

Abstract
Reported herein is the first stereoselective synthesis of (2S,3R)-4-[bis-(tert-butyloxy)phosphinyl]-2-[(9H-fluoren-9-ylmethoxy)carbonyl]amino-3-methylbutanoic acid [(N-Fmoc, O,O-(bis-(tert-butyl))-Pmab, 4] as a hydrolytically-stable phosphothreonine mimetic bearing orthogonal protection compatible with standard solid-phase protocols. The synthetic approach used employs Evans’ oxazolidinone for chiral induction. Also presented is the application of 4 in the solid-phase synthesis of polo-like kinase 1 (Plk1) polo box domain (PBD)-binding peptides. These Pmab-containing peptides retain PBD binding efficacy similar to a parent pThr containing peptide. Reagent 4 should be a highly useful reagent for the preparation of signal transduction-directed peptides.
Phosphorylation of proteins facilitates critical protein-protein binding interactions that may result in signal propagation or modulation of enzyme activity.1–4 Changes in normal post-translational modification of proteins through phosphorylation of tyrosine, serine and threonine residues is a central paradigm in oncogenic transformation.5–7 In light of this, development of kinase-directed signal transduction inhibitors is a promising approach toward new anticancer therapeutics.8–10 Synthetic phosphopeptides based on shortened sequences derived from phosphoproteins, can retain significant binding affinities and they can serve as competitive antagonists of cognate protein-protein interactions. In this fashion they can provide initial starting points for the design of peptidomimetic-based therapeutics. Typically, a key component of the recognition provided by phosphoamino acids is derived from the phosphoryl group itself.11 However the hydrolytic lability of phosphoryl esters to phosphatases limits the use of phosphopeptides in cellular contexts. Development of hydrolytically-stable mimetics, in which the labile phosphoryl ester oxygen has been replaced non-hydrolyzable methylene or difluoromethylene groups, offers one approach to circumvent this limitation. Peptides containing metabolically stable analogues have proven to be useful biological tools that may serve as potential leads for further therapeutic design.12–17
Although a significant body of literature exists concerning the development and application of phosphotyrosyl (pTyr) mimetics,18 fewer examples can be found dealing with mimetics of phosphothreonine (pThr, 1, Figure 1). Stereoselective synthesis of the pThr mimetic (2S,3R)-2-amino-3-methyl-4-phosphonobutanoic acid (Pmab, 2) has been reported using Schollkopf’s bislactim ether. This has provided derivatized Pmab bearing O,O-(bis-allyl) protection of the phosphonic acid group along with N-Fmoc protection.19 Synthesis of the corresponding 4,4-difluoro analogue (F2Pmab, 3) bearing O,O-(bis-ethyl) phosphonic acid and N-Boc protection groups, has been approached using both (R)-isopropylideneglycerol as a chiral synthon20 and Oppolzer’s sultam chiral auxiliary.21 To date, there have been no stereoselective syntheses reported of Pmab bearing orthogonal O,O-[bis-(tert-butyl)] phosphonic acid and N-Fmoc groups. This protection scheme would allow facile use in standard solid-phase protocols on acid-labile resins. Therefore, we report herein the first synthesis of (2S,3R)-4-[bis-(tert-butyloxy)phosphinyl]-2-[(9H-fluoren-9-ylmethoxy)carbonyl]amino-3-methylbutanoic acid [(N-Fmoc, O,O-(bis-(tert-butyl))-Pmab, 4] by a route using Evans’ oxazolidinone for chiral induction. We also present the application of this reagent in the solid-phase synthesis of a biologically active peptide.
Figure 1.
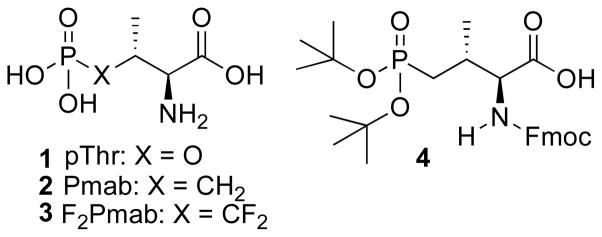
Structures of pThr and pThr mimetics discussed in the text.
Chemistry
Stereoselective synthesis of orthogonally-protected Pmab (4) began with the Swern oxidation of tert-butyldimethylsilyl (TBDMS) mono-protected (2E)-2-butene-1,4-diol 522 followed by sodium chlorite oxidation. This provided acid 6 with Z-double bond geometry (previously reported as the E-isomer23) (Scheme 1). Acid 6 was coupled with the Evan’s chiral auxiliary, (4R)-4-phenyl-2-oxazolidinone24 and the Z-double bond geometry was isomerized by treatment with tri-n-butyl phosphine in THF to give the desired E-isomer (7). Both α and β stereogenic centers of 9 were constructed by a tandem sequence consisting of an asymmetric Cu(I)-catalyzed 1,4-Michael addition of methylmagnesium chloride followed by electrophilic α-bromination.25 The crude (2R)-bromide was then converted to the corresponding (2S)-azide by nuclephilic SN2 replacement using sodium azide. A single (2S,3R)-diastereomer (9) was obtained by column chromatographic purification and crystallization. Assignment of absolute stereochemistries was based on well-established literature precedence.25–27 Removal of the TBDMS group by treatment with catalytic p-toluenesulfonic acid was followed by cyclization to release the Evan’s auxiliary group and provide the 5-membered lactone. The azide was reduced by hydrogenation in a mixture of AcOH and MeOH and protected in situ to provide the lactone 10 as well as the ring-open alcohol 11 in a 1 to 4 ratio. Lactone 10 was further converted to 11 (Scheme 1).
Scheme 1.

Reagents and conditions: (a) 1. Oxalyl chloride, DMSO, DCM, −78 °C, 2 hrs; 2. NaClO2, KH2PO4, 2-methyl-2-butene, tert-butanol/H2O, rt, overnight, 97% for 2 steps. (b) 1. Trimethylacetyl chloride, triethylamine, THF, −78 °C - 0 °C, 20 min; 2. (R)-(+)-Phenyl-2-oxazolidione lithium salt, THF, −78 °C - 0 °C, overnight, 100%. (c) Tributylphosphine, THF, rt, 1 hr, 84%. (d) 1. Methylmagnesium chloride, copper(I) bromide•dimethyl sulfide, dimethyl sulfide/THF, −78 °C - −40 °C, 2 hr; 2. NBS, −78 °C, 1.5 hr; 3. NaN3, DMF, 0°C, 2 hr, 79% for 2 steps. (e) 1. p-Toluenesulfonic acid monohydrate, MeOH, rt, 6 hr; 2. 1 atm H2, 10% Pd•C (10%), MeOH/AcOH, rt, overnight; 3. Benzyl chloroformate, NaHCO3, THF/H2O, 0 °C, 4 hr, 49% for 11 over 3 steps.
