitro-fatty acid (9- and 10-nitro-octadeca-9-cis-enoic acid; OA-NO2) on PknG activity. Fatty acid nitroalkenes are electrophilic species produced during inflammation and metabolism that react with nucleophilic residues of target proteins (i.e., Cys and His), modulating protein function and subcellular distribution in a reversible manner. Here, we show that OA-NO2 inhibits kinase activity by covalently adducting PknG remote from the catalytic domain. Mass spectrometry-based analysis established that cysteines located at Rbx are the specific targets of the nitroalkene. Cys-nitroalkylation is a Michael addition reaction typically reverted by thiols. However, the reversible OA-NO2-mediated nitroalkylation of the kinase results in an irreversible inhibition of PknG. Cys adduction by OA-NO2 induced iron release from the Rbx domain, revealing a new strategy for the specific inhibition of PknG. These results affirm the relevance of the Rbx domain as a target for PknG inhibition and support that electrophilic lipid reactions of Rbx-Cys may represent a new drug strategy for specific PknG inhibition.
Keywords: PknG, Ser/Thr kinase, Nitrated fatty acids, Nitroalkene, Rubredoxin, Mycobacterium tuberculosis
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis, is a major public health problem that causes more than one million deaths every year (http://www.who.int/tb/publications/global_report/gtbr12_executivesummary.pdf). Several factors comprise the efficacy of the available pharmacological treatments, including the emergence of drug-resistant strains, the lack of new drugs, and the bacilli’s ability to persist inside the host macrophages by inhibiting phagosome maturation. One of the most promising strategies for drug discovery in tuberculosis is to interfere with bacterial and host signal transduction mechanisms [1,2].
Genomic studies of M. tuberculosis reveal the presence of 11 “eukaryotic-like” Ser/Thr protein kinases [3]. Among them, PknG, gains significance by regulating critical processes in mycobacterial survival [1,4,5]. Functional roles for PknG include regulation of metabolic processes and interference of signaling pathways from the infected host cell [4,5]. The inactivation of the pknG gene decreases cell viability and virulence in animal models, and suggests a central role in controlling intracellular glutamate/glutamine levels [6]. It was further demonstrated that PknG participates in the regulation of glutamate metabolism via the phosphorylation of an endogenous substrate (GarA), with similar functions reported for PknG in the related actinomycete Corynebacterium glutamicum [5,7]. PknG also is a virulence factor that mediates M. tuberculosis survival within host cells by preventing macrophage phagosome-lysosome fusion [4]. In addition, inhibition of PknG activity yields bacteria more susceptible to intracellular degradation [4]. Due to the critical cellular processes that it controls, PknG inhibition has emerged as an attractive strategy for drug discovery. The main challenge is to achieve selectivity for PknG inhibition, since the catalytic mechanisms and the active Ser/Thr protein kinase fold are conserved from prokaryotes to eukaryotes.
M. tuberculosis PknG is a multidomain protein. The conserved canonical catalytic kinase domain is flanked by N- and C-terminal domains having undefined functional roles. The C-terminal domain of PknG contains a tetratricopeptide repeat motif (TPR), a domain that participates in protein–protein interactions in both eukaryotic and prokaryotic cells [8]. The TPR domain influences intermolecular interactions, as defined by the crystal structure of PknG. Notably, whether and how dimerization is linked to enzyme activity is currently unknown [8]. The N-terminal sequence preceding the kinase domain contains both autophosphorylation sites and a rubredoxin-like domain (Rbx) [5]. This latter domain is typified by an iron ion coordinated to four conserved cysteine residues that participates in electron transfer reactions [9,10]. The N-terminal sequence of PknG contains two CXXCG motifs involved in metal binding in Rbx domains. The crystal structure of a PknG construct confirmed the presence of a Rbx-like arrangement interacting with the kinase domain without occluding active site accessibility [8]. The role of the Rbx domain in PknG is still uncertain. Metal binding site disruption by simultaneous mutations of multiple key cysteine residues has a remarkable effect on PknG activity [8,11], pointing to a relevant functional or structural role of Rbx domain. This finding encouraged us to evaluate the effect of an electrophilic fatty acid nitroalkene on PknG activity.
Electrophilic unsaturated fatty acids are generated by diverse enzymatic and redox-related mechanisms, yielding signaling mediators that induce anti-inflammatory and chemotherapeutic responses [12]. In particular, nitration confers unsaturated fatty acids with an electrophilic reactivity that mediates the reversible nitroalkylation of proteins at nucleophilic Cys and His residues. This thiol-reversible posttranslational modification modulates protein function and distribution [13]. The reactivity of these molecules is directed by the electrophilic carbon β to the electron-withdrawing NO2 group, allowing reversible Michael addition [12–14]. Compared with other electrophilic lipids, nitro-fatty acids (NO2-FA) reversibly react with thiols at a greater rate constant [12,13,15].
Herein unique structural features of PknG are exploited to inhibit kinase activity by modification of its non-catalytic Rbx domain. These results reveal a novel mechanism for inducing kinase inhibition by iron release from the Rbx domain on covalent cysteine modification by nitrated fatty acids.
Materials and methods
Materials and chemicals
9-Octadecenoic acid (oleic acid; OA) was purchased from Nu-Check Prep (Elysian, MN). 9- and 10-nitro-octadeca-9-cis-enoic acid (OA-NO2) were prepared as previously described [16]. GSH, DTT, bathophenanthrolinedisulfonic acid disodium salt (BPS), iodoacetamide (IAM), and 8-anilino-1-naphthalenesulfonic acid ammonium salt (ANS) were from Sigma (St. Louis, MO). C18-Omix Pipette tips for sample preparation were from Varian (Lake Forest, CA). Sequencing grade modified trypsin was from Promega (Madison, WI).
Protein production and purification
Full-length PknG was overexpressed in Escherichia coli BL21 (DE3) cells grown for 24 h at 30 °C without IPTG and supplemented with 100 μM FeCl3. PknG was purified as described before [5].
Site-directed mutagenesis of PknG was performed by PCR on pET-28a-Δ74PknG using a QuikChange II site-directed mutagenesis kit (Agilent Technologies). The sequence of the construct (PknG 74-406, Δ74/TPR) was verified by DNA sequencing. PknG Δ74/TPR was overexpressed in E. coli BL21(DE3) cells grown at 30 °C until OD600 =0.6, and then ON at 14 °C after addition of 1 mM IPTG. PknGΔ74/TPR purification was performed as previously described [5].
GarA (Rv1827) expression was performed in E. coli BL21(AI). Cells were grown in LB medium supplemented with 0.1% glucose and 10 μg/μL tetracycline at 37 °C until OD600 =0.6, and then for 18 h at 22 °C after addition of 1 mM IPTG and 0.02% arabinose. The cells were harvested by centrifugation and resuspended in buffer A (5 mM NaH2PO4, 50 mM Na2HPO4, 500 mM NaCl, 5% glycerol, 25 mM imidazole, pH 8.0) supplemented with Complete protease inhibitor cocktail (Roche). GarA was first purified by metal-affinity chromatography on a HisTrap Ni2+-IMAC column (GE Healthcare) equilibrated in buffer A, using a linear imidazole gradient (20–400 mM). The GarA-containing fractions were dialyzed against buffer B (25 mM Tris-HCl, 150 mM NaCl, 5% glycerol, 1 mM DTT, pH 7.6), and the His6 tag was removed by incubation for 24 h at 18 °C in the presence of His6-tagged TEV endoprotease at a 1:30 ratio followed by separation on Ni-NTA agarose column (Qiagen). The untagged GarA was then further purified by size-exclusion chromatography on a Superdex 75 26/60 column (GE Healthcare) equilibrated in buffer B without DTT.
