

Abstract
While many p53-deficient cell types are impaired in global genomic nucleotide excision repair of cyclobutane pyrimidine dimers (CPDs), human epidermal keratinocytes expressing human papillomavirus type 16 E6 and E7 are p53 deficient and yet maintain repair of CPD. We hypothesized that the p53 homolog, p63, may participate in governing global repair instead of p53 in keratinocytes. Following ultraviolet radiation (UVR) of E6/E7 keratinocytes, depletion of p63 but not of p73 impaired global genomic repair of CPD relative to control cells. In all cases, repair of pyrimidine(6-4)pyrimidone photoproducts, the other major UVR-induced DNA lesions, was unaffected. In E6/E7 keratinocytes treated with p63 small interfering RNA, reduced global repair of CPD was associated not with reduced levels of messenger RNA-encoding DNA damage recognition proteins but rather with decreased levels of DDB2 and XPC proteins, suggesting that p63 posttranscriptionally regulates levels of these proteins. These results indicate that global repair may be regulated at multiple levels and suggest a novel role for p63 in modulating repair of DNA damage in human keratinocytes. The results may provide insight into mechanisms of genomic stability in epithelia infected with oncogenic human papilloma viruses and may further explain the lack of increased skin cancer incidence in Li–Fraumeni syndrome.
Introduction
Nucleotide excision repair (NER) is a highly conserved process for removing bulky or distorting DNA lesions. The importance of NER in the response to solar ultraviolet radiation (UVR) is illustrated by the disease xeroderma pigmentosum in which mutations in certain NER proteins dramatically increase the risk of non-melanoma skin cancer due to impairment of the global genomic repair (GGR) subpathway of NER in which repair in non-transcribed DNA is defective. In many cell types, p53 has been shown to regulate GGR by transcriptionally activating certain genes, including XPC and DDB2 that encode key DNA damage recognition proteins (1–3). In several cell types, a p53 deficiency impairs GGR of certain DNA lesions, including the most common UVR-induced lesions, cyclobutane pyrimidine dimers (CPDs) (4). Paradoxically, Li–Fraumeni syndrome, which is most frequently due to inactivating mutations in one p53 allele, is characterized by a predisposition to many types of internal malignancies associated with the loss of the remaining wild-type allele, but neither photosensitivity nor non-melanoma skin cancers has been reported (5). This clinical phenotype suggests that keratinocytes may possess alternative keratinocyte-specific mechanisms to maintain GGR even if p53 is lost.
Although epidermal keratinocytes are the most appropriate cells in which to study DNA repair following UVR, relatively little is known about the mechanisms by which these cells regulate repair. Recent evidence suggests that keratinocytes possess responses to UVR that are distinct from other cell types (6,7). We have reported that human keratinocytes are proficient in maintaining GGR when p53 is absent or reduced, and while induction of XPC and DDB2 is reduced in p53-deficient keratinocytes, basal expression of these genes and of XPC protein is preserved (8,9). These results contrast with other types of p53-deficient cells in which XPC and DDB2 expression are reduced and GGR of CPD is impaired (10–12).
The p53 family includes p53 as well as two homologs known as p63 and p73 that also affect some p53-dependent processes (13,14). Unlike p53, the p63 gene possesses two promoters resulting in two N-terminal variants (ΔN and TA) as well as alternative splice sites producing α, β and γ C-terminal variants. The resulting p63 isoforms are differentially expressed in various tissues, including epidermis, and certain p63 isoforms such as ΔNp63α are highly expressed in epidermal keratinocytes. p63 clearly plays a critical role in epidermal development and homeostasis both in mice and humans (15–17), but it is still unclear if p63 plays a role in carcinogenesis. While p63 has been reported to be absent or reduced in some human cancers, including basal cell carcinomas (18), it has more frequently been reported to be overexpressed in various tumors (19,20), including non-melanoma skin cancers (21–24). Similarly, conflicting data from mice heterozygous for p63 have not resolved whether p63 is a tumor suppressor or oncogene (25–27).
There is evidence, however, that p63 is modulated by and affects the cellular response to genotoxic stress. Following UVR or treatment with other DNA-damaging agents, ΔNp63 isoforms decrease (28–31), whereas TAp63 isoforms have been reported to increase following UVR (32,33). ΔNp63α levels have also been reported to predict the clinical response of tumors to cisplatin (34). While p63 has been reported to be important in regulating the apoptotic response following DNA damage (19,28,31,35), its roles in other cellular pathways that respond to genotoxic stress have not yet been defined. We hypothesized that p63 may be a tissue-specific candidate for maintaining GGR selectively in keratinocytes, even in the absence of p53. We have measured GGR in a human keratinocyte model using two independent methods to reduce p63. The results indicate that a simultaneous reduction of p53 and p63, but not of p53 and p73, compromises GGR of CPD in human keratinocytes and that this effect is associated with reductions in DDB2 and XPC levels.