It is of note that alchohol 11 can also be prepared from L-aspartic acid through the known bis-methyl ester 13.28 Selectively reduction of the γ-carboxyl of 13 using DIBAL provided the alcohol 14 (Scheme 2). Key to this reaction was the use of substrate concentrations less than 0.03 M. Similar to above, N-deprotection of 14 by hydrogenation in a mixture of AcOH and MeOH and subsequent Cbz protection gave the lactone 10 and the alcohol 11 in a 1 to 4 ratio.
Scheme 2.

Reagents and conditions: (a) DIBAL, THF, −40 °C - 0 °C, 4 hr, 61%. (b) 1. 1 atm H2, 10% Pd•C (10%), MeOH/AcOH, rt, overnight; 2. Benzyl chloroformate, NaHCO3, THF/H2O, 0 °C, 4 hr.
Swern oxidation of alcohol 11 gave the corresponding aldehyde (15). This aldehyde was subjected to a phospho-Mukaiyama aldol reaction with freshly-prepared di-tert-butyltrimethylsilyl phosphite (17) to yield the aldehyde 17 (Scheme 3).29, 30 Subsequent treatment with citric acid gave the free alcohol (18), which was derivatized as the phenylthiocarbonate 19 and subjected to Barton-McCombie deoxygenation to yield 20.31 Hydrolysis of the methyl ester, then hydrogenation and re-protection using Fmoc-OSu provided the orthogonally protected Pmab derivative 4.
Scheme 3.

Reagents and conditions: (a) Oxalyl chloride, DMSO, DCM, −78 °C, 2 hrs. (b) 16, DCM, rt, 3 hr. (c) citric acid, MeOH/H2O, rt, overnight, 88% over 3 steps. (d) O-phenylchlorothionoformate, DMAP (cat.) and N, N-diisopropylethylamine, DCM, r.t, overnight. (e) Tributyltin hydride, AIBN, toluene, 100 °C, 20 min, 58% over 2 steps. (f) 1. LiOH, THF/H2O, rt, overnight; 2. 1 atm H2, 10% Pd•C (10%), MeOH, rt, overnight; 3. FmocOSu, NaHCO3, dioxane/ H2O, rt, overnight, 100% over 3 steps.
Application of reagent 4 to the synthesis of Polo Box Domain - binding peptides
The polo-like kinase 1 (Plk1) functions as an important mitotic regulator that phosphorylates serine and threonine residues.32 Its over-expression in a number of cancers33 and its association with poor prognosis have made it a potential anticancer therapeutic target.34, 35 A main focus of Plk1 inhibitor development has been directed at the kinase catalytic domain.36–43 However, Plk1 contains modular C-terminal “Polo-box domains” (PBDs) that bind specific phosphoserine and phosphothreonine-containing sequences to provide critical localization of Plk1.32, 44, 45 Competitive PBD binding antagonists could serve as inhibitors of Plk1 function that are distinct from kinase-directed agents.44 A starting point for the development of PBD-binding antagonists is given by short pThr-containing peptides modeled on consensus binding sequences derived from the p-Thr78 region (p-T78) of the PBD-binding protein, PBIP1.47, 48 By examining various PBD-binding phosphpeptides, it has recently been shown that a 5–mer phosphopeptide “PLHSpT” (21, Figure 2) specifically interacts with the Plk1 PBD with high affinity (Kd = 0.45 μM).49 In order to provide phosphatase–stable peptides for in vivo studies, F2Pmab (3) was also incorporated into a 6-mer T78 peptide, “PLHSTA”, to give the corresponding peptide 25.49 (Note: The 6-mer sequence “PLHS-F2Pmab-A” (25) was synthesized due to inefficient synthesis of the 5-mer sequence, “PLHS-F2Pmab”). It was found that 25 showed much weaker PBD-binding affinity than the respective p-T78 peptide, “PLHSpTA”, and it exhibited significant toxicity in cell-based experiments. The toxicity can potentially be attributed to the highly acidic CF2PO3H moiety. Therefore, using solid-phase techniques and standard Fmoc-based protocols, we employed reagent 4 to synthesize the Pmab-containing peptides 23 and 24 (Figure 2).
Figure 2.

Structures of synthetic peptides used in the study.
To examine the ability of Pmab- and F2Pmab-containing peptides to inhibit PBD-dependent interactions, Plk1 PBD-binding inhibition assays were conducted in the presence of various concentrations of synthetic peptides. It was found that “PLHS-Pmab” (23) inhibits the interaction of the Plk1 PBD with a biotinylated 9-mer p-T78 peptide [Biotin-Cys-(CH2)5-CO-DPPLHSpTAI-NH2] as effectively as the wild-type peptide, “PLHSpT” (22, Figure 3A). In contrast, the peptide, “PLHS-F2Pmab-A” (25, Figure 2), inhibits the interaction at a somewhat reduced level. Replacement of the critical (pThr-1) Ser residue with an alanine (equivalent to S77A mutation) is known to significantly attenuate PBD binding affinity.49 The non-phosphorylated control peptide “PLHST” (21, Figure 2) and the S77A mutants of the Pmab- and the F2Pmab-containing peptides (24 and 26, respectively, Figure 2), did not inhibit PBD binding even at 1000-fold higher molar concentrations (Figure 3A).
Figure 3.

Measurement of the ability of synthetic peptides 21 – 24 to inhibit PBD-dependent interactions. (A) PBD-binding inhibition assays were carried out in the presence of different concentrations of the indicated inhibitory peptides.49 The level of the remaining interaction between a biotinylated p-T78 peptide and full-length Plk1 was quantified by optical density (O.D.) at 450 nm (error bars represent standard deviation). (B) Representative images of green fluorescence in EGFP plasmid-containing HeLa cells following microinjection with PLHS-Pmab (23) or the PBD-binding defective peptides, PLHST (21) and PLHA-Pmab (24), are shown (procedure described in the Experimental Section). Note induction of mitotically-arrested, rounded-up, morphologies associated with the PBD-binding competent PLHS-Pmab.