Protein kinase assay
Protein phosphorylation reactions were performed in 50 mM Hepes buffer, pH 7.0, containing 2 mM MnCl2 and 100 μM ATP. Activity of PknG was assayed using recombinant GarA as substrate. The molar ratio of kinase:substrate ranged from 1:10 to 1:20. Reaction mixtures were incubated 30 min at 37 °C and substrate phosphorylation was evaluated by MALDI-TOF MS. The autophosphorylation activity of PknG was assessed by incubation of the enzyme in the presence of 2 mM MnCl2 and 100 μM ATP for 40 min at 37 °C. The samples were then digested with trypsin and phosphopeptides were detected by MS.
The effect of OA-NO2-PknG preincubation times on kinase inhibition was assayed. PknG and OA-NO2 were incubated and at different time points aliquots of treated and control enzymes were removed for kinase activity determination. Activity assay was performed using Kinase Glo® Plus Luminescent Kinase Assay (Promega) according to the manufacturer guidelines. Briefly, activity was tested using GarA as substrate (kinase:substrate ratio was 1:25) and remaining ATP was quantified by luminescence after 30 min incubation time at 37 °C. For each time point the inhibition relative to control enzyme is plotted.
MS analysis
Proteolytic digestion was carried out by incubating the proteins with trypsin in 70 mM ammonium bicarbonate, pH 8.0, for 12 h at 37 °C (enzyme–substrate ratio 1:10). Mass spectra of peptides mixtures were acquired in a 4800 MALDI TOF/TOF instrument (Applied Biosystems) in positive ion reflector or linear mode using a matrix solution of α-cyano-4-hydroxycinnamic acid in 0.2% trifluoroacetic acid in acetonitrile-H2O (50%, v/v) and were externally calibrated using a mixture of standard peptides (Applied Biosystems). The molecular mass of the native and phosphorylated GarA was determined using a sinapinic acid matrix (10 mg/mL in acetonitrile-H2O 50%, 0.2% trifluoroacetic acid). Alternatively, a linear ion trap mass spectrometer (LTQ Velos, Thermo) coupled on line with a nano-liquid chromatography system (easy-nLC, Proxeon-Thermo) was used for peptide mixtures analysis. Peptides were separated on a reversed-phase column (EASY-column™ 100 mm, ID 75 μm, 3 μm, C18-A2 from Proxeon) and eluted with a linear gradient of acetonitrile 0.1% formic acid (0–60% in 60 min) at a flow rate of 400 nL/min. Electrospray voltage was 1.40 kV and capillary temperature was 200 °C. Peptides were detected in the positive ion mode using a mass range of 300–2000 in the data-dependent triple play MS2 mode (full scan followed by zoom scan and MS/MS of the top 5 peaks in each segment).
Exposure of PknG to OA-NO2 and other Cys-alkylating reagents
Native PknG (ranging from 5 to 10 μM) in 70 mM ammonium bicarbonate, pH 8.0, was incubated for 10 min at 25 °C with OA-NO2 (0–100 μM) or IAM (0–500 μM) and kinase activity was immediately measured. PknG treatment with OA-NO2 was also performed in 50 mM sodium bicarbonate, pH 7.2, or 50 mM Hepes buffer, pH 7.3. As a control, PknG was exposed to OA-NO2 vehicle (methanol) under the same conditions. In addition for OA-NO2 experiments, kinase activity in the presence of equivalent concentration of OA was determined. In some experiments, after nitroalkene treatment PknG was incubated with DTT (42 mM) or GSH (24 mM) for 10 min at 25 °C and enzymatic activity was redetermined. The activity of control PknG in the presence and absence of the thiol containing reagents was also assayed. Previous to enzymatic digestions, excess of DTT was removed by immobilization of PknG on reverse-phase Poros 10 R2 beads (Applied Biosystems).
PknG tryptic peptides were isolated by reverse-phase HPLC (Vydac® C18; 2.1 × 100 mm) and fractions including cysteine-containing peptides were selected after mass analysis by MALDI-TOF. Selected fractions were dried, resuspended in 70 mM ammonium bicarbonate, pH 8.0, and treated with OA-NO2 (1:4) for 10 min at 25 °C. Peptide modification was analyzed by MALDI MS and ESI MS.
The experiments using rabbit muscle GAPDH as model protein were carried out as previously reported [13].
Iron determination
Control and OA-NO2-treated PknG were loaded onto OMIX C4 pipette tips (Agilent Technologies) and flow through was collected. Non-bound fraction was incubated for 15 min at 25 °C with 40 mM DTT and iron was determined spectrophotometrically using BPS as previously described [17].
ANS fluorescence
Concentration of ANS was determined using the molar extinction coefficient ε=5000 M−1 cm−1 at 350 nm [18]. PknG exposed to OA-NO2 under the same experimental conditions stated above was diluted at 1 μM, dialyzed to remove excess OA-NO2, and incubated with 16 μM ANS. As controls, PknG natively folded and thermally denatured was incubated with ANS. Fluorescence was collected on a Cary Eclipse fluorescence spectrophotometer (Varian, Inc.) with the excitation wavelength set on 350 nm and emission between 370 and 650 nm.
Circular dichroism (far-UV) spectroscopy
Far-UV circular dichroism spectra were performed on OA-NO2-treated and OA-treated PknG. CD spectra were recorded between 190 and 260 nm on an Aviv 215 spectropolarimeter (Aviv Biomedical), using a cylindrical cell with a 0.02 cm path length and an averaging time of 1 s per step, with protein samples at 0.5 mg/mL in 25 mM Tris-HCl, 100 mM NaCl, glycerol 5%, pH 8.0. Five consecutive scans from each sample were merged to produce an averaged spectrum and corrected using buffer baselines measured under the same conditions. Data were normalized to the molar peptide bond concentration and path length and expressed as mean residue ellipticity ([θ] degree · cm2 · dmol−1).
Bioinformatics procedures
Protein sequence homologs to PknG were searched at NCBI’s NR database using CS-Blast [19], Psi-Blast [20], and HHsenser [21]. All searches were run performing both gapped and ungapped alignments, in order to selectively detect proteins carrying both Rbx and Ser/Thr protein kinase domains. Significant hits (Blast E values < 1e−10) found with all methods with sequence coverage > 75% were kept. Multiple sequence alignments (MSAs) were computed with Mafft [22], T-Coffee [23], and Prank [24]. Such MSAs were manually analyzed in order to detect sequences with N-terminal Rbx motifs (all had the kinase domain). Distance-based phylograms for a subset of 652 sequences (lengths between 500 and 800 amino acids, pairwise identities <76%) were computed with BioNJ [25]. For proteins with the Rbx domain, maximum-likelihood phylogenetic trees were built by way of PhyML [26].