Materials and methods
Cells
Normal human keratinocytes (NHKs) were isolated from neonatal foreskins using standard techniques (8,9) and cultured in keratinocyte serum-free medium (Invitrogen, Carlsbad, CA) containing 5 ng/ml epidermal growth factor and 5 μg/ml bovine pituitary extract and used within two passages. Transformed human epidermal keratinocytes expressing human papillomavirus (HPV) type 16 E6 and E7 genes (CRL-2309, American Type Culture Collection, Manassas, VA) were grown in medium supplemented with 35 ng/ml epidermal growth factor and 5 μg/ml bovine pituitary extract.
Small interfering RNA treatment
Cells (5 × 105) were plated 24 h prior to transfection. Thirty minutes prior to transfection, medium was replaced with serum-free medium containing no epidermal growth factor or bovine pituitary extract. Cells were treated with either a non-targeting sequence or small interfering RNAs (siRNAs) (Dharmacon, Lafayette, CO) targeting two distinct sequences encoding the DNA-binding domain common to all p63 isoforms: siRNA DB1 targets 5′-AACCAUGAGC-UGAGCCGUGAAU-3′ (36) and siRNA DB2 targets 5′-GUCCCUAUAACA-CAGACCA-3′, which is similar to a previously reported target except for an additional base at the 3′ end (37). An siRNA targeting 5′-AACCACGAG-CUCGGGAGGGAC-3′ found in all p73 isoforms was obtained commercially (Santa Cruz Biotechnology, Santa Cruz, CA). Cells were transfected with 123 nM siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. After 4 h, transfection medium was replaced by fresh medium containing all supplements and incubated for an additional 20 h.
Ultraviolet irradiation
As described previously, cells were irradiated with 10 J/m2 with a germicidal lamp emitting predominantly at 254 nm (8,9).
Measurement of messenger RNA levels
XPA, XPC, DDB2, glyceraldehyde-3-phosphate dehydrogenase and peptidyl prolyl isomerase messenger RNA (mRNA) levels were assayed by quantitative real-time reverse transcription–polymerase chain reaction, as described previously (26). TAp63, ΔNp63, TAp73 and ΔNp73 mRNA levels were determined similarly, using the following primers (Oligos Etc., Wilsonville, OR) with location in the mRNA sequence denoted by position of the 5′ end relative to the translation start site: 5′-GACCTGAGTGACCCCATGTG-3′ (TAp63 forward, position 187), 5′-TGCCCAGACTCAATTTAGTGAG-3′ (ΔNp63 forward, position 21), 5′-AGAGAGAGCATCGAAGGTGGAG-3′ (common p63 reverse, ΔN position 198, TA position 363), 5′-CCACCTGGAGGGCATGAC-TA-3′(TAp73 forward, position 156), 5′-ATGCTGTACGTCGGTGACCC-3′ (ΔNp73 forward, position 1) and 5′-TTGTGCGTAGGGCGAGTGG-3′ (common p73 reverse, ΔN position 156, TA position 303 TA). Results are the average of four independent experiments assayed in duplicate.
Western immunoblotting
For detection of all proteins except XPC and DDB2, cells were extracted and analyzed as described previously (8,9). For analysis of XPC and DDB2, cells were lysed and total protein was extracted and quantified as reported (38). Extracts containing equivalent amounts of total protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, blotted and probed as described previously (8), using monoclonal antibodies against p63 (1:200, clone 4A4, Santa Cruz Biotechnology), p53 (1:500, clone DO-1, Santa Cruz Biotechnology), XPC (clone 3.26, 1:1000, GeneTex, San Antonio, TX), β-actin (clone AC-74, 1:3000, Sigma, St. Louis, MO) and a rabbit polyclonal antibody to DDB2 (1:500, gift from J.M. Ford). The chemiluminescence signal from bands on western blots was imaged (LAS 3000 Luminescence Image Analyzer, FujiFilm Co., Tokyo, Japan) and quantified (Image Gauge, version 4.22, FujiFilm Co.). Plotted results are the average of two or three independent experiments as indicated in figure captions.
GGR assay
GGR was assayed as described previously (8,9). Briefly, cells were prelabeled with methyl-[3H]thymidine (Amersham, Piscataway, NJ), irradiated and allowed to repair for up to 24 h. DNA damage was probed with an immunoblot assay using monoclonal antibodies against CPD (TDM-2) and pyrimidine(6-4)pyrimidone photoproducts (6-4PP) (6-4M2) (MBL International, Woburn, MA). Individual samples were subjected to scintillation counting to correct for replication. Results are the average of at least three independent biological experiments each assayed in triplicate.
Statistical analysis
Experimental and control samples were compared using unpaired t-tests assuming equal variances (Kaleidagraph, version 3.6, Synergy Software, Reading, PA).