Evidence suggests that the PBD plays critical roles in the proper sub-cellular localization and mitotic functions of Plk1. Disruption of PBD-dependent Plk1 functions by expressing a dominant-negative form of PBD results in a mitotic arrest that ultimately leads to apoptotic cell death.50 To investigate the effects of inhibiting Plk1 PBD interactions peptides 21, 23 and 24 were introduced into HeLa cells. In order to overcome poor membrane permeability of the negatively charged Pmap-containing peptides, microinjection was employed. The Pmab-containing peptide (23), but not the non-phosphorylated peptide 21 or the respective S77A mutant (24), induced mitotically arrested, rounded-up, morphology in approximately 50% of the microinjected, green fluorescent protein (GFP)-positive population (Figure 3B). These results demonstrate that inhibition of PBD function by the Pmab-containing p-T78 mimetic peptide is sufficient to interfere with the mitotic functions of Plk1.
Conclusions
Although a significant body of literature exists concerning the development and application of pTyr mimetics, fewer examples can be found dealing with mimetics of pThr. Presented herein is the first stereoselective synthesis of the hydrolytically-stable phosphothreonine mimetic Pmab (4), bearing (O,O)-bis-tert-butyl protection of the phosphonic acid group along with N-Fmoc derivatization. This orthogonal protection scheme allows facile use in standard solid-phase protocols on acid-labile resins, where side chain protecting groups can be removed during TFA-mediated resin cleavage. Our synthetic approach to Pmab utilizes Evans’ oxazolidinone for chiral induction. We also present the application of 4 in the solid-phase synthesis of biologically active peptides directed against the Plk1 PBD. Here we show that Pmab-containing peptides retain PBD binding efficacy similar to a parent pThr-containing peptide, while retaining the ability to inhibit PBD-dependent interactions in whole cells. In summary, reagent 4 should be a highly useful reagent for the preparation of signal transduction-directed peptides.
Experimental Procedures
General
All experiments involving moisture-sensitive compounds were conducted under dry conditions (positive argon pressure) using standard syringe, cannula, and septa apparatus. Solvents: All solvents were purchased anhydrous (Aldrich) and used directly. HPLC-grade hexanes, EtOAc, CH2Cl2, and MeOH were used in chromatography. TLC: analytical TLC was performed on Analtech precoated plates (Uniplate, silica gel GHLF, 250 microns) containing a fluorescence indicator; NMR spectra were recorded using a Varian Inova 400 MHz spectrometer. The coupling constants are reported in Hertz, and the peak shifts are reported in the δ (ppm) scale. Low resolution mass spectra (ESI) was measured with Agilent 1200 LC/MSD-SL system, and high resolution mass spectra (ESI or APCI) was measured by UCR Mass Spectrometry Facility, Department of Chemistry, University of California, 3401 Watkins Dr., Riverside CA, 92521. Optical rotations were measured on a Jasco P-1010 polarimeter at 589 nm. IR spectra were obtained neat with a Jasco FT-IR/615 spectrometer.
ELISA-based PBD-binding inhibition assays
An ELISA-based PBD-binding inhibition assay was carried out using an immobilized biotinylated 9-mer p-T78 peptide [Biotin-Cys-(CH2)5-CO-DPPLHSpTAI-NH2] and cellular lysates expressing HA-EGFP-Plk1 as described in reference 49. Results are shown in Figure 3A.
Cell microinjection and confocal microscopy
Similar to procedures described in reference 49, HeLa cells were arrested at the G1/S boundary by double thymidine treatment and released into fresh medium. Six hours after release, the cells were microinjected with a mixture of 3 mM of peptides 21, 23 or 24 and 30 ng/μL of pEGFP-C1 vector and the cells were then photographed 15 h after G1/S release. Co-injected EGFP plasmid provided a convenient marker to identify the microinjected cells. Results are shown in Figure 3B.
Synthetic
(E)-4-[(Tert-butyldimethylsilyl)oxy]-2-buten-1-ol (5)
To a solution of (2E)-2-butene-1,4-diol (8.22 mL, 0.10 mol) and imidazole (8.50 g, 0.125 mol) in DMF (50 mL) at 0 °C, was added tert-butyldimethylsilyl chloride (7.50 g, 0.050 mol) in several portions over 10 minutes. The resulting mixture was warmed to room temperature and stirred (2 h), then poured into H2O (200 mL) and extracted with EtOAc (2 × 150 mL). The organic layer was washed (brine), dried (Na2SO4) and purified by silica gel column chromatography (hexanes: EtOAc) to yield 5 as a colorless oil (9.0 g, 89% yield). 1H NMR (400 MHz, CDCl3) δ 5.60 – 5.50 (m, 2 H), 4.18 (m, 2 H), 4.09 (m, 2 H), 2.76 (br, 1 H), 0.83 (s, 9 H), 0.01 (s, 6 H). 13C NMR (100 MHz, CDCl3) δ 136.3, 135.4, 64.8, 63.8, 31.1, 23.6.
Z-4-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-2-butenoic acid (6)
To a solution of oxalyl chloride (3.55 mL, 40.8 mmol) in CH2Cl2 (100 mL) at −78° C, was added a solution of DMSO (5.80 mL, 81.7 mmol) in CH2Cl2 (40 mL) and the mixture was stirred (15 minutes). Alcohol 5 (5.50 g, 27.2 mmol) in dry CH2Cl2 (40 mL) was added over 5 minutes, then the mixture was stirred at −75° C (2 h) then triethylamine (31 mL, 0.22 mol) was added. The mixture was warmed room temperature and saturated NH4Cl (50 mL) was added and the mixture was extracted with Et2O (2 × 100 mL). The combined organic layers were washed (brine), dried (Na2SO4) and evaporated to yield the intermediate aldehyde as a pale yellow liquid. Without purification, to a mixture of the aldehyde, potassium phosphate monobasic (5.55 g, 40.8 mmol) and 2-methyl-2-butene (14.4 mL, 136 mmol) in tert-butanol (150 mL) and H2O (30 mL) at 0° C, was added sodium chlorite (9.23 g, 81.6 mmol, 80% technical grade) in several portions over 10 minutes. The mixture was warmed to room temperature slowly and stirred (night). After cooling to 0° C, a solution of sodium bisulfate (31.8 g, 0.30 mol) in H2O (100 mL) was added slowly and the mixture was stirred (30 minutes) and extracted with EtOAc (2 × 150 mL). The combined organic layer was washed (brine), dried (Na2SO4) and purified by silica gel column chromatography (hexanes: EtOAc) to yield acid 6 as a colorless oil (5.70 g, 97% yield). 1H NMR (400 MHz, CDCl3) δ 6.40 (dt, J = 11.6, 4.6 Hz, 1 H), 5.68 (dt, J = 12.0, 2.6 Hz, 1 H), 4.65 (dd, J = 4.6, 2.4 Hz, 2 H), 0.83 (s, 9 H), 0.00 (s, 6 H). 13C NMR (100 MHz, CDCl3) δ 176.0, 159.7, 123.0, 67.1, 31.0, 23.0, 0.00. APCI (−VE) m/z: 215.2 (M − H)−. HR-ESI MS cacld for C10H19O3Si (M − H)−: 215.1109, Found: 215.1103.