Results
PknG inhibition by OA-NO2
To evaluate whether the electrophilic OA-NO2 has an effect on PknG kinase activity, the enzyme was treated with different concentrations of OA-NO2 (below its critical micelle concentration) and activity was measured using a recombinant protein substrate, GarA. Native and phosphorylated GarA were detected by MALDI-TOF MS as ions of m/z 17145 and m/z 17222, respectively [27]. The mass shift corresponds to the incorporation of one phosphate group (80 Da, Fig. 1A). Kinase activity of PknG exposed to micromolar concentrations of nitro-oleic acid (molar excess ranging from 1:1 to 1:3) was significantly inhibited. The ability to phosphorylate GarA was decreased after 10 min incubation of the kinase with OA-NO2 (50 μM, molar ratio OA-NO2:PknG 5:1) (Fig. 1A). The same extent of enzyme inhibition was observed when PknG was exposed to OA-NO2 under different experimental conditions (ammonium bicarbonate, pH 8.0, sodium bicarbonate, pH 7.2, and Hepes, pH 7.3). As controls, PknG was treated with equivalent concentrations of oleic acid or methanol (vehicle) and no effect on kinase activity was observed (data not shown).
Fig. 1.

OA-NO2 inhibits PknG kinase activity. (A) GarA phosphorylation by PknG. Linear MALDI-TOF mass spectrum of GarA (m/z 17145, dashed line), GarA phosphorylated by PknG using a molar ratio PknG:GarA of 1:20 (m/z 17222, gray line), and GarA phosphorylated by PknG pretreated with 50 μM OA-NO2 for 10 min (m/z 17143, black line). (B) PknG autophosphorylation. Upper panel: linear MALDI-TOF spectrum of native PknG after tryptic digestion showing basal autophosphorylation pattern. Unphosphorylated peptide with seq. 10–60 (m/z 5395.8), monophosphorylated (m/z 5475.4), and diphosphorylated species (m/z 5555.1) are detected as expected. Middle panel: linear MALDI-TOF spectrum of PknG after incubation with ATP and Mn2+ and further typtic digestion (autophoshorylation positive control). Diphosphorylated peptide (m/z 5555.0) is the most intense ion observed while unphosphorylated and monophosphorylated ions were almost undetectable. Lower panel: linear MALDI-TOF spectrum of tryptic digestion of PknG treated with 30 μM OA-NO2 for 10 min before autophosporylation reaction. The detection of all three forms of the phosphorylatable peptide indicates that OA-NO2 impairs conversion into the fully phosphorylated species. The spectra are representative of three independent experiments. (C) Dose-dependent inhibition of PknG by OA-NO2. PknG (8 μM) was incubated with OA-NO2 ranging from 0 to 80 μM in 70 mM ammonium bicarbonate, pH 8.0, at 25 °C. As a control, PknG was incubated with vehicle under the same experimental conditions. After 10 min of incubation enzyme activities were determined. Samples were analyzed in triplicates. (D) Time-dependent inhibition of PknG by OA-NO2. PknG (10 μM) was incubated with 50 μM OA-NO2 (treated PknG) or vehicle (control PknG) in 70 mM ammonium bicarbonate, pH 8.0, at 25 °C. At the indicated time points aliquots of control and treated PknG were removed, and enzyme inhibition was determined using Kinase Glo® by comparison with control activity. Samples were analyzed in triplicates.
The effect of OA-NO2 on PknG autophosphorylation was also evaluated to confirm whether the inhibition observed reflects a general loss of the kinase activity as opposed to a substrate-specific effect (Fig. 1B). PknG can autophosporylate its N-terminal sequence at specific Thr residues [5]. The spectrum obtained in the linear mode after tryptic digestion of native PknG showed unphosphorylated (m/z 5395.8), monophosphorylated (m/z 5475.4), and diphosphorylated (m/z 5555.1) ions (sequence 10–60) as previously described (Fig. 1B, upper panel) [5]. When native PknG was incubated with ATP under phosphorylation conditions and then digested, the most intense ion observed corresponded to the diphosphorylated specie while unphosphorylated and monophosphorylated ions were almost undetectable (Fig. 1B, middle panel). On the other hand, OA-NO2 (30 μM) impairs the conversion of the un- and monophosphorylated forms of the kinase into the fully phosphorylated species, indicating that autocatalytic activity is also inhibited by OA-NO2 (Fig. 1B, lower panel).
The effect of OA-NO2 on kinase activity was tested using inhibitor concentrations ranging from 0 to 80 μM. There was a dose-dependent inactivation of PknG on preincubation of the enzyme with different OA-NO2 concentrations for 10 min (Fig. 1C). Under these conditions, 50% of enzyme inhibition was reached with 35 μM OA-NO2.
To further characterize the effect of OA-NO2 on PknG, the time course of enzyme inhibition was studied. PknG and OA-NO2 (or vehicle control) were mixed for time periods ranging from 0 to 30 min, and kinase activity was then measured using a commercial luminescent kinase assay (Kinase Glo®). Under these conditions the control activity varies less than 10% during the 30 min incubation time. The percentage of remaining kinase activity was calculated with respect to control activity for the same time point. The exposure of PknG to OA-NO2 decreased kinase activity in a time-dependent manner (Fig. 1D). The dependence of PknG inhibition on enzyme-OA-NO2 reaction times suggests that the effect of OA-NO2 is mediated by a covalent modification of PknG.
Cysteines in the Rbx domain are the molecular targets of OA-NO2
In order to identify the PknG residues that account for the inhibition of the kinase, samples obtained by treatment with OA-NO2 under different experimental conditions were digested with trypsin and peptide mixtures were analyzed by MS (Fig. 2). MALDI-TOF mass spectra of OA-NO2-treated samples (OA-NO2 concentration up to 35 μM) showed loss of only two peptides, each one containing a different CXXCG motif of the Rbx domain: m/z 1292.56 and 3530.62 (considering the intramolecular disulfide bond) (Figs. 2A and B). No other peptide showed a significant change in signal intensity compared with control spectra (data not shown). These observations were confirmed under all experimental conditions used (ammonium bicarbonate, pH 8.0, sodium bicarbonate, pH 7.2 and Hepes, pH 7.3).
Fig. 2.

Modification sites of PknG by OA-NO2. (A) MALDI-TOF mass spectrum of Cys-containing peptides generated by tryptic digestion of untreated PknG. Upper panel: peak m/z 1292.56 corresponds to sequence FC106WNC109GRPVGR with an intramolecular disulphide bridge. Middle panel: peak m/z 3530.62 corresponds to sequence GASEGWC128PYC131G-SPYSFLPQLNPGDIVAGQYEVK with an intra molecular disulphide bridge. Lower panel: peak m/z 1813.92 (GC156IAHGGLGWIYLALDR) is indicated. (B) MALDI-TOF mass spectrum of Cys-containing peptides generated by tryptic digestion of OA-NO2-treated PknG. Upper panel: peak m/z 1292.57 is partially consumed and a new peak appears showing a mass increment of 198/200/202 Da. Inset: zoom in showing the 198/200/202 pattern and isotopic distribution. Middle panel: peak m/z 3530.61 is also partially consumed and again a new signal appears showing a mass increment of 198/200/202 Da. Inset: zoom in showing the 198/200/202 mass increment pattern. Lower panel: signal intensity of m/z 1813.93 is unchanged on treatment with OA-NO2, and in agreement with this observation there were no new signals in this m/z range (the arrows indicate the expected m/z values: +327 for OA-NO2 modification and +198/200/202 for the previously experimentally observed mass shift). (C) MALDI-TOF MS/MS spectrum of ion at m/z 3730.60 obtained from tryptic digestion of OA-NO2-treated PknG. Sequence 122–154 showing main observed fragments (natives or modified). The spectra are representative of five independent experiments.