Results
PI3 kinase inhibition reduces ΔNp63 and impairs GGR in E6/E7 keratinocytes
A human genetic model for simultaneous p53 and p63 deficiency is unavailable, and changes in CPD repair kinetics in potential rodent models are challenging to measure due to the normally poor GGR of CPD in rodent cells [(39) and data not shown]. Thus, to evaluate the effect of p63 on GGR, it was necessary to reduce p63 in p53-deficient human keratinocytes. A transformed human epidermal keratinocyte cell line that expresses the HPV16 E6 and E7 oncoproteins and is p53 deficient, yet has normal GGR activity, was chosen as the basic model system for subsequent p63 depletion (8). As shown in Figure 1A, relative to NHKs, E6/E7 keratinocytes had almost undetectable p53 protein levels, though we have also shown previously that XPC levels are not reduced in E6/E7 (8). Since ΔNp63α, the most abundant p63 isoform in epidermal keratinocytes, appears to be reduced by phosphoinositide 3 kinase (PI3 kinase) inhibition (40), reducing ΔNp63α with the PI3 kinase inhibitor, LY294002, was explored initially in the E6/E7 keratinocyte model. As previously reported (40), LY294002 reduced ΔNp63α mRNA levels in E6/E7 keratinocytes to levels that were only 10% of those observed in control cells (Figure 1B). In addition, consistent with prior reports (41), ΔNp63α protein levels were not significantly affected by E6 and E7 viral oncoproteins when compared with levels in normal keratinocytes (Figure 1C). Following treatment with LY294002, both normal and E6/E7 keratinocytes exhibited levels of ΔNp63α protein that were 20–30% of diluent-treated control cells, similar to results previously reported (40). In both normal and E6/E7 cells, the reduction in ΔNp63α persisted following UVR.
Fig. 1.
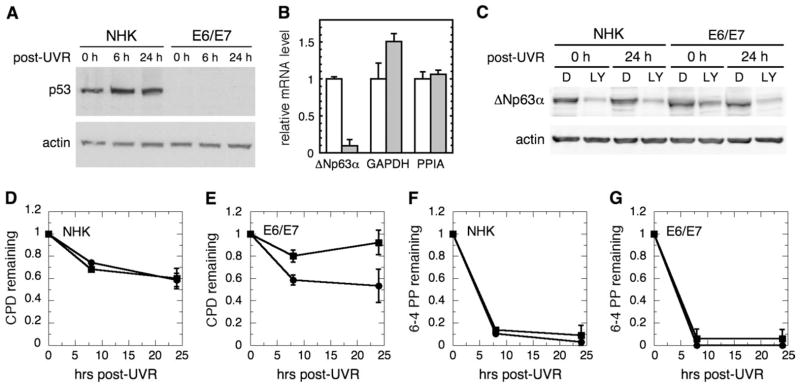
PI3 kinase inhibition reduces p63 levels and GGR. (A) Western immunoblotting of p53 in NHKs and E6/E7 keratinocytes following UVR. β-Actin was a loading control. (B) E6/E7 keratinocytes, treated with dimethyl sulfoxide (white bars) or 40 μM LY294002 (gray bars) for 24 h, were analyzed by quantitative reverse transcription–polymerase chain reaction for ΔNp63, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and peptidyl prolyl isomerase (PPIA) mRNA. (C) NHKs or E6/E7 keratinocytes were treated with dimethyl sulfoxide (D) or LY294002 (LY) for 24 h, irradiated, harvested at 0 or 24 h after UVR and analyzed for ΔNp63α by western immunoblotting, using β-actin as a loading control. (D–G) NHKs (panels D and F) or E6/E7 keratinocytes (E6/E7, panels E and G) were treated with either dimethyl sulfoxide (filled circles) or LY294002 (filled squares) for 24 h, irradiated and allowed to repair for up to 24 h. DNA was analyzed by an immunoblot assay for either CPD (D and E) or 6-4PP (F and G). Error bars represent standard deviations of at least three independent experiments.
To determine if the reduced ΔNp63α levels were associated with alterations in GGR, cells were pretreated with LY294002 or diluent, irradiated and assayed for repair. As shown in Figure 1D, relative to diluent-treated control cells, GGR of CPD was unaffected by LY294002 in NHKs, suggesting that reducing p63 alone in a normal p53 background does not affect GGR. In contrast, while p53-deficient E6/E7 keratinocytes treated with diluent repaired similarly to normal keratinocytes (8), E6/E7 cells treated with LY294002 repaired <10% of CPD by 24 h (Figure 1E, P < 0.01). The repair kinetics of 6-4 photoproducts (6-4PP), the other major UVR-induced DNA lesion, in both normal and E6/E7 keratinocytes treated with LY294002 were indistinguishable from control cells (Figure 1F and G).