(4R)-3-[(2Z)-[4-[(1,1-Dimethylethyl)dimethylsilyl]oxy]-1-oxo-2-buten-1-yl]-4-phenyl-2-oxazolidinone (7)
To a solution of acid 6 (6.0 g, 28.2 mmol) in THF (40 mL) at −78 °C, was added triethylamine (4.00 mL, 28.2 mmol) followed by trimethylacetyl chloride (3.46 mL, 28.2 mmol) drop-wise. The mixture was warmed to 0 °C over 20 minutes, then the anhydride mixture was cooled to −78 °C. Separately, to a solution of (R)-(+)-phenyl-2-oxazolidione (Aldrich) (4.60 g, 28.2 mmol) in THF (40 mL) at −78 °C was carefully added n-BuLi (2.50 M in THF, 11.3 mL, 28.2 mmol) and the mixture was stirred (30 minutes) then transferred to the anhydride solution at −78° C. The final reaction mixture was warmed to room temperature and stirred (over night). The mixture was diluted with EtOAc (200 mL), washed (H2O and brine), dried (Na2SO4), and purified by silica gel column chromatography (hexanes: EtOAc) to yield 7 as a colorless oil (10.2 g, 100% yield). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.20 (m, 5 H), 7.10 (dt, J = 11.6, 2.6 Hz, 1 H), 6.50 (dt, J = 12.0, 4.6 Hz, 1 H), 5.44 (dd, J = 8.8, 4.0 Hz, 1 H), 4.68 – 4.59 (m, 3 H), 4.22 (dd, J = 8.8, 4.0 Hz, 1 H), 0.85 (s, 9 H), 0.00 (s, 6 H). 13C NMR (100 MHz, CDCl3) δ 169.3, 160.6, 158.8, 144.3, 134.5, 134.0, 131.0, 122.0, 75.2, 67.9, 62.8, 31.1, 23.4, 0.00. ESI (+VE) m/z: 384.1 (M + Na)+. HR-ESI cacld for C19H28NO4Si (M +Na)+: 362.1782, Found: 362.1789.
(4R)-3-[(2E)-[4-[(1,1-Dimethylethyl)dimethylsilyl]oxy]-1-oxo-2-buten-1-yl]-4-phenyl-2-oxazolidinone (8)
To a solution of 7 (5.00 g, 13.9 mmol) in anhydrous THF (70 mL) at room temperature was added tributylphosphine (0.34 mL, 1.39 mmol). The resulting solution was stirred at room temperature (60 minutes), then diluted with EtOAc (200 mL), washed (H2O and brine), dried (Na2SO4), and purified by silica gel column chromatography (hexanes: EtOAc) to yield 8 as a white solid (4.20 g, 84% yield). [α]20D −54.5 (c 1.40, CHCl3). mp. 79–81 °C. 1H NMR (400 MHz, CDCl3) δ 7.48 (dt, J = 15.2, 2.4 Hz, 1 H), 7.30 – 7.21 (m, 5 H), 7.02 (dt, J = 15.2, 3.4 Hz, 1 H), 5.39 (dd, J = 8.6, 3.8 Hz, 1 H), 4.60 (t, J = 8.8 Hz, 1 H), 4.28 (dd, J = 3.4, 2.2 Hz, 2 H), 4.17 (dd, J = 8.8, 4.0 Hz, 1 H), 0.85 (s, 9 H), 0.00 (s, 6 H). 13C NMR (100 MHz, CDCl3) δ 170.0, 159.0, 155.4, 144.5, 134.6, 134.1, 131.4, 124.1, 75.3, 68.1, 63.2, 31.3, 23.8, 0.00. IR (KBr) νmax: 2927, 2855, 1759, 1693, 1324, 1201, 1104, 951, 834, 715 cm−1. ESI (+VE) m/z: 384.1 (M + Na)+. HR-ESI cacld for C19H28NO4Si (M +Na)+: 362.1782, Found: 362.1790.
(4R)-3-[(2S,3R)-[2-Azido-4[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-methyl-1-oxo-butyl)]-4-phenyl-2-oxazolidinone (9)
To a solution of copper(I) bromide dimethyl sulfide complex (2.56 g, 12.45 mmol) in dimethyl sulfide (20 mL) and THF (30 mL) at −78 °C was added a solution of methylmagnesium chloride (3.0 M in THF, 5.50 mL, 16.4 mmol). The suspension was stirred at −78 °C (20 minutes), then warmed to 0 °C (20 minutes) and cooled to −78 °C. The mixture was then transferred to a pre-cooled (−78 °C) solution of 8 (1.80 g, 4.98 mmol) in THF (16.0 mL) and CH2Cl2 (8.0 mL) using a cannula. The resulting mixture was kept at −78 °C (60 minutes) then warmed to −40 °C (60 minutes) and cooled again to −78 °C. To the mixture was added a pre-cooled (−78 °C) solution of N-bromosuccinimide (4.45 g, 25.0 mmol) in THF (50 mL) and the mixture was stirred at −78 °C (90 minutes). The reaction was quenched by addition of saturated NaHSO3 (50 mL), extracted with EtOAc (100 mL × 2). The combined organic phase was washed (H2O and brine), dried (Na2SO4), and purified by silica gel column chromatography (hexanes: EtOAc) to yield the requisite α-bromo-containing intermediate as a white solid (1.93 g). To a solution of the α-bromo compound (1.93 g) in DMF (25 mL) at 0 °C, was added sodium azide (1.00 g, 15.4 mmol) and the mixture was stirred (2 h). The mixture was diluted with EtOAc (150 mL), washed (H2O and brine), dried (Na2SO4), and purified by silica gel column chromatography (hexanes: EtOAc) then crystallized (EtOAc: petroleum ether) to yield azide 9 as a white solid (1.65 g, 79% yield). [α]20D −73.0 (c 1.10, CHCl3). mp. 80–82 °C. 1H NMR (400 MHz, CDCl3) δ 7.40 – 7.30 (m, 5 H), 5.49 (dd, J = 8.8, 4.0 Hz, 1 H), 5.17 (d, J = 8.8 Hz, 1 H), 4.75 (t, J = 9.0 Hz, 1 H), 4.34 (dd, J = 8.8, 4.0 Hz, 1 H), 3.65 (dd, J = 10.2, 5.4 Hz, 1 H), 3.48 (dd, J = 10.2, 3.4 Hz, 1 H), 2.14 (m, 1 H), 0.89 (s, 9 H), 0.83 (d, J = 6.8 Hz, 3 H), 0.03 (s, 3 H), 0.00 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 175.7, 158.7, 143.9, 134.8, 134.6, 131.9, 75.7, 69.3, 66.7, 63.4, 43.5, 31.4, 23.8, 19.4, 0.00. IR (KBr) νmax: 2930, 2359, 2106, 1786, 1710, 1206, 1097, 833, 778 cm−1. ESI (+VE) m/z: 441.1(M + Na)+. HR-ESI MS cacld for C20H31N4O4Si (M +H) +: 419.2109, Found: 419.2114.