The expected mass shift for the incorporation of a molecule of OA-NO2 is 327 Da [13]. However, under this experimental conditions no peptide was detected with this mass increment. Analysis of MALDI-TOF/TOF mass spectra of OA-NO2-treated samples showed the systematic appearance of a cluster of peaks with 198, 200, and 202 Da greater masses than the CXXCG-containing peptides (Fig. 2B, upper and middle panel, inset). MS/MS spectra of the observed ions (m/z 1492.54 and 3730.59) confirmed that these ions corresponded to modified peptides containing the sequences 105–115 and 122–154, respectively. Fig. 2C shows the MS/MS spectra of the precursor ion corresponding to the modified sequence 122–154 (GASEGWC128PYC131GSPYSFLPQLNPGDIVAG-QYEVK) with a mass increment of 200 Da. The presence of native y-ion series up to y23 together with the detection of the modified y27 (+200 Da) indicates that any of the residues in between (i.e., 128CPYC131) might be modified by OA-NO2 (Fig. 2C). Based on the presence of small signals corresponding to unmodified y24, y24+200 Da, unmodified y26, and y26+200 Da we postulate that ion m/z=3730.59 is actually a mixture of singly modified peptides where C128 or C131 have been individually modified by OA-NO2 (Table S1). Similarly, MS/MS analysis of the other modified Rbx peptide (m/z 1492.54) showed that only daughter ions that contain the CWNC residues appeared with a modified mass (data not shown). Although there was not enough sequence information to identify the modified residue(s) within this motif, based on the previously reported reactivity of the nitrated fatty acid toward nucleophilic residues these results support that the Cys residues of those peptides are the main target of OA-NO2 [13].
To further characterize this modification and the observed atypical mass shift, all three PknG native tryptic peptides that contained Cys residues (m/z 1292.57, m/z 1813.92, and m/z 3530.60) were isolated by RP-HPLC and then treated with OA-NO2. The peptide with the sequence 155–171 (m/z 1813.92) is the only tryptic peptide of PknG that contains a single Cys. The modification pattern previously observed was found for all those three Cys-containing peptides treated with OA-NO2 (Fig. 3). The mass shift for the peptide containing a single Cys was 196/198/200 Da. This observation is in agreement with the observed mass shift of 198/200/202 Da for the CXXCG containing peptide, with respect to the native peptide with an intramolecular disulfide bond. These results indicate that the presence of a single Cys residue is enough to generate the observed mass shift in MALDI-TOF MS analyses. In contrast, the analysis of those same peptides by electrospray ionization-MS showed the expected mass increment of 327 Da, but not the 198/200/202 Da pattern (Figs. 4A and B). MS/MS spectra of the modified peptides unequivocally identified Cys as the residue modified by OA-NO2 (Figs. 4B and C). In aggregate, these results support the hypothesis of MALDI ionization-induced decomposition of Cys-nitro-fatty acid adducts. However, MALDI MS/MS spectra did not provide enough information to interpret the structure of the modification responsible for the 198/200/202 Da mass shift.
Fig. 3.

Treatment of isolated Cys-containing peptides from PknG with OA-NO2. (A) MALDI-TOF mass spectrum of peptides generated by tryptic digestion of native PknG. Peptides were purified by reverse-phase chromatography prior to MS analysis. Upper panel: peak m/z 1294.63 (FC106WNC109GRPVGR); middle panel: peak m/z 1815.04 (GC156IAHGGLGWIYLALDR); and lower panel: peak m/z 3532.80 (GASEGWC128PYC131GSPYSFLPQLNPGDIVAGQYEVK). The reduced peptides containing the CXXCG sequence are detected after the chromatographic step but are spontaneously converted into the intermolecular disulphide bridged oxidized form. (B) MALDI-TOF mass spectrum of peptides isolated in (A), treated with OA-NO2. Upper panel: a new peak appears showing a mass shift of 198/200/202 Da relative to m/z 1292.63; middle panel: a new peak appears showing a mass shift of 196/198/200 Da relative to m/z 1814.94; and lower panel: a new peak appears showing a mass shift of 198/200/202 Da relative to m/z 3530.61.
Fig. 4.

MS/MS spectra of OA-NO2 modified peptide 155–171. (A) MALDI ionization. MS/MS spectrum of ion at m/z 2014.31 (native peptide +200 Da) obtained using MALDI-TOF MS. (B) ESI ionization. MS/MS spectrum of ion at m/z 2141.85 (native peptide +327 Da) obtained using LC-MS. (C) List of theoretical m/z values of fragment ions from peptide of m/z 2141.85 (GC156IAHGGLGWIYLALDR modified by OA-NO2). y- and b-ions detected by LC-MS are highlighted in bold.
The PknG sequence contains a unique non-rubredoxin Cys (C156). Noticeably, no consumption of the peptide containing this free Cys was observed on treatment of PknG with OA-NO2 (Figs. 2A and B, lower panel). In agreement with this observation, no appearance of a new signal with the previously observed mass shift (198/200/202 Da) or the theoretical expected mass shift (+327 Da) was observed. To gain further insight into the reactivity of PknG-Cys residues, iodoacetamide was used to model cysteine alkylation. No modification of Rbx cysteines was observed for IAM concentrations up to 150 μM (Fig. S1). Conversely, the unbound Cys156 was alkylated by IAM under similar conditions, revealing a different reactivity of OA-NO2 vs other alkylating reagents for Cys residues in PknG. When treating PknG, nitro-fatty acid-modified cysteines tightly bound to the metal ion and inhibited the enzyme, while treatment with IAM only resulted in modification of free Cys without affecting kinase activity (Fig. S1). Thus Rbx-Cys, typically reported as non-reactive with alkylating reagents, is the principal target of reaction with an electrophilic nitroalkene.
At higher concentrations of OA-NO2 (50–80 μM; molar ratio PknG:OA-NO2 from 1:5 to 1:10) the modification of several His residues occurred at very low yields. In agreement with this observation, there was no significant consumption of His-containing peptides (Fig. S2 and Table 1). In contrast to cysteine modification, alkylated histidine showed the expected mass shift of 327 Da when analyzed by MALDI MS and ESI MS. MS/MS analysis of these peptides confirmed that the incorporation of the nitrated fatty acid occurred at a His residue (Table 1 and Fig. S2). To confirm that inhibition is a consequence of Cys and not His modification, studies were repeated using an active PknG construct where N-terminal and TPR domains were removed (PknGΔ74/TPR). This truncated construct preserves only His185 out of the seven previously modified His residues. After the exposure of PknGΔ74/TPR to OA-NO2 the enzyme is fully inhibited, the Cys-containing peptides from the Rbx domain appeared alkylated and another set of His residues was detected at low yields (Table S2 and Fig. S3).
Table 1.