Deficient GGR associated with targeted p63 knockdown in E6/E7 keratinocytes
Because PI3 kinase inhibition could have effects unrelated to p63 function, RNA interference was employed as a more specific method to reduce p63. Two separate siRNA (DB1 and DB2) targeting DNA-binding domain sequences that are common to all isoforms of p63 mRNA but not present in either p53 or p73 mRNA sequences were used independently to further ensure that results were not due to sequence-specific off-target effects (36,37). Transfection of fluorescently labeled siRNA into E6/E7 keratinocytes resulted in >90% of the cells exhibiting fluorescence after 24 h (data not shown). In contrast, similar experiments with NHKs indicated that <50% of these cells were transfectable under our conditions and did not result in significant p63 reduction (data not shown), and thus normal keratinocytes were not used further in siRNA experiments. Following siRNA transfection, siRNAs DB1 and DB2 each reduced ΔNp63 mRNA isoforms by at least 60% and TAp63 mRNA isoforms by 75–85% (Figure 2A). mRNA levels of both TAp73 and ΔNp73 isoforms as well as of glyceraldehyde-3-phosphate dehydrogenase were not comparably affected by the p63 siRNA, consistent with previous reports that p73 protein levels in keratinocytes are unaffected by p63 siRNA (42). Similarly, DB1 and DB2 reduced basal ΔNp63α protein levels by at least 75% further validating both of these siRNAs as effective inhibitors of p63 levels in keratinocytes (Figure 2B).
Fig. 2.

Impairment of GGR by p63 siRNA. (A) E6/E7 keratinocytes were transfected with either a non-targeting sequence (NTS, white bar) or one of two siRNAs targeting p63—DB1 (gray bar) or DB2 (black bar)—for 24 h prior to quantitative reverse transcription–polymerase chain reaction determination of ΔNp63, TAp63, TAp73, ΔNp73 and glyceraldehyde-3-phosphate dehydrogenase mRNA levels. (B–D) Following treatment with NTS or p63 siRNA (DB1 or DB2), cells were irradiated and harvested immediately or up to 24 h after UVR. (B) ΔNp63α protein levels were measured by western immunoblotting, using β-actin as a loading control. (C and D) An immunoblot assay for (C) CPD or (D) 6-4PP was performed on DNA harvested from cells treated with either NTS (filled circles) or DB1 (filled squares) or DB2 (filled triangle). Error bars in panels (A), (C) and (D) denote standard deviation of three independent experiments.
GGR kinetics were then measured in cells that were treated with each p63 siRNA for 24 h, irradiated and assayed for repair. As shown in Figure 2C, E6/E7 keratinocytes treated with a non-targeting sequence repaired ~35% of CPD by 8 h and slightly >50% of CPD by 24 h after UVR, comparable with repair activity observed previously for normal keratinocytes as well as other keratinocytes deficient in p53 alone [see Figure 1 and (8)]. In contrast, cells treated with either of the p63 siRNA repaired <20% of CPD by 8 h (P ≤ 0.04) and only 20–30% of CPD by 24 h (P = 0.01) after UVR, representing a 40–50% decrease in overall repair activity of CPD during the first 24 h following UVR. In all cases, repair of 6-4PP was unaffected by the loss of p63 (Figure 2D).
In contrast, an siRNA directed against all p73 isoforms resulted in specific reductions in both TAp73 and ΔNp73 mRNA (Figure 3A), yet did not alter GGR kinetics for either CPD or 6-4PP relative to the controls (Figure 3B and C).
Fig. 3.
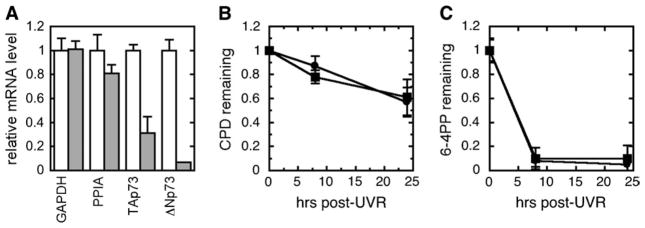
Normal GGR following p73 siRNA. Following treatment with non-targeting sequence (NTS) or p73 siRNA for 24 h, cells were irradiated and harvested immediately or 24 h after UVR. (A) Expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), peptidyl prolyl isomerase (PPIA), TAp73 and ΔNp73 mRNA by quantitative reverse transcription–polymerase chain reaction following treatment by NTS (white bar) or p73 siRNA (gray bar). (B and C) An immunoblot assay for (B) CPD or (C) 6-4PP was performed on DNA harvested from cells treated with either NTS (filled circles) or p73 siRNA (filled squares). Error bars denote standard deviations of three independent experiments.