[(3S,4R)-Tetrahydro-4-methyl-2-oxo-3-furanyl]-carbamic acid phenylmethyl ester (10) and (2S,3R)-4-hydroxy-N-(phenylmethoxycarbonyl)-L-valine methyl ester (11)
To a solution of 9 (600 mg, 1.44 mmol) in MeOH (20 mL) at room temperature was added p-toluenesulfonic acid monohydrate (14 mg, 0.07 mmol). The solution was stirred at room temperature (6 h), then diluted with EtOAc (150 mL), washed (H2O and brine), dried (Na2SO4), and purified by silica gel column chromatography (hexanes: EtOAc) to yield the intermediate azide-containing lactone as a colorless liquid (270 mg, containing a small amount EtOAc).1H NMR (400 MHz, CDCl3) δ 4.36 (dd, J = 8.8, 6.4 Hz, 1 H), 4.26 (d, J = 7.2 Hz, 1 H), 4.02 (dd, J = 9.2, 4.0 Hz, 1 H), 2.75 (m, 1 H), 1.13 (d, J = 7.2 Hz, 3 H). A suspension of the this lactone and Pd•C (10%, 60 mg) in MeOH (9.0 mL) and acetic acid (1.0 mL) was stirred under H2 (1 atmosphere) at room temperature (overnight). The catalyst was removed by filtration though a celite pad under argon and the filtrate was concentrated. The residue was re-dissolved in THF (10.0 mL) containing H2O (10 mL) and then cooled to 0 °C. To this was added benzyl chloroformate (0.32 mL, 2.25 mmol) and NaHCO3 (840 mg, 10.0 mmol) and the mixture was stirred (4 h). The mixture was diluted with EtOAc (150 mL), washed (H2O and brine), dried (Na2SO4), and purified by silica gel column chromatography (hexanes: EtOAc) to yield 10 as a white crystalline solid (90 mg, 25% yield over 3 steps) and 11 as a viscous colorless oil (250 mg, 49% yield over 3 steps). For (10): mp. 125–127 °C. 1H NMR (400 MHz, CDCl3) δ 7.40 – 7.30 (m, 5 H), 5.33 (m, 1 H), 5.10 (s, 2 H), 4.53 (t, J = 6.8 Hz, 1 H), 4.35 (dd, J = 9.2, 5.2 Hz, 1 H), 4.05 (d, J = 9.2 Hz, 1 H), 2.92 (m, 1 H), 0.95 (d, J = 7.2 Hz, 3 H). 13C NMR (100 MHz, CDCl3) δ 174.5, 156.1, 135.9, 128.5, 128.3, 128.1, 72.4, 67.3, 54.5, 34.1, 12.7. ESI (+VE) m/z: 272.1 (M + Na)+. HR-ESI cacld for C13H16NO4 (M + H) +: 250.1074, Found: 250.1081.
For (11): 1H NMR (400 MHz, CDCl3) δ 7.29 – 7.28 (m, 5 H), 5.98 (d, J = 8.4 Hz, 1 H), 5.04 (s, 2 H), 4.34 (m, 1 H), 3.65 (s, 3 H), 3.54 (dd, J = 11.2, 4.4 Hz, 1 H), 3.44 (dd, J = 11.2, 6.0 Hz, 1 H), 2.92 (s, 1 H), 2.14 (m, 1 H), 0.92 (d, J = 7.2 Hz, 3 H). ESI (+VE) m/z: 304.2 (M + Na)+. HR-ESI MS cacld for C14H20NO5 (M + H) +: 282.1336, Found: 282.1343.
(2S,3R)-4-hydroxy-N-(9-Phenylfluoren-9-yl)-N-benzyl-L-valine methyl ester (14)
To a solution of 13 (4.00 g, 7.91 mmol) in anhydrous THF (260 mL) at −40 °C, was added DIBAL (1.0 M in Hexanes, 19.8 mL, 19.8 mmol). The mixture was stirred for 4 hr (−40 °C – 0 °C) before cooled down to −78 °C, quenched by acetone (10 mL), warmed to r.t., stirred with 1 N KH2PO4 (500 mL) and sodium potassium tartrate (30.0 g) overnight, filtered through the celite. The filtrate was extracted with EtOAc, washed (H2O and brine), dried (Na2SO4), and purified by silica gel column chromatography (hexanes: EtOAc) to yield alcohol 14 as a white wax (2.30 g, 61% yield, quantitive yield based on recovered starting material) and recycled 13 as a white wax (1.60 g). 1H NMR (400 MHz, CDCl3) δ 7.76 – 7.60 (m, 8 H), 7.35 – 7.20 (m, 10 H), 4.70 (AB, JAB = 13.6 Hz, 1 H), 4.38 (AB, JAB = 13.6 Hz, 1 H), 3.84 (dd, J = 10.8, 3.6 Hz, 1 H), 3.33 (dd, J = 10.8, 6.4 Hz, 1 H), 3.04 (d, J = 8.4 Hz, 1 H), 2.93 (s, 3 H), 1.40 (m, 1 H), 0.34 (d, J = 6.8 Hz, 3 H). 13C NMR (100 MHz, CDCl3) δ 171.7, 148.3, 144.8, 144.0, 142.0, 141.3, 139.7, 129.7, 128.6, 128.4, 128.0, 127.7, 127.3, 127.2, 127.1, 127.0, 125.3, 120.2, 80.3, 65.5, 63.3, 50.6, 36.3, 14.2. ESI (+VE) m/z: 478.2 (M + H)+. HR-ESI MS cacld for C32H32NO3 (M + H) +: 478.2377, Found: 478.2385.