Identification of OA-NO2-modified residues in PknG.
| OA-NO2:PknG ratioa
|
||||
|---|---|---|---|---|
| Low | High | |||
| Peptide from-to | Modified residue | Assigned sequenceb (observed m/z) Peptides containing modified cysteine residues (Δm=198/200/202Da) |
||
| 105–111 | C106 or C109 | FCWNCGR (1081.31/1083.30/1085.33) | x | x |
| 105–115 | C106 or C109 | FCWNCGRPVGR (1490.51/1492.54/1496.55) | x | x |
| 122–154 | C128 or C131 | GASEGWCPYCGSPYSFLPQLNPGDIVAGQYEVK (3728.55/3730.59/3732.60) Peptides containing modified histidine residues (Δm=327 Da) |
x | x |
| 182–199 | H185 | GLVHSGDAEAQAMAMAER (2171.09) | x | |
| 420–443 | H430 or H440 | STFGVDLLVAHTDVYLDGQVHAEK (2941.56) | x | |
| 488–510 | H488 | HGALDADGVDFSESVELPLMEVR (2813.41) | x | |
| 558–573 | H558 | HFTEVLDTFPGELAPK (2128.15) | x | |
| 635–640 | H635 | HFTTAR (1059.61) | x | |
| 698–714 | H702 or H711 | ASTNHILGFPFTSHGLR (2182.21) | x | |
| 733–743 | H733 | HRYTLVDMANK (1674.91) | x | |
Low OA-NO2:PknG ratio means a molar ratio in between 1:1 and 3:1 ([OA-NO2] between 8 and 30 μM). High OA-NO2:PknG ratio means a molar ratio 10:1 ([OA-NO2] =80 μM).
The sequence and alkylation sites were confirmed by MS/MS analysis.
Using both enzyme constructs, the Cys-Rbx domain encompasses the only OA-NO2-modified residues using concentrations up to 35 μM that can render detectable loss of PknG activity. Altogether, these data indicate that Cys residues at the Rbx domain are the targets of OA-NO2 and account for enzyme inhibition.
Irreversible PknG inhibition by the reversible OA-NO2-mediated nitroalkylation of the kinase
We have previously reported that nitrated fatty acids inhibit GAPDH activity by modification of nucleophilic amino acid residues in a thiol-reversible manner [13]. Herein, the reversibility of PknG nitroalkylation was analyzed. Treatment of nitroalkylated PknG with DTT (42 mM) or GSH (24 mM) did not restore kinase activity (Fig. 5A). Under the same conditions GAPDH activity, used as a control, was restored (Fig. 5A). PknG control activity was measured in the presence of the thiol-containing reagents. Neither DTT nor GSH have an effect per se on kinase activity under these experimental conditions.
Fig. 5.
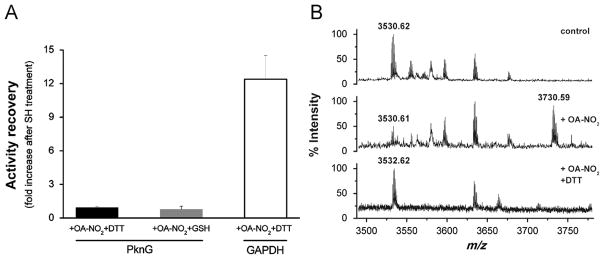
Irreversible PknG inhibition by reversible OA-NO2-mediated nitroalkylation. (A) PknG was inhibited by incubation with OA-NO2 (50 μM) in 70 mM ammonium bicarbonate, pH 8.0, at 25 °C for 10 min, as previously described. Kinase activity of PknG inhibited by OA-NO2 was measured (PknG+OA-NO2). Subsequently, nitroalkylated samples were treated with DTT (42 mM) or GSH (24 mM) for 15 min at 25 °C and kinase activity was remeasured (PknG+OA-NO2+RSH). Activity recovery was expressed as a ratio between [activity of PknG+OA-NO2+RSH]/[activity PknG+OA-NO2] where 1 means no activity recovery after thiol-containing agent treatment. GAPDH was used as a positive control: after DTT treatment 90% of the initial activity was recovered representing a 12-fold increase in the enzyme activity after the RSH treatment. Treatment of control PknG samples with 42 mM DTT or 24 mM GSH showed that these concentrations of the thiol reagents had no effect on kinase activity per se. Three independent experiments were performed. (B) MALDI-TOF mass spectrum of peptides generated by tryptic digestion of PknG. Upper panel: untreated PknG; middle panel: PknG exposed to OA-NO2; and lower panel: idem middle panel+42 mM DTT. In each panel the native or nitroalkylated form of the peptide with the sequence 122–154 that includes the Rbx-Cys 128 and 131 is shown. All the spectra are representative of three independent experiments.
To evaluate the reversibility of the modification of His and Cys residues in PknG, OA-NO2-treated samples were exposed to DTT and GSH before protein digestion and MS analysis. The peptide containing two Rbx-Cys residues (sequence 122–154) showed no change in mass after nitroalkylation and DTT treatment, supporting reversibility of this reaction of OA-NO2. In addition, native cysteine-containing peptides were fully recovered after treatment with DTT (Fig. 5B). Moreover, there were no detectable Cys- or His-nitrated fatty acid adducts after treatment with DTT or GSH (data not shown). Although nitroalkylation was reversible as expected, the effect of OA-NO2 on PknG activity was irreversible (Fig. 5A). These results indicate that OA-NO2 is inactivating PknG by an unusual mechanism involving an irreversible change in PknG structure or function that persists after Cys nitroalkylation is reversed.
OA-NO2 induces iron release from the Rbx domain
The rubredoxin domain contains an iron ion coordinated by the sulfur atoms of four conserved cysteine residues, forming an almost regular tetrahedron where Fe3+ is typically very stable [28,29]. To evaluate the effect of Cys nitroalkylation on the metal center of the Rbx domain, the amount of iron released on OA-NO2 treatment was measured using a specific ligand that allows for spectrophotometric determinations. Non-protein-bound iron present in control and OA-NO2-treated PknG samples was recovered, reduced with DTT, and quantified spectrophotometrically. The results showed that PknG nitroalkylation led to iron release from the Rbx domain (Fig. 6). These data showed that 70% of the iron present in the Rbx domain was released after protein exposure to 50 μM OA-NO2. As a positive control, total iron in native protein was determined after Rbx domain disruption by protein digestion (data not shown).
Fig. 6.

OA-NO2 induces iron release from Rbx domain. Non-protein-bound iron present in control and OA-NO2-treated PknG samples was recovered, reduced with DTT, and quantified as Fe2+ using bathophenantrolinedisulfonic acid. The complex absorbs at 535 nm (ε535 nm [BPS · Fe2+]=22140 M−1 cm−1 [17]). Iron present on native PknG samples (dashed line) or from OA-NO2-treated PknG (solid line) is shown.
PknG inhibition by OA-NO2 is not the consequence of a global change in PknG structure
In order to address if the effect of OA-NO2 could be mediated by an unspecific global distortion of protein tridimensional structure as a consequence of the introduction of a quite large hydrophobic molecule, global changes in protein structure were analyzed by different approaches. Enzyme was first treated with OA-NO2 and then ANS, a fluorescent probe that binds to hydrophobic patches on proteins with a concomitant change in emission spectrum (Fig. 7A). The same spectra were obtained with native and OA-NO2-modified PknG, supporting that the hydrophobic surfaces of the protein were not affected by nitroalkylation (Fig. 7A). This suggests that the enzyme inhibition was not due to large protein structural changes. In order to discard minor changes in secondary structure circular dichroism experiments were performed on PknG treated with OA-NO2 or OA (Fig. 7B). The comparison of the spectra showed no difference between both samples. Overall, these results suggest that PknG inhibition by OA-NO2 is not due to a general structural modification as a result of Cys nitroalkylation and iron release from the Rbx domain.