XPC and DDB2 mRNA levels are not affected by loss of p63 in E6/E7 keratinocytes
Since we have previously observed that, unlike fibroblasts, keratinocytes appear to maintain their basal levels of XPC and DDB2 expression regardless of their p53 status (8), we explored the possibility that p63 also transcriptionally activates XPC or DDB2 in order to account for impaired GGR in keratinocytes that are deficient in both p53 and p63. E6/E7 keratinocytes were treated with siRNAs or with a non-targeting sequence, irradiated with UVR and then analyzed by quantitative reverse transcription–polymerase chain reaction (Figure 4). In contrast to previously reported results for normal keratinocytes and as previously observed in E6/E7 keratinocytes (8,9), E6/E7 cells treated with non-targeting control sequences exhibited poor or no induction of DDB2, XPC or XPA mRNA levels following UVR. Relative to cells treated with non-targeting control sequences, neither of the p63 siRNAs altered basal or induced mRNA levels of any NER genes examined (P > 0.05); an apparent increase relative to the control in XPC and DDB2 mRNA at 24 h only in DB1-treated cells was not statistically significant. These results suggest that the loss of GGR associated with p63 reductions is not predominantly due to a loss of transcriptional activity of NER genes.
Fig. 4.

Levels of mRNA encoding NER proteins following UVR. E6/E7 keratinocytes were treated for 24 h with a non-targeting sequence (NTS, white bars) or either of two distinct p63 siRNA (DB1, gray bars or DB2, black bars), and then irradiated and harvested at 0 and 24 h after UVR for quantitative reverse transcription–polymerase chain reaction to measure relative mRNA levels of XPC, DDB2 and XPA as well as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and peptidyl prolyl isomerase (PPIA). Results have been normalized to the initial level of a particular mRNA in NTS-treated cells. Error bars denote standard deviations of four independent experiments.
Reduced DDB2 and XPC protein levels in E6/E7 keratinocytes deficient in p63
Since transcription of NER genes did not appear to change in association with reduced p63 and impaired GGR, we next examined actual XPC and DDB2 protein levels following UVR in E6/E7 keratinocytes treated with p63 siRNA. As shown in Figure 5A, DDB2 levels decreased slightly in control cells during the first 24 h following UVR, similar to previous findings in other cell types (43). However, both DB1 and DB2 siRNA treatment decreased basal levels of DDB2 to levels <50% of non-targeting sequence controls, and this deficiency persisted during the 24 h following UVR. It should be noted that under the lysis conditions used, >80% of the DDB2 signal was found in the initially insoluble fraction that was then solubilized and analyzed and that this fraction is presented in Figure 5A. The initially soluble fraction following lysis did not exhibit detectable differences in DDB2 levels following p63 siRNA treatment or following UVR (data not shown).
Fig. 5.

p63 siRNA results in reduced levels of DDB2 and XPC. E6/E7 keratinocytes were treated with p63 siRNAs (DB1 or DB 2) or a non-targeting sequence (NTS) for 24 h prior to irradiation. Cell lysates were collected at 0 and 24 h following UVR and assayed for (A) DDB2 or (B) XPC proteins by western immunoblotting, using β-actin as a loading control. Representative blots are shown on the left, and averaged levels of DDB2 or XPC normalized to actin from two (DDB2) or three (XPC) independent experiments are plotted on the right. Error bars denote standard deviations. Symbols above bars denote P < 0.04 (#) and P < 0.01 (*).
XPC levels were more modestly affected by p63 siRNA (Figure 5B). Following treatment with the non-targeting sequence control, cells exhibited moderately elevated levels of XPC by 24 h. However, cells treated with either p63 siRNA exhibited basal XPC levels that were initially 75–85% of control cells treated with a non-targeting sequence, and then were <50% of those in control cells by 24 h after UVR.
Discussion
p63 is a critical gene in both epidermal development and maintenance (15,44). In addition, p63 has been reported to be important for the apoptotic response independent of p53 (35), and the ΔNp63 isoform in particular has been shown to be critical for preventing apoptosis in keratinocytes following UVR as well as in other cell types following DNA damage (19,28,31,35). However, a role for p63 in tumorigenesis is still unclear. Although p63 was originally anticipated to function as a tumor suppressor similarly to p53, there is also evidence that p63 functions as an oncogene (27,45). Recently, disparate reports have emerged regarding the tumor predisposition of mice heterozygous for p63 and p63/p53 (25,26,46).
Our results suggest that, in addition to modulating apoptosis, p63 has yet another role in the response to DNA damage. Using p53-deficient E6/E7 human keratinocytes, we used both a small molecule PI3 kinase inhibitor as well as two separate siRNA targeting sequences that are common to all p63 isoforms to demonstrate that simultaneous depletion of p63 and p53 in human keratinocytes is associated with impaired GGR of CPD. The repair results suggest that, even in the absence of p53, p63 functions to support GGR activity of CPD in human keratinocytes. While both LY294002 and p63 siRNAs substantially reduced ΔNp63α levels, they did not completely abolish them, and it is possible that further reductions in p63 levels would further impair GGR. Since both of our siRNAs targeted all p63 isoforms, it is also still unclear which isoforms of p63 support GGR in the absence of p53. In addition, we assessed whether p73 may play a role in repair since p63’s apoptotic effects have been shown to be p73-dependent in head and neck squamous cell carcinomas (20). Although p73 has been implicated in the apoptotic response to DNA damage (35), we observed no effect on p73 mRNA levels when p63 is reduced, consistent with previous reports that p73 protein is unaffected by p63 siRNA in keratinocytes (42). Further, p73 siRNA had no detectable effect on GGR of CPD or 6-4PP following UVR.