(2S,3R)-4-[Di-(tert-butyl)-oxyphosphinyl]-4-hydroxy-N-phenylmethoxycarbonyl)-L-valine methyl ester (18)
To a solution of of oxalyl chloride (0.96 mL, 10.1 mmol) in CH2Cl2 (40 mL) at −78° C, was added a solution of DMSO (1.60 mL, 20.2 mmol) in CH2Cl2 (5 mL) and the mixture was stirred (15 minutes). To this was added alcohol 11 (0.63 g, 2.24 mmol) in dry CH2Cl2 (5 mL) over 5 minutes and the mixture was stirred at −75° C (2 h). triethylamine (8.40 mL, 53.8 mmol) was added and the mixture was warmed to room temperature. To this was added saturated NH4Cl (50 mL) and the mixture was extracted with Et2O (100 mL × 2) and the combined organic phase was washed (brine), dried (Na2SO4), and purified by silica gel column chromatography (hexanes: EtOAc). Aldehyde 15 was obtained as a viscous colorless oil (450 mg, 96% yield based on recovered starting material) along with starting alcohol 11 (160 mg). To a solution of di-tert-butyl phosphite (0.30 mL, 1.50 mmol) and triethylamine (0.21 mL, 1.50 mmol) in CH2Cl2 (5 mL) at 0° C, was added chlorotrimethylsilane (0.19 mL, 1.50 mmol) and the mixture was stirred (5 minutes) and then transfer to a solution of aldehyde 15 (300 mg, 1.08 mmol) in CH2Cl2 (5 mL) at room temperature and the mixture was stirred (3 h). The mixture was diluted with EtOAc (150 mL), washed (brine), dried (Na2SO4) and concentrated. The resulting crude silyl-protected 17 was re-dissolved in MeOH (10 mL), to this was added H2O (1.0 mL) and citric acid (200 mg) and the mixture was stirred at room temperature (over night). The mixture was diluted with EtOAc (200 mL), washed (saturated NaHCO3 and brine), dried (Na2SO4) and purified by silica gel column chromatography (hexanes: EtOAc) to yield 18 as a white wax epimeric at the γ-carbon (450 mg, 88% yield). 1H NMR (400 MHz, CDCl3) δ 7.30 – 7.29 (m, 5 H), 6.30 (d, J = 8.0 Hz, 0.7 H), 5.30 (m, 0.3 H), 5.10 – 5.05 (m, 2 H), 4.30 (m, 0.7 H), 4.09 (m, 0.3 H), 3.75 – 3.55 (m, 4 H), 2.51 (m, 0.7 H), 1.51 – 1.40 (m, 18 H), 1.15 – 1.00 (m, 3 H). 13C NMR (100 MHz, CDCl3) δ 172.3, 156.7, 136.4, 128.4, 128.0, 70.5, 68.8, 67.3, 66.8, 59.8, 53.9, 53.1, 52.2, 36.6, 35.4, 30.3, 24.1, 14.7, 11.5, 9.4. ESI (+VE) m/z: 496.2 (M + Na)+. HR-ESI MS cacld for C22H36NO8NaP (M + Na) +: 496.2071, Found: 496.2065.
(2S,3R)-4-[Di-(tert-butyl)-oxyphosphinyl]-N-(phenylmethoxycarbonyl)-L-valine methyl ester (20)
A solution of alcohol 18 (250 mg, 0.53 mmol), O-phenylchlorothionoformate (215 μL, 1.60 mmol), 4-(dimethylamino) pyridine (DMAP) (15 mg, 0.20 eq.) and N,N-diisopropylethylamine (363 μL, 2.10 mmol) in anhydrous CH2Cl2 (8.0 mL) was stirred at room temperature (overnight). The mixture was diluted with EtOAc (100 mL), washed (sat. NaHCO3 and brine), dried (Na2SO4) and purified by silica gel column chromatography (hexanes: EtOAc) to give the intermediate thiocarbonate 19 as a pale brown wax (225 mg). Crude 19 was dissolved in toluene (10 mL) and to this was added tributyltin hydride (0.42 mL, 1.59 mmol) and azobisisobutyronitrile (AIBN) (one spatula tip). The mixture was maintained at 100 °C (20 minutes), then cooled to room temperature and concentrated under vacuum. The residue was purified by silica gel column chromatography (hexanes: EtOAc) to give 20 as viscous colorless oil (140 mg, 58% yield for 2 steps). [α]20D + 2.4 (c 0.85, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.27 – 7.20 (m, 5 H), 5.80 (d, J = 8.4 Hz, 1 H), 5.07 (AB, JAB = 12.4 Hz, 1 H), 5.02 (AB, JAB = 12.4 Hz, 1 H), 4.23 (m, 1 H), 3.67 (s, 3 H), 2.33 (m, 1 H), 1.69 – 1.10 (m, 20 H), 1.05 (d, J = 6.8 Hz, 3 H). 13C NMR (100 MHz, CDCl3) δ 172.1, 156.2, 136.3, 128.4, 128.1, 82.1, 66.9, 59.4, 52.2, 32.2, 30.3, 29.6, 27.8, 26.8, 17.5, 13.5. IR (KBr) νmax: 2976, 1720, 1535, 1322, 1252, 975 cm−1. ESI (+VE) m/z: 480.3 (M + Na)+. HR-ESI MS cacld for C22H36NO7NaP (M +Na)+: 480.2122, Found: 480.2126.