Fig. 7.

OA-NO2 modification does not generate a global change in PknG structure. (A) Fluorescence of PknG-ANS complexes was collected using excitation wavelength set on 350 nm and emission between 370 and 650 nm. PknG natively folded is represented as solid black line; PknG exposed to OA-NO2 is shown as solid gray line; and PknG thermally denatured is indicated as dashed black line. (B) Far UV-CD spectra of OA-NO2-treated PknG (black circles) and OA-treated PknG (white diamonds).
Rbx and kinase domain co-occurrence is restricted to few Actinomycetales
The specificity of OA-NO2 reaction with Cys residues in the Rbx domain of PknG raised the possibility of a selective inhibition of certain kinases containing this domain. A bioinformatics analysis informed about the co-occurrence of Rbx and kinase domains. Multiple sequence alignments of PknG orthologs showed a minority of PknG-like kinases harboring the CXXCG motif linked to Rbx domains (Fig. 8). In a subset of 652 PknG-like kinases, the Rbx domain was present in less than 10% (shown in red in Fig. 8). The same trend was observed in more thorough data sets; e.g., from 1294 proteins, just 88 had the N-terminal motif. The co-occurrence of Rbx and kinase domains in different species was also analyzed. Fig. 8 displays a maximum-likelihood phylogenetic tree for a set of 52 sequences with sequence identities below 90% and above 30%. At sequence identity levels above 25%, all but two PknG homologs carrying both domains were confined to few suborders of the Actinomycetales. The two exceptions were Acetivibrio cellulolyticus (Firmicutes) and Ktedonobacter racemifer (Chloroflexi).
Fig. 8.

Rbx and kinase domain co-occurrence is restricted to few Actinomycetales. Left: Distance-based tree of 652 sequence homologs with pairwise identities <76%. The sequences were aligned with Mafft [22] and the tree was built with BioNJ [39]. Zoom in the clade containing sequences harboring both kinase and Rbx domains. They were multiply aligned using T-Coffee [23] for a maximum-likelihood reconstruction with PhyML [26]. The two non actynobacterial sequences (A. cellulolyticus and K. racemifer) were omitted. Right: Maximum-likelihood phylogenetic reconstruction for a set of 52 dissimilar homologous proteins. Species names and accession numbers are shown. Branch support values are shown in red, omitting a number of internal nodes for clarity. Scale bar indicates average substitutions per site.
Conservation of the catalytic domain, as well as variability of the N-terminus, has been described for a number of PknGs [30,31]. However, the presence or absence of the Rbx domain and sequence conservation levels had not been analyzed. Notably, (a) the joint occurrence of kinase and Rbx domains is restricted to bacteria from the Actynomycetales order, including pathogenic and nonpathogenic mycobacteria and (b) the sequences indicated by a dot in Fig. 8 correspond to different species of Corynebacterium lacking the CXXCG motifs of the Rbx domain.
Discussion
The analysis of the M. tuberculosis genome sequence predicted the presence of 11 eukaryotic-like Ser/Thr protein kinases denoted as pknA to pknL [3]. The importance of these enzymes in myco-bacterial physiology and virulence has only recently been appreciated [1,32]. In particular, one of these enzymes, PknG, regulates critical processes in M. tuberculosis. Previous data support that PknG regulates glutamate metabolism in M. tuberculosis through the phosphorylation of GarA, an intermediate regulator of three metabolic enzymes [5,33]. PknG is also an important virulence factor that inhibits phagosome maturation in infected macrophages through an unknown mechanism [4,11]. In agreement with this, inhibition of PknG activity yields bacteria more susceptible to macrophage killing.
In this scenario, PknG inhibitors represent a promising drug development strategy, with a few PknG inhibitors of moderate activity previously described [2,8,34]. These inhibitors are directed toward the kinase catalytic site. For example, AX20017 displays some degree of specificity toward PknG, based on structural features of its active site [8]. It is already appreciated that active “eukaryotic-like” kinase folding is highly conserved even among different kingdoms [35], making inhibitor specificity problematic. An alternative strategy for selective kinase inhibition is to target other protein domains besides active site. Herein, we report that the inhibition of PknG kinase activity by OA-NO2 occurs by reversible alkylation of specific Cys residues of the Rbx domain, outside the catalytic domain.
The combination of Rbx and kinase domains has not been described for any other proteins than PknG-like kinases. PknG orthologs are present in all mycobacterial genomes sequenced to date, as well as in other related actinomycetes [31]. While the kinase domain is very well preserved, some other domains are not. In particular, the N-terminal Rbx domain appears in few PknG-like kinases from Actinomycetales (Fig. 8). Moreover, PknG ortholog from Corynebacterium spp, with the equivalent function toward glutamate metabolism, lacks the Rbx domain. This highlights the relevance of nitrated fatty acids as potential selective inhibitors acting on a subset of Rbx-containing PknG-like enzymes among prokaryotic and eukaryotic kinases.
The reported structure of a truncated form of PknG shows that the rubredoxin-like domain is closely associated with the N-terminal lobe of the catalytic domain of PknG, facing the kinase active site (Scheme 1). Thus, the modification of the Rbx domain could directly affect the active kinase conformation. Herein, the modification of Rbx-Cys by a nitroalkene was reversible and leads to a permanent effect on enzyme activity because of concomitant loss of an iron atom. Nitroalkylation of PknG did not induce a global change in structure (Fig. 7). In support of this, the conversion of bacterial rubredoxins to the apo form is followed by only minor structural changes [28], suggesting that minor local structural changes and/or the loss of iron could be mediating a regulatory role of kinase activity. To confirm the effect of iron loss on kinase activity, we attempted to reincorporate the iron ion into OA-NO2-treated PknG. Nevertheless, we were not able to reassemble the Rbx domain by OA-NO2 removal by thiol reagents in the presence of iron salts. In addition, previous attempts to generate apo-Rbx-PknG under nondenaturating conditions failed, and thus did not permit more definitive conclusions. Elucidating the effect of iron release on more detailed Rbx structure and function will require further investigation. Significantly, PknG is one of the two soluble kinases present in M. tuberculosis lacking an extracellular sensor domain. The signals that activate/inactivate PknG and the mechanism for activity regulation are still unknown.
Scheme 1.

Overall fold of PknG kinase and rubredoxin domains. The catalytic domain of M. tuberculosis PknG (PDB code 2PZI) shown in ribbon representation (colored from blue to red) interacts with the Rbx domain (ribbon+surface representation). The four Cys residues of the Rbx domain and the bound metal (red) are shown. The figure was drawn with Pymol.