While it is possible that the E6 and E7 proteins confer an occult predisposition to a repair deficiency that is unmasked by p63 depletion, the quantitatively similar results from both PI3 kinase inhibition and two separate p63 siRNA suggest that the particular mechanism of p63 reduction in the E6/E7 background is unlikely to be important. Nevertheless, it will be important to confirm these results in other p53-deficient keratinocytes. It is also still unclear whether loss of p63 without loss of p53 impairs GGR, although the inability of LY294002 inhibitor to reduce GGR in normal keratinocytes suggests that loss of at least ΔNp63α alone is insufficient to reduce GGR. In addition, several human syndromes in which p63 but not p53 is mutated are characterized by developmental defects, but neither photosensitivity not tumorigenesis, further suggesting that loss of p63 function alone is insufficient to confer an NER defect (16,47).
One mechanism by which p53 regulates GGR in non-keratinocyte cells appears to involve direct transcriptional activation of certain genes, including DDB2 and XPC that encode key DNA damage recognition proteins (1,2,48). While both TAp63 and ΔNp63 have been reported to directly transactivate various target genes, the lack of decreased DDB2, XPC and XPA mRNA levels upon p63 knockdown in a p53-deficient cell suggests that neither p63 nor p53 directly regulates these genes in keratinocytes. These results contrast with those from fibroblasts, and it is possible that keratinocytes possess alternative mechanisms for regulating transcription of NER genes.
Although an effect of p63 on transcriptional activity of XPC and DDB2 genes was not observed, reduced GGR of CPD was associated with reduced levels of DDB2 and to a lesser extent of XPC proteins in E6/E7 keratinocytes transfected with p63 siRNA. These data suggest that p63 may affect GGR indirectly by supporting the steady state levels of DDB2 and XPC. While the decrease in basal XPC levels was not dramatic, it may be biologically significant since XPC participates in a rate-limiting step in NER and, at least in HeLa cells, is one of the least abundant NER proteins (49). Levels and activities of both DDB2 and XPC appear to be regulated by multiple mechanisms, including modification by ubiquitin and small ubiquitin-like modifier (38,50–52), and p63 may affect such posttranslational processes.
Because we and others have observed UVR-associated reductions in ΔNp63α in control human keratinocytes (28–30), it may seem surprising that p63 is involved in maintaining global repair of UVR-induced CPD. There may be several potential explanations for this apparent paradox. First, the physiological decrease in ΔNp63α following UVR is smaller than the reductions mediated by p63 siRNA and may not be sufficient to affect GGR. Second, while the ΔNp63α isoform has been most easily studied following UVR, the behavior of other p63 isoforms is less clear, though others have reported that UVR induces TAp63 (32,33). Finally, both DDB2 and XPC, as well as other proteins involved in the UVR response, also decrease initially following UVR, and therefore a decrease in a repair-related protein following UVR may still be consistent with a role in repair, though the significance of the dynamics in protein levels remains unclear (38,43,51).
The E6 and E7 oncoproteins are essential in the pathogenesis of HPV infection and transformation of stratified squamous epithelia. It may be advantageous for the propagation of certain types of HPV that inactivation of p53 does not also impair NER in keratinocytes due to the presence of p63. In addition, although this work was conducted with E6/E7 keratinocytes, p63-dependent maintenance of NER to prevent UVR-induced mutations despite loss of p53 in human keratinocytes but not in other tissues suggests a more detailed explanation for the Li–Fraumeni syndrome phenotype in which internal malignancies but not epidermal carcinogenesis are increased (5).
Acknowledgments
Funding
Department of Veterans Affairs Medical Research Service; National Institutes of Health (CA105958).
We thank J. Ford for providing anti-DDB2 antibodies, S. Pennypacker for assistance with cell culture and C. Largman, J. Ford and P. Hanawalt for helpful discussions.
Abbreviations
- CPD
cyclobutane pyrimidine dimer
- GGR
global genomic repair
- HPV
human papillomavirus
- mRNA
messenger RNA
- NER
nucleotide excision repair
- NHK
normal human keratinocyte
- 6-4PP
pyrimidine(6-4) pyrimidone photoproduct
- siRNA
small interfering RNA
- UVR
ultraviolet radiation
Footnotes
Conflict of Interest Statement: None declared.
