

Abstract
Peptide binding to class II MHC protein is commonly viewed as a combination of discrete anchor residue preferences for pockets 1, 4, 6/7, and 9. However, previous studies have suggested cooperative effects during the peptide binding process. Investigation of the DRB1*0901 binding motif demonstrated a clear interaction between peptide binding pockets 6 and 9. In agreement with prior studies, pockets 1 and 4 exhibited clear binding preferences. Previously uncharacterized pockets 6 and 7 accommodated a wide variety of residues. However, although it was previously reported that pocket 9 is completely permissive, several substitutions at this position were unable to bind. Structural modeling revealed a probable interaction between pockets 6 and 9 through β9Lys. Additional binding studies with doubly substituted peptides confirmed that the amino acid bound within pocket 6 profoundly influences the binding preferences for pocket 9 of DRB1*0901, causing complete permissiveness of pocket 9 when a small polar residue is anchored in pocket 6 but accepting relatively few residues when a basic residue is anchored in pocket 6. The β9Lys residue is unique to DR9 alleles. However, similar studies with doubly substituted peptides confirmed an analogous interaction effect for DRA1/B1*0301, a β9Glu allele. Accounting for this interaction resulted in improved epitope prediction. These findings provide a structural explanation for observations that an amino acid in one pocket can influence binding elsewhere in the MHC class II peptide binding groove.
Peptides of variable length are bound by MHC class II on the surface of APC and recognized by CD4+ T cells. The docking of a peptide with the peptide binding cleft of MHC class II protein is facilitated by peptide binding pockets, which include polymorphic residues (1). For this reason, a given class II protein prefers specific amino acid residues within a given binding pocket while excluding others. Published crystal structures (2-4) provide a basis for the view that peptide binding preferences are a combination of anchor residue preferences at distinct pocket positions. Epitope prediction algorithms typically assume that individual pocket preferences are independent of other pockets, such that the peptide binding preferences of a given MHC class II allele can be modeled as a linear combination of the individual pocket preferences (5). However, rigorous studies of peptide binding to class II MHC have suggested both negative and positive cooperative effects between both pocket and solvent exposed residues (6, 7). Clearly, an accurate understanding of binding motifs is critical for effective epitope prediction, for rational modifications to peptides or protein, for use as vaccines, and so forth. Understanding peptide binding preferences also provides a key component for MHC-based disease associations, because only certain “susceptible” alleles will bind and present the relevant peptides.
The peptide binding motifs for a number of class MHC class II alleles have been investigated by a variety of methodologies. However, the binding motifs for many important alleles remain unknown or only partially characterized. Among these, the peptide binding motif for DRA1/B1*0901 (DR0901)3 is interesting because of its prevalence in Asian populations and its association with type 1 diabetes (8-12) and gelatin allergy (13). This motif has been investigated previously (14, 15). However, these studies could only define binding characteristics for pockets 1 and 4. The initial objective of this study was to identify the complete peptide binding motif of DR0901 using an in vitro peptide competition assay complimented by structural modeling. This information was used to verify T cell epitopes within antigenic peptides identified by tetramer-guided epitope mapping. During the course of this work, we observed surprising differences between our results and previous reports for pocket 9. This led us to examine possible interactions between pockets 6 and 9, first for DR0901 and subsequently for DRA1/B1*0301 (DR0301).
Materials and Methods
Peptides and MHC class II protein
Panels of 20-mer peptides with overlapping sequences spanning the influenza A/Puerto Rico/8/34 nucleoprotein (NP), influenza A/Puerto Rico/8/34 matrix protein (M1), influenza B/Hong Kong/330/2001 hemagglutinin (Flu B HA), influenza A/Panama/2007/99 hemagglutinin (HA Pan), influenza A/New Caledonia/20/99 hemagglutinin (HA NC), tetanus toxin H chain (TT), and tetanus toxin L chain (TTL) proteins were synthesized on polyethylene pins with 9-fluorenylmethoxycarbonyl chemistry by Mimotopes. The biotinylated reference tetanus toxoid peptide TT 503–515 (LNFNYSLDKIIVD) and sperm whale myoglobin 137–148 peptide (LFRKDIAAKYKE) were synthesized with an Applied Biosystems 433A Peptide Synthesizer (Applied Biosystems). These sequences included two Fmoc-6-aminohexanoic acid spacers between the peptide sequence and an N-terminal biotin label. A panel of 66 13-mer peptides (48 with single substitutions and 18 with double substitutions) with sequences based on the TT 503–515 sequence and a second panel of 27 peptides (with single or double substitutions) with sequences based on sperm whale myoglobin 137–148 was synthesized on polyethylene pins with 9-fluorenylmethoxycarbonyl chemistry by Mimotopes. Peptides were dissolved in DMSO at 20 mg/ml and subsequently diluted as needed. Recombinant DR0901 and DR0301 proteins were produced as described previously (16). Briefly, soluble DR0901 or DR0301 was purified from insect cell culture supernatants by affinity chromatography and dialyzed against phosphate storage buffer (pH 6.0).