Fatty acid nitroalkenes are electrophilic species produced during inflammation and metabolism that react with nucleophilic amino acid residues of target proteins (i.e., Cys and His residues), reversibly modulating protein function and subcellular distribution. Nitroalkene reactivity is dictated by the electrophilic character of the β-carbon proximal to the alkenyl NO2 group (Scheme 2) [16]. Nitroalkene fatty acids react with nucleophilic residues via a Michael addition to generate covalent adducts that can be reversed by thiol-containing molecules. This typically restores the native protein function [13]. Rbx-Cys are viewed as poor nucleophiles that do not react with low molecular weight alkylating reagents. However, mass spectrometry-based analysis confirmed that cysteines located at Rbx domain are the targets for nitroalkene reaction with PknG. While the kinase sequence contains reactive Cys and His residues, this is a novel reactivity of nitroalkenes with Rbx-Cys, wherein irreversible inhibition is achieved by reversible nitroalkylation of a redox-sensitive non-catalytic domain. The molecular basis of the selectivity of this reaction requires additional investigation.
Scheme 2.

Nitrated oleic acid (OA-NO2). Two regioisomers of OA-NO2 were synthesized by nitrosenylation of oleic acid yielding 9- and 10-nitro-9-cis-octade-cenoic acids. Taken from [16].
Xanthine oxidoreductase (XOR) inhibition by OA-NO2 was the first irreversible inhibition reported for fatty acid nitroalkenes. As in the case of PknG, XOR inhibition was not reversed by thiol reagents [15]. At that time, we postulated that the inhibition of XOR was mediated via: (1) an irreversible covalent reaction between OA-NO2 and XOR or (2) a reaction of the nitroalkene with the dithiolene of the pterin moiety and the concomitant loss of the molybdenum atom, with no direct evidence supporting either contention [15]. Inhibition of other enzymes by nitro-fatty acids was also previously reported [13,36]. OA-NO2 inhibition of both GAPDH and PknG is achieved within the same range of micromolar concentrations. It is important to note that OA-NO2 is considered a potent inhibitor of GAPDH, a key enzyme of intermediate metabolism that, due to a catalytic Cys residue, has also been postulated to be a redox sensor. OA-NO2 is almost an order of magnitude more reactive toward GAPDH than hydrogen peroxide and peroxynitrite [13].
Covalent inhibitors display time-dependent inhibition and their potency must be characterized by the inactivation rate for different inhibitor concentrations. In the case of OA-NO2, the determination of an inhibition constant for PknG is difficult for several reasons. OA-NO2 concentration cannot be readily increased over its critical micelle concentration and increasing inhibitor concentration also increases the number of nonspecific covalent modifications, complicating kinetic analysis. There is a remarkable effect on kinase activity, with micromolar concentrations of OA-NO2 at PknG:OA-NO2 ratios of 1:3 and 1:5. Even though this does not allow us to evaluate inhibition constants, the data point to a potent effect of OA-NO2 on kinase activity.
Analysis of Cys-alkylated peptides showed an unexpected mass shift by MALDI-TOF/TOF analysis. We hypothesized that laser ionization induces photodecomposition of cysteine-OA-NO2 adducts, generating the observed pattern with a mass shift of 198/200/202 Da. In support of this, decomposition of nitro-compounds during MALDI analysis has been previously reported [37]. In contrast to cysteine modification, alkylated histidine showed the expected mass shift of 327 Da, indicating that the mass increment of 198/200/202 Da is a fingerprint of Cys modification when detected by MALDI MS. This can also explain why Cys-OA-NO2 adducts were not detected in previous MALDI analyses [13]. MALDI MS/MS spectra of the modified peptides with this atypical mass increments did not show fragments of the 198/200/202 Da modification, thus not allowing a structural characterization.
This study exploited the unique structural characteristics of the multidomain protein kinase PknG for specific inhibition of enzymatic activity, revealing a new mechanism for PknG inhibition involving the Rbx domain rather than the catalytic domain. Electrophilic fatty acids thus represent a class of mediators that can induce the specific inhibition of a small subset of kinases containing a Rbx domain. Many in vivo studies have been performed with nitro oleic acid and are recently reviewed in Schopfer et al. [38]. This warrants further study of these PknG inhibitors in more complex model systems.
Supplementary Material
Acknowledgments
This work was supported in part by the Agencia Nacional de Investigación e Innovación (Uruguay), project FCE2009_2479 (to R.D. and C.B.) and NIH R01-HL058115, R01-HL64937, P01-HL103455 (B.A.F.), and R01 AT-006822 (F.J.S.). We are grateful to Dr. Bruno Baron (Biophysics of Macromolecules and their Interactions Platform, Institut Pasteur) for his technical assistance with our CD studies. We thank the Agencia Nacional de Investigación e Innovación (ANII, Uruguay) and AMSUD-Pasteur Program for research fellowships (to M.G.). B.A.F. and F.J.S. acknowledge financial interest in Complexa, Inc.
Abbreviations
- ANS
8-anilino-1-naphthalenesulfonic acid ammonium salt
- BPS
bathophenanthroline disulfonate
- ESI
electrospray ionization
- IAM
iodoacetamide
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- LC
liquid chromatography
- OA oleic acid
9-octadecenoic acid
- OA-NO2 nitro-oleic acid
9- and 10-nitro-9-cis-octade-caenoic acids
- Rbx
rubredoxin
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2013.06.021.
References
- 1.Wehenkel A, Bellinzoni M, Grana M, Duran R, Villarino A, Fernandez P, Andre-Leroux G, England P, Takiff H, Cervenansky C, Cole ST, Alzari PM. Mycobacterial Ser/Thr protein kinases and phosphatases: physiological roles and therapeutic potential. Biochim Biophys Acta. 2008;1784:193–202. doi: 10.1016/j.bbapap.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Szekely R, Waczek F, Szabadkai I, Nemeth G, Hegymegi-Barakonyi B, Eros D, Szokol B, Pato J, Hafenbradl D, Satchell J, Saint-Joanis B, Cole ST, Orfi L, Klebl BM, Keri G. A novel drug discovery concept for tuberculosis: inhibition of bacterial and host cell signalling. Immunol Lett. 2008;116:225–231. doi: 10.1016/j.imlet.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 4.Walburger A, Koul A, Ferrari G, Nguyen L, Prescianotto-Baschong C, Huygen K, Klebl B, Thompson C, Bacher G, Pieters J. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 2004;304:1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 5.O’Hare HM, Duran R, Cervenansky C, Bellinzoni M, Wehenkel AM, Pritsch O, Obal G, Baumgartner J, Vialaret J, Johnsson K, Alzari PM. Regulation of glutamate metabolism by protein kinases in mycobacteria. Mol Biol. 2008;70:1408–1423. doi: 10.1111/j.1365-2958.2008.06489.x. [DOI] [PubMed] [Google Scholar]
- 6.Cowley S, Ko M, Pick N, Chow R, Downing KJ, Gordhan BG, Betts JC, Mizrahi V, Smith DA, Stokes RW, Av-Gay Y. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol Biol. 2004;52:1691–1702. doi: 10.1111/j.1365-2958.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- 7.Niebisch A, Kabus A, Schultz C, Weil B, Bott M. Corynebacterial protein kinase G controls 2-oxoglutarate dehydrogenase activity via the phosphorylation status of the OdhI protein. J Biol Chem. 2006;281:12300–12307. doi: 10.1074/jbc.M512515200. [DOI] [PubMed] [Google Scholar]