References
- 1.Adimoolam S, et al. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci USA. 2002;99:12985–12990. doi: 10.1073/pnas.202485699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hwang BJ, et al. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA. 1999;96:424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh T, et al. Impaired regulation of tumor suppressor p53 caused by mutations in the xeroderma pigmentosum DDB2 gene: mutual regulator interactions between p48DDB2 and p53. Mol Cell Biol. 2003;23:7540–7553. doi: 10.1128/MCB.23.21.7540-7553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ford JM. Regulation of DNA damage recognition and nucleotide excision repair: another role for p53. Mutat Res. 2005;577:195–202. doi: 10.1016/j.mrfmmm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Olivier M, et al. Li-Fraumeni and related syndromes: correlation between tumor type, family structure and TP53 genotype. Cancer Res. 2003;63:6643–6650. [PubMed] [Google Scholar]
- 6.D’Errico M, et al. Apoptosis and efficient repair of DNA damage protect human keratinocytes against UVB. Cell Death Differ. 2003;10:754–756. doi: 10.1038/sj.cdd.4401224. [DOI] [PubMed] [Google Scholar]
- 7.D’Errico M, et al. Differential role of transcription-coupled repair in UVB-induced response of human fibroblasts and keratinocytes. Cancer Res. 2005;65:432–438. [PubMed] [Google Scholar]
- 8.Ferguson BE, et al. Proficient global nucleotide excision repair in human keratinocytes but not fibroblasts deficient in p53. Cancer Res. 2005;65:8723–8729. doi: 10.1158/0008-5472.CAN-05-1457. [DOI] [PubMed] [Google Scholar]
- 9.Oh DH, et al. Differentiating human keratinocytes are deficient in p53 but retain global nucleotide excision repair following ultraviolet radiation. DNA Repair. 2005;4:1149–1159. doi: 10.1016/j.dnarep.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Ford JM, et al. Human fibroblasts expressing the human papillomavirus E6 gene are deficient in global genomic nucleotide excision repair and sensitive to ultraviolet irradiation. Cancer Res. 1998;58:599–603. [PubMed] [Google Scholar]
- 11.El-Mahdy MA, et al. p53-degradation by HPV-16 E6 preferentially affects the removal of cyclobutane pyrimidine dimers from non-transcribed strand and sensitizes mammary epithelial cells to UV-radiation. Mutat Res. 2000;459:135–145. doi: 10.1016/s0921-8777(99)00066-x. [DOI] [PubMed] [Google Scholar]
- 12.Therrien JP, et al. Human cells compromised for p53 function exhibit defective global and transcription-coupled nucleotide excision repair, whereas cells compromised for pRb function are defective only in global repair. Proc Natl Acad Sci USA. 1999;96:15038–15043. doi: 10.1073/pnas.96.26.15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang A, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:306–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 14.Kaghad M, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 15.Koster MI, et al. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004;18:126–131. doi: 10.1101/gad.1165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunner HG, et al. The p63 gene in EEC and other syndromes. J Med Genet. 2002;39:337–381. doi: 10.1136/jmg.39.6.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senoo M, et al. p63 is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129:523–536. doi: 10.1016/j.cell.2007.02.045. [DOI] [PubMed] [Google Scholar]
- 18.Dellavalle RP, et al. CUSP/p63 expression in basal cell carcinoma. Exp Dermatol. 2002;11:203–208. doi: 10.1034/j.1600-0625.2002.110302.x. [DOI] [PubMed] [Google Scholar]
- 19.Fomenkov A, et al. RACK1 and stratifin target ΔNp63α for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle. 2004;3:e27–e37. doi: 10.4161/cc.3.10.1155. [DOI] [PubMed] [Google Scholar]
- 20.Rocco JW, et al. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 21.Reis-Filho JS, et al. p63 expression in normal skin and usual cutaneous carcinomas. J Cutan Pathol. 2002;29:517–523. doi: 10.1034/j.1600-0560.2002.290902.x. [DOI] [PubMed] [Google Scholar]
- 22.Di Como CJ, et al. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res. 2002;8:494–501. [PubMed] [Google Scholar]
- 23.Nylander K, et al. Differential expression of p63 isoforms in normal tissues and neoplastic cells. J Pathol. 2002;198:417–427. doi: 10.1002/path.1231. [DOI] [PubMed] [Google Scholar]
- 24.Wrone DA, et al. The expression of p63 in actinic keratoses, seborrheic keratosis, and cutaneous squamous cell carcinomas. Dermatol Surg. 2004;30:1299–1302. doi: 10.1111/j.1524-4725.2004.30403.x. [DOI] [PubMed] [Google Scholar]