Tetramer reagents
To prepare MHC class II tetramers, DR0901 or DR0301 protein was biotinylated at a sequence-specific site using biotin ligase (Avidity) before dialysis into phosphate storage buffer. The biotinylated DR0901 monomer was loaded with 0.2 mg/ml peptide by incubating at 37°C for 72 h in the presence of 2.5 mg/ml n-octyl-β-d-glucopyranoside and 1 mM Pefabloc SC (Sigma-Aldrich). Peptide-loaded monomers were subsequently conjugated as tetramers using R-PE streptavidin (BioSource International) at a molar ratio of 8:1.
Human subjects
Healthy DR0901 and DR0301 subjects were recruited with written consent as part of an Institutional Review Board-approved study.
Tetramer-guided epitope mapping
The tetramer-guided epitope mapping procedure was conducted as previously described (17) for each protein. PBMC were isolated from the blood of vaccinated healthy DR0901 or DR0301 subjects by Ficoll underlay and CD4+ T cells isolated using the Miltenyi CD4+ T cell isolation kit (Miltenyi Biotec). Cells from the CD4− fraction were incubated in 48-well plates (3 × 106 cells/well) for 1 h and then washed, leaving adherent cells as APC. After adding 2 million CD4+ T cells/well, each well was stimulated with a pool of five consecutive peptides. After 14 days, 100 μl of resuspended cells was stained with pooled peptide PE-conjugated tetramers for 60 min at 37°C. Subsequently, cells were stained with CD4-PerCP (BD Biosciences), CD3-FITC, and CD25-allophycocyanin mAbs (eBioscience) and analyzed by flow cytometry. Cells from pools that gave positive staining were analyzed again using the corresponding individual peptide tetramers.
Peptide binding competition assay
Various concentrations of each test peptide were incubated at pH 5.4 in competition with either 0.01 mM biotinylated TT 503–515 (DR0901) or sperm whale myoglobin 137–148 (DR0301) peptide in triplicate wells containing HLA-DR0901 or HLA-DR0301 protein as described previously (18). After washing, the biotinylated peptide was traced using europium-conjugated streptavidin (PerkinElmer) and quantified using a Victor2 D time-resolved fluorometer (PerkinElmer). Peptide binding curves were normalized by dividing each curve by its maximum counts and then fitted by nonlinear regression with a sigmoidal dose-response curve model using Prism software (version 4.03; GraphPad Software). IC50 binding values (the concentration needed to reduce binding of the biotinylated reference peptide by 50%) and their corresponding error bounds were calculated from the resulting curves using Prism software. Relative binding affinity (RBA) values were calculated as the IC50 value of the substituted peptide divided by the IC50 value of the nonsubstituted peptide. KD values were calculated as described by Ferrante and Gorski (7) based on the IC50 value and the direct binding of biotinylated peptide to DR0901 or DR0301. The ratio of two IC50 values indicates the fold difference in peptide binding affinity.
To isolate the effect of pocket 9 residues alone for dually substituted peptides, adjusted RBA values (RBAadj) were calculated as the IC50 value of the dually substituted peptide divided by the IC50 value of the singly substituted (or nonsubstituted) peptide with a matching pocket 6 residue. The resulting value reflects the influence of the pocket 9 residue, including any cooperative effect. Cooperative effects were calculated essentially as described by Ferrante and Gorski (7), taking the ratio of expected the effect of the two single substitutions (calculated) to the effect of the double substitution (observed) in terms of ΔKD. Values greater than or less than 1 indicate the presence of negative cooperativity (better binding than expected) or positive cooperativity (worse binding than expected), respectively.
Molecular modeling
Models of DR0901 with the TT 503–515 peptide and its variants were prepared on Silicon Graphics Indigo 2 and Octane work stations using the program Insight II, version 2000 (Accelrys), essentially as described previously (19). Energy minimization was performed at pH 5.4, the experimental pH used for binding studies. The crystal structure of HLA-DRB1*0101 in complex with the HLA-A2 peptide (2) was used as the base molecule for all simulation studies. Figures were drawn with the aid of WebLabViewer version 3.5 and DSViewer Pro version 6.0 of Accelrys using previously published formatting and color conventions (19).
Epitope prediction
To predict epitopes, an array of binding coefficients (Cp) was developed for each pocket based on the observed binding of single (at pockets 1, 4, 6, 7, or 9) and double (at pockets 6 and 9) amino acid substituted versions of the tetanus toxin 503–515 peptide sequence. These Cp values are summarized in supplementary Table I.4 Because all possible amino acids were not measured for any of the pockets, missing values in the data set were estimated based on the observed values for chemically similar amino acids. Peptide binding affinities were calculated based on the following formula: RBA =Cp1 × Cp4 × Cp6 × Cp7 × Cp9.