- 8.Scherr N, Honnappa S, Kunz G, Mueller P, Jayachandran R, Winkler F, Pieters J, Steinmetz MO. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2007;104:12151–12156. doi: 10.1073/pnas.0702842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sieker LC, Stenkamp RE, LeGall J. Rubredoxin in crystalline state. Methods Enzymol. 1994;243:203–216. doi: 10.1016/0076-6879(94)43016-0. [DOI] [PubMed] [Google Scholar]
- 10.van Beilen JB, Neuenschwander M, Smits TH, Roth C, Balada SB, Witholt B. Rubredoxins involved in alkane oxidation. J Bacteriol. 2002;184:1722–1732. doi: 10.1128/JB.184.6.1722-1732.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiwari D, Singh RK, Goswami K, Verma SK, Prakash B, Nandicoori VK. Key residues in Mycobacterium tuberculosis protein kinase G play a role in regulating kinase activity and survival in the host. J Biol Chem. 2009;284:27467–27479. doi: 10.1074/jbc.M109.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schopfer FJ, Batthyany C, Baker PR, Bonacci G, Cole MP, Rudolph V, Groeger AL, Rudolph TK, Nadtochiy S, Brookes PS, Freeman BA. Detection and quantification of protein adduction by electrophilic fatty acids: mitochondrial generation of fatty acid nitroalkene derivatives. Free Radic Biol Med. 2009;46:1250–1259. doi: 10.1016/j.freeradbiomed.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Batthyany C, Schopfer FJ, Baker PR, Duran R, Baker LM, Huang Y, Cervenansky C, Branchaud BP, Freeman BA. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schopfer FJ, Cole MP, Groeger AL, Chen CS, Khoo NK, Woodcock SR, Golin-Bisello F, Motanya UN, Li Y, Zhang J, Garcia-Barrio MT, Rudolph TK, Rudolph V, Bonacci G, Baker PR, Xu HE, Batthyany CI, Chen YE, Hallis TM, Freeman BA. Covalent peroxisome proliferator-activated receptor gamma adduction by nitro-fatty acids: selective ligand activity and anti-diabetic signaling actions. J Biol Chem. 2010;285:12321–12333. doi: 10.1074/jbc.M109.091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley EE, Batthyany CI, Hundley NJ, Woodcock SR, Bonacci G, Del Rio JM, Schopfer FJ, Lancaster JR, Jr, Freeman BA, Tarpey MM. Nitro-oleic acid, a novel and irreversible inhibitor of xanthine oxidoreductase. J Biol Chem. 2008;283:36176–36184. doi: 10.1074/jbc.M802402200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blair D, Diehl H. Bathophenanthrolinedisulphonic acid and bathocuproine-disulphonic acid, water soluble reagents for iron and cooper. Talanta. 1961;7:163–174. [Google Scholar]
- 18.Weber G, Young LB. Fragmentation of bovine serum albumin by pepsin. I. The origin of the acid expansion of the albumin molecule. J Biol Chem. 1964;239:1415–1423. [PubMed] [Google Scholar]
- 19.Biegert A, Soding J. Sequence context-specific profiles for homology searching. Proc Natl Acad Sci USA. 2009;106:3770–3775. doi: 10.1073/pnas.0810767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soding J, Remmert M, Biegert A, Lupas AN. HHsenser: exhaustive transitive profile search using HMM-HMM comparison. Nucleic Acids Res. 2006;34:W374–378. doi: 10.1093/nar/gkl195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Briefings Bioinformatics. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 23.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 24.Loytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci USA. 2005;102:10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. System Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 26.Guindon S, Delsuc F, Dufayard JF, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009;537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 27.Villarino A, Duran R, Wehenkel A, Fernandez P, England P, Brodin P, Cole ST, Zimny-Arndt U, Jungblut PR, Cervenansky C, Alzari PM. Proteomic identification of M. tuberculosis protein kinase substrates: PknB recruits GarA, a FHA domain-containing protein, through activation loop-mediated interactions. J Mol Biol. 2005;350:953–963. doi: 10.1016/j.jmb.2005.05.049. [DOI] [PubMed] [Google Scholar]
- 28.Bonomi F, Iametti S, Kurtz DM, Jr, Ragg EM, Richie KA. Direct metal ion substitution at the [M(SCys)4]2P site of rubredoxin. J Biol Inorg Chem. 1998;3:595–505. [Google Scholar]
- 29.Messerschmidt A, Huber R, Poulos T, Wieghardt K, editors. Handbook of metalloproteins. Hoboken, NJ: Wiley; 2001. [Google Scholar]
- 30.Fiuza M, Canova MJ, Zanella-Cleon I, Becchi M, Cozzone AJ, Mateos LM, Kremer L, Gil JA, Molle V. From the characterization of the four serine/threonine protein kinases (PknA/B/G/L) of Corynebacterium glutamicum toward the role of PknA and PknB in cell division. J Biol Chem. 2008;283:18099–18112. doi: 10.1074/jbc.M802615200. [DOI] [PubMed] [Google Scholar]
- 31.Narayan A, Sachdeva P, Sharma K, Saini AK, Tyagi AK, Singh Y. Serine threonine protein kinases of mycobacterial genus: phylogeny to function. Physiol Genomics. 2007;29:66–75. doi: 10.1152/physiolgenomics.00221.2006. [DOI] [PubMed] [Google Scholar]
- 32.Alber T. Signaling mechanisms of the Mycobacterium tuberculosis receptor Ser/Thr protein kinases. Curr Opin Struct Biol. 2009;19:650–657. doi: 10.1016/j.sbi.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nott TJ, Kelly G, Stach L, Li J, Westcott S, Patel D, Hunt DM, Howell S, Buxton RS, O’Hare HM, Smerdon SJ. An intramolecular switch regulates phosphoindependent FHA domain interactions in Mycobacterium tuberculosis. Sci Signal. 2009;2ra12 doi: 10.1126/scisignal.2000212. [DOI] [PubMed] [Google Scholar]
- 34.Anand N, Singh P, Sharma A, Tiwari S, Singh V, Singh DK, Srivastava KK, Singh BN, Tripathi RP. Synthesis and evaluation of small libraries of triazolylmethoxy chalcones, flavanones and 2-aminopyrimidines as inhibitors of mycobacterial FAS-II and PknG. Bioorg Med Chem. 2012 doi: 10.1016/j.bmc.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Ortiz-Lombardia M, Pompeo F, Boitel B, Alzari PM. Crystal structure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J Biol Chem. 2003;278:13094–13100. doi: 10.1074/jbc.M300660200. [DOI] [PubMed] [Google Scholar]
- 36.Bonacci G, Schopfer FJ, Batthyany CI, Rudolph TK, Rudolph V, Khoo NK, Kelley EE, Freeman BA. Electrophilic fatty acids regulate matrix metalloproteinase activity and expression. J Biol Chem. 2011;286:16074–16081. doi: 10.1074/jbc.M111.225029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarver A, Scheffier NK, Shetlar MD, Gibson BW. Analysis of peptides and proteins containing nitrotyrosine by matrix-assisted laser desorption/ionization mass spectrometry. J Am Soc Mass Spectrom. 2001;12:439–448. doi: 10.1016/S1044-0305(01)00213-6. [DOI] [PubMed] [Google Scholar]
- 38.Schopfer FJ, Cipollina C, Freeman BA. Formation and signaling actions of electrophilic lipids. Chem Rev. 2012;111:5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desper R, Gascuel O. Getting a tree fast: Neighbor Joining, FastME, and distance-based methods. Curr Protoc Bioinformatics. 2006;Chapter 6(Unit 6):3. doi: 10.1002/0471250953.bi0603s15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.