- 25.Flores ER, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 26.Keyes WM, et al. p63 heterzygous mutant mice are not prone to spontaneous or chemically induced tumors. Proc Natl Acad Sci USA. 2006;103:8435–8440. doi: 10.1073/pnas.0602477103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills AA. p63: oncogene or tumor suppressor. Curr Opin Genet Dev. 2006;16:38–44. doi: 10.1016/j.gde.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Liefer KM, et al. Down-regulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Res. 2000;60:4016–4020. [PubMed] [Google Scholar]
- 29.Marchbank A, et al. The CUSP ΔNp63α isoform of human p63 is downregulated by solar-simulated ultraviolet radiation. J Dermatol Sci. 2003;32:71–74. doi: 10.1016/s0923-1811(03)00040-9. [DOI] [PubMed] [Google Scholar]
- 30.Westfall MD, et al. Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation of ΔNp63α. Cell Cycle. 2005;4:710–716. doi: 10.4161/cc.4.5.1685. [DOI] [PubMed] [Google Scholar]
- 31.Lee H, et al. A dominant negative form of p63 inhibits apoptosis in a p53-independent manner. Biochem Biophys Res Commun. 2006;344:166–172. doi: 10.1016/j.bbrc.2006.03.128. [DOI] [PubMed] [Google Scholar]
- 32.Katoh I, et al. p51A (TAp63γ), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene. 2000;19:3126–3130. doi: 10.1038/sj.onc.1203644. [DOI] [PubMed] [Google Scholar]
- 33.Okada Y, et al. p53 gene family p51(p63)-encoded, secondary trans-activator p51B(TAp63α) occurs without forming an immunoprecipitable complex with MDM2, but responds to genotoxic stress by accumulation. Exp Cell Res. 2002;276:194–200. doi: 10.1006/excr.2002.5535. [DOI] [PubMed] [Google Scholar]
- 34.Zangen R, et al. ΔNp63α levels correlate with clinical tumor response to cisplatin. Cell Cycle. 2005;4:1313–1315. doi: 10.4161/cc.4.10.2066. [DOI] [PubMed] [Google Scholar]
- 35.Flores ER, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 36.Harmes DC, et al. Positive and negative regulation of ΔN-p63 promoter activity by p53 and ΔN-p63-α contributes to differential regulation of p53 target genes. Oncogene. 2003;22:7607–7616. doi: 10.1038/sj.onc.1207129. [DOI] [PubMed] [Google Scholar]
- 37.Kurata S, et al. p53/p63 controls subunit α3 of the major epidermis integrin anchoring the stem cells to the niche. J Biol Chem. 2004;279:50069–50077. doi: 10.1074/jbc.M406322200. [DOI] [PubMed] [Google Scholar]
- 38.Sugasawa K, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 39.Tang JY, et al. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol Cell. 2000;5:737–744. doi: 10.1016/s1097-2765(00)80252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbieri CE, et al. ΔNp63α expression is regulated by the phosphoinositide 3-kinase pathway. J Biol Chem. 2003;278:51408–51414. doi: 10.1074/jbc.M309943200. [DOI] [PubMed] [Google Scholar]
- 41.Roth J, et al. Failure of viral oncoproteins to target the p53-homologue p51A. J Gen Virol. 1999;80:3251–3255. doi: 10.1099/0022-1317-80-12-3251. [DOI] [PubMed] [Google Scholar]
- 42.DeYoung MP, et al. Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Res. 2006;66:9362–9368. doi: 10.1158/0008-5472.CAN-06-1619. [DOI] [PubMed] [Google Scholar]
- 43.Rapic-Otrin V, et al. Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res. 2002;30:2588–2598. doi: 10.1093/nar/30.11.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKeon F. p63 and the epithelial stem cell: more than status quo? Genes Dev. 2004;18:465–469. doi: 10.1101/gad.1190504. [DOI] [PubMed] [Google Scholar]
- 45.Yang A, et al. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002;18:90–95. doi: 10.1016/s0168-9525(02)02595-7. [DOI] [PubMed] [Google Scholar]
- 46.Perez-Losada J, et al. p63 and p73 do not contribute to p53-mediated lymphoma suppressor activity in vivo. Oncogene. 2005;24:5521–5524. doi: 10.1038/sj.onc.1208799. [DOI] [PubMed] [Google Scholar]
- 47.Brunner HG, et al. p63 gene mutations and human developmental syndromes. Am J Hum Genet. 2002;112:284–290. doi: 10.1002/ajmg.10778. [DOI] [PubMed] [Google Scholar]
- 48.Tan T, et al. p53 binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol Cell Biol. 2002;22:3247–3254. doi: 10.1128/MCB.22.10.3247-3254.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Araujo SJ, et al. Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol. 2001;21:2281–2291. doi: 10.1128/MCB.21.7.2281-2291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wittschieben BO, et al. DDB complexities. DNA Repair. 2003;2:1065–1069. doi: 10.1016/s1568-7864(03)00113-7. [DOI] [PubMed] [Google Scholar]
- 51.Wang QE, et al. DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res. 2005;33:4023–4034. doi: 10.1093/nar/gki684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X, et al. A kinase-independent function of c-Abl in promoting proteolytic destruction of damaged DNA binding proteins. Mol Cell. 2006;22:489–499. doi: 10.1016/j.molcel.2006.04.021. [DOI] [PubMed] [Google Scholar]