(2S,3R)-4-[Di-(tert-butyl)-oxyphosphinyl]-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-valine (4)
To a solution of 20 (140 mg, 0.31 mmol) in THF (3.0 mL) and H2O (3.0 mL) at 0 °C, was added LiOH•H2O (26 mg, 0.62 mmol) and the mixture was stirred room temperature (over night). The THF was removed by rotary evaporation and the residual aqueous phase was neutralized by addition of saturated aqueous NH4Cl (20 mL) and extracted with EtOAc (3 × 50 mL). The combinded organic extract was washed with H2O (50 mL), brine (50 mL), dried (Na2SO4) and concentrated. The resulting residue was dissolved in MeOH (20 mL) and hydrogenated (1 atmosphere H2) over 10% Pd•C (40 mg) at room temperature (over night). The catalyst was removed by filtration and the filtrate was concentrated. The resulting residue was dissolved in dioxane (5.0 mL) and H2O (5.0 mL) and 9-fluorenylmethyl-succinimidyl carbonate Fmoc-OSu (173 mg, 0.465 mmol) and NaHCO3 (62 mg, 0.62 mmol) were added and the mixture was stirred at room temperature (over night). The reaction mixture was neutralized by addition of saturated NH4Cl (20 mL) and extracted with EtOAc (3 × 50 mL). The combined EtOAc layer was washed with H2O (50 mL), brine (50 mL), dried (Na2SO4) and purified by silica gel column chromatography (CH2Cl2: MeOH) to yield 4 as a white wax (166 mg, quantitative yield over 3 steps). [α]20D + 16.5 (c 0.65, CHCl3). 1H NMR (400 MHz, DMS)-d6) δ 7.88 (d, J = 7.6 Hz, 2 H), 7.70 (d, J = 7.2 Hz, 2 H), 7.41 (t, J = 7.4 Hz, 2 H), 7.31 (t, J = 7.6 Hz, 2 H), 4.30– 4.19 (m, 4 H), 3.84 (m, 1 H), 2.31 (m, 1 H), 1.80 – 1.55 (m, 2 H), 1.42 (s, 18 H), 0.96 (d, J = 6.4 Hz, 3 H). 13C NMR (100 MHz, CDCl3) δ 155.8, 143.9, 140.7, 127.6, 127.0, 125.1, 120.0, 80.7, 65.4, 60.5, 46.7, 31.5, 30.0, 16.9. ESI (+VE) m/z: 554.2 (M + Na)+. HR-ESI MS cacld for C28H38NO7NaP (M +Na)+: 554.2278, Found: 554.2277.
Peptide Synthesis 21–24
Fmoc-Thr(PO(OBzl)OH)-OH and other Fmoc protected amino acids were purchased from Novabiochem. Peptides were synthesize on NovaSyn®TGR resin (Novabiochem, cat. no. 01-64-0060) using standard Fmoc solid-phase protocols in N-Methyl-2-pyrrolidone (NMP). 1-O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) (5.0 eq.), hydroxybenzotriazole (HOBT) (5.0 eq.) and N,N-Diisopropylethylamine (DIPEA) (10.0 eq.) were used as coupling reagents. The N-terminal was acetylated by 1-Acetylimidazole. The final resin was washed with N, N-dimethylforamide (DMF), methanol, dichloromethane and ether then dried under vacuum (over night). Peptides were cleaved from resin (200 mg) by treatment with 5 mL of trifluoroacetic acid: triisbutylsilane: H2O (90: 5: 5) (4 h). The resin was filtered off and the filtrate was concentrated under vacuum, then precipitated with ether and the precipitate washed with ether. The resulting solid was dissolved in 50% aqueous acetonitrile 5 mL) and purified by reverse phase preparative HPLC using a Phenomenex C18 column (21 mm dia x 250 mm, cat. no: 00G-4436-P0) with a linear gradient from 0% aqueous acetonitrile (0.1% trifluoroacetic acid) to 50% acetonitrile (0.1% trifluoroacetic acid) over 35 minutes at a flow rate of 10.0 mL/minute. Peptide 21: ESI (+VE) m/z: 595.3 (M + H)+. Peptide 22: ESI (+VE) m/z: 675.3 (M + H)+. Peptide 23: ESI (+VE) m/z: 673.3 (M + H)+. Peptide 24: ESI (+VE) m/z: 657.3 (M + H)+. Analytical HPLC [By using Phenomenex C18 column (4.60 mm dia x 250 mm, cat. no: 00G-4435-E0) with a linear gradient from 5% aqueous acetonitrile (0.1% trifluoroacetic acid) to 100% acetonitrile (0.1% trifluoroacetic acid) over 25 minutes at a flow rate of 1.0 mL/minute.] indicated the purity of peptide 21: 100%, peptide 22: 100%, peptide 23: 87%, peptide 24: 83%.
Acknowledgments
This Work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health.
Footnotes
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Notes and references
- 1.Pawson T. Cell. 2004;116:191. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 2.Smerdon SJ, Yaffe MB. Handbook Cell Signaling. 2004;1:505. [Google Scholar]
- 3.Ladbury JE. Protein-protein recognition in phosphotyrosine-mediated intracellular signaling. Protein Rev. 2005;3:165. [Google Scholar]
- 4.Mayer BJ. Methods Mol Biol. 2006;332:79. doi: 10.1385/1-59745-048-0:79. [DOI] [PubMed] [Google Scholar]
- 5.Hubbard SR, Till JH. Annu Rev Biochem. 2000;69:373. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 6.Blume-Jensen P, Hunter T. Nature (London, U K) 2001;411:355. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 7.Tsatsanis C, Spandidos DA. Ann N Y Acad Sci. 2004;1028:168. doi: 10.1196/annals.1322.019. [DOI] [PubMed] [Google Scholar]
- 8.Adjei AA, Hidalgo M. J Clin Oncol. 2005;23:5386. doi: 10.1200/JCO.2005.23.648. [DOI] [PubMed] [Google Scholar]
- 9.Krause DS, Van Etten RA. N Engl J Med. 2005;353:172. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 10.Bianco R, Melisi D, Ciardiello F, Tortora G. Eur J Cancer. 2006;42:290. doi: 10.1016/j.ejca.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 11.Yaffe MB. Nat Rev Mol Cell Biol. 2002;3:177. doi: 10.1038/nrm759. [DOI] [PubMed] [Google Scholar]
- 12.Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Mol Cell Biol. 1995;15:2809. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKinsey TA, Chu ZL, Ballard DW. J Biol Chem. 1997;272:22377. doi: 10.1074/jbc.272.36.22377. [DOI] [PubMed] [Google Scholar]
- 14.Bex F, Murphy K, Wattiez R, Burny A, Gaynor RB. J Virol. 1999;73:738. doi: 10.1128/jvi.73.1.738-745.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng W, Zhang Z, Ganguly S, Weller Joan L, Klein David C, Cole Philip A. Nat Struct Biol. 2003;10:1054. doi: 10.1038/nsb1005. [DOI] [PubMed] [Google Scholar]
- 16.Zheng W, Schwarzer D, Lebeau A, Weller Joan L, Klein David C, Philip Cole. J Biol Chem. 2005;280:10462. doi: 10.1074/jbc.M412283200. [DOI] [PubMed] [Google Scholar]
- 17.Foss Frank W, Jr, Snyder Ashley H, Davis Michael D, Rouse M, Okusa Mark D, Lynch Kevin R, Macdonald Timothy L. Bioorg Med Chem. 2007;15:663. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burke Terrence R, Jr, Lee K. Acc Chem Res. 2003;36:426. doi: 10.1021/ar020127o. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz M, Ojea V, Shapiro G, Weber HP, Pombo-Villar E. Tetrahedron Lett. 1994;35:4551. [Google Scholar]
- 20.Berkowitz DB, Eggen M, Shen Q, Shoemaker RK. J Org Chem. 1996;61:4666. doi: 10.1021/jo9604752. [DOI] [PubMed] [Google Scholar]
- 21.Otaka A, Mitsuyama E, Kinoshita T, Tamamura H, Fujii N. J Org Chem. 2000;65:4888. doi: 10.1021/jo000169v. [DOI] [PubMed] [Google Scholar]
- 22.Ziegler FE, Lim H. J Org Chem. 1984;49:3278. [Google Scholar]
- 23.Tilley SD, Reber KP, Sorensen EJ. Org Lett. 2009;11:701. doi: 10.1021/ol802768p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans DA, Chapman KT, Bisaha J. J Am Chem Soc. 1988;110:1238. [Google Scholar]
- 25.Liu F, Zha HY, Yao ZJ. J Org Chem. 2003;68:6679. doi: 10.1021/jo0340152. [DOI] [PubMed] [Google Scholar]
- 26.Nicolas E, Russell KC, Knollenberg J, Hruby VJ. J Org Chem. 1993;58:7565–7571. [Google Scholar]
- 27.Lung FD, Li G, Lou BS, Hruby VJ. Synth Commun. 1995;25:57–61. [Google Scholar]
- 28.Humphrey JM, Bridges RJ, Hart JA, Chamberlin AR. J Org Chem. 1994;59:2467. [Google Scholar]
- 29.Evans DA, Hurst KM, Takacs JM. J Am Chem Soc. 1978;100:3467. [Google Scholar]
- 30.Wiemann A, Frank R, Tegge W. Tetrahedron. 2000;56:1331. [Google Scholar]
- 31.Barton DHR, McCombie SW. J Chem Soc, Perkin Trans 1. 1975;16:1574. [Google Scholar]
- 32.Lowery DM, Lim D, Yaffe MB. Oncogene. 2005;24:248. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- 33.Eckerdt F, Yuan J, Strebhardt K. Oncogene. 2005;24:267. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 34.Goh KC, Wang H, Yu N, Zhou Y, Zheng Y, Lim Z, Sangthongpitag K, Fang L, Du M, Wang X, Jefferson AB, Rose J, Shamoon B, Reinhard C, Carte B, Entzeroth M, Ni B, Taylor ML, Stuenkel W. Drug Dev Res. 2004;62:349. [Google Scholar]
- 35.Strebhardt K, Ullrich A. Nature Rev Cancer. 2006;6:321. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 36.McInnes C, Mezna M, Fischer PM. Curr Top Med Chem. 2005;5:181. doi: 10.2174/1568026053507660. [DOI] [PubMed] [Google Scholar]
- 37.Strebhardt K. ACS Chem Biol. 2006;1:683. doi: 10.1021/cb600462z. [DOI] [PubMed] [Google Scholar]
- 38.Peters U, Cherian J, Kim JH, Kwok BH, Kapoor TM. Nat Chem Biol. 2006;2:618. doi: 10.1038/nchembio826. [DOI] [PubMed] [Google Scholar]
- 39.Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, Noro N, Furuta M, Emmitte KA, Gilmer TM, Mook RA, Cheung M. Mol Cancer Ther. 2007;6:450. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- 40.Santamaria A, Neef R, Eberspacher U, Eis K, Husemann M, Mumberg D, Prechtl S, Schulze V, Siemeister G, Wortmann L, Barr FA, Nigg EA. Mol Biol Cell. 2007;18:4024. doi: 10.1091/mbc.E07-05-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanan EJ, Fucini RV, Romanowski MJ, Elling RA, Lew W, Purkey HE, VanderPorten EC, Yang W. Bioorg Med Chem Lett. 2008;18:5186. doi: 10.1016/j.bmcl.2008.08.091. [DOI] [PubMed] [Google Scholar]
- 42.Fucini RV, Hanan EJ, Romanowski MJ, Elling RA, Lew W, Barr KJ, Zhu J, Yoburn JC, Liu Y, Fahr BT, Fan J, Lu Y, Pham P, Choong IC, VanderPorten EC, Bui M, Purkey HE, Evanchik MJ, Yang W. Bioorg Med Chem Lett. 2008;18:5648. doi: 10.1016/j.bmcl.2008.08.095. [DOI] [PubMed] [Google Scholar]
- 43.Emmitte KA, Adjebang GM, Andrews CW, Badiang-Alberti JG, Bambal R, Chamberlain SD, Davis-Ward RG, Dickson HD, Hassler DF, Hornberger KR, Jackson JR, Kuntz KW, Lansing TJ, Mook RA, Nailor KE, Pobanz MA, Smith SC, Sung CM, Cheung M. Bioorg Med Chem Lett. 2009;19:1694. doi: 10.1016/j.bmcl.2009.01.094. [DOI] [PubMed] [Google Scholar]
- 44.Sillje HHW, Nigg EA. Science (Washington, DC, United States) 2003;299:1190. [Google Scholar]
- 45.van de Weerdt BCM, Littler DR, Klompmaker R, Huseinovic A, Fish A, Perrakis A, Medema RH. Biochim Biophys Acta, Mol Cell Res. 2008;1783:1015. doi: 10.1016/j.bbamcr.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 46.Reindl W, Yuan J, Kraemer A, Strebhardt K, Berg T. Chem Biol (Cambridge, MA, U S) 2008;15:459. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 47.Cheng KY, Lowe ED, Sinclair J, Nigg EA, Johnson LN. EMBO J. 2003;22:5757. doi: 10.1093/emboj/cdg558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elia AEH, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. Cell (Cambridge, MA, United States) 2003;115:83. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 49.Yun SM, Moulaei T, Lim D, Bang JK, Shenoy SR, Park JE, Liu F, Kang YH, Liao C, Soung NK, Lee S, Yoon DY, Lim Y, Lee DH, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR, Jr, Yaffe MB, Wlodawer A, Lee KS. Nature Struct Biol. 2009;16:876. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanisch A, Wehner A, Nigg EA, Sillje HH. Mol Biol Cell. 2006;17:448. doi: 10.1091/mbc.E05-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]