In this formula, each Cp refers to the observed or estimated binding coefficient. For standard predictions (RBA), the “0” value of Cp9 was used regardless of the amino acid found in pocket 6. For corrected predictions (CRBA), the value of Cp9 was taken from the appropriate column (“−1” value, “0” value, “1” value, or “0.5” value) using a conditional lookup based on the identity of the amino acid found in pocket 6. For both methods, the RBA for a given peptide was determined by scanning across every possible binding register and taking the highest observed RBA value.
Results
Tetramer-guided epitope mapping of DR0901-restricted epitopes
Tetramer-guided epitope mapping was used to identify DR0901-restricted epitopes within the NP, M1, Flu B HA, HA Pan, HA NC, TT, and TTL proteins. Overlapping peptides (20 aa long with a 12 residue overlap) spanning each protein were synthesized and divided into pools as described in Materials and Methods. CD4+ T cells from multiple DR0901 subjects were stimulated with pooled peptides and analyzed using the corresponding pooled peptide tetramers after 2 wk in culture. Pools with tetramer-positive populations (having a population at least 0.5% above background) were analyzed again using individual peptide tetramers to reveal the antigenic peptides within each pool. Representative results for pooled peptide tetramer analysis are shown in Fig. 1A. For TT, positive staining was observed for six peptide pools. Subsequent results for individual peptide tetramer analysis are shown in Fig. 1B. Positive staining was observed for seven individual peptides. Similar analysis was completed for the remaining proteins, identifying a total of 31 peptides that contain DR0901-restricted epitopes. Among these, the core region of the TT 498–517 peptide, residues 503–515, was chosen as a study peptide to determine the binding preferences of DR0901.
FIGURE 1.

Tetramer-guided epitope mapping of TT epitopes. A, Pool mapping: T cells from a DR0901 donor were stimulated with overlapping peptide mixtures spanning the TT and stained using pooled peptide-loaded tetramers after 2 wk. Peptide pools 2, 7, 8, 9, 10, and 19 gave positive staining. B, Individual peptide mapping: T cells from the tetramer-positive panels shown in A were stained again using tetramers loaded with individual peptide from the corresponding peptide pools. Peptides p6, p34, p35, p36, p45, p50, and p91 were identified as peptides containing DR0901-restricted epitopes.
Binding register of the study peptide
To facilitate subsequent study of binding preferences, the binding register of the study peptide (TT 503–515) was confirmed using a set of arginine-substituted peptides. Replacing anchor residues with this basic amino acid is known to disrupt peptide binding (20). As shown in Fig. 2, arginine substitution at residues 505F, 508S, and 513I led to substantially diminished binding. These findings suggest that those positions represent primary anchor residues for binding to DR0901. The spacing of these residues establishes 505F as the P1 anchor, 508S as the P4 anchor, and 513I as the P9 anchor. Substitutions at the remaining positions had only minor influences on binding.
FIGURE 2.
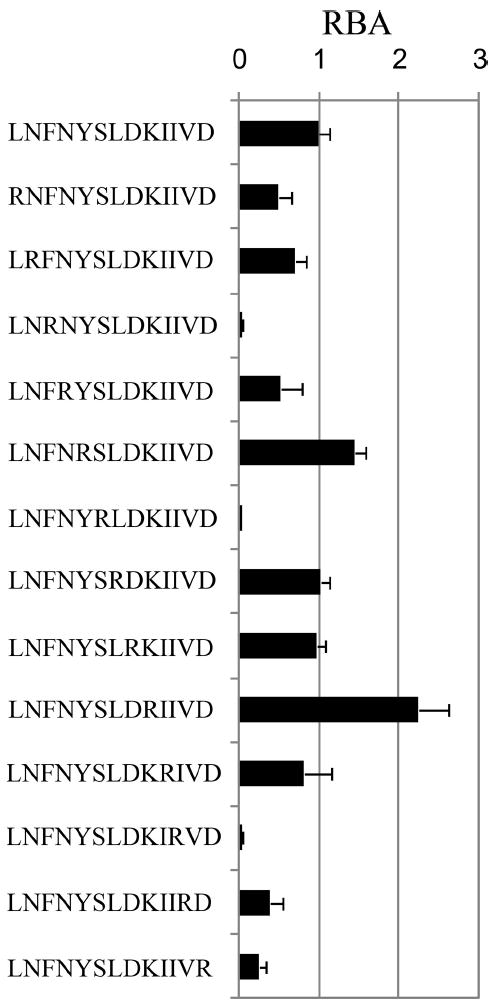
Binding register of the test peptide. RBA for arginine substituted derivatives of tetanus toxoid 503–515 peptide are shown. RBA values were calculated as the IC50 value of the substituted peptide divided by the IC50 value of the unsubstituted peptide based on binding curves (europium counts vs test peptide concentration) after normalization (as described in Materials and Methods). Arginine substitutions at pockets 1, 4, and 9 led to markedly reduced binding affinities.
Binding of peptides with single substitutions
Using this knowledge of the binding register, a panel of TT 503–515-derived peptides with single amino acid substitutions was designed to determine the amino acid preferences for the binding pockets of DR0901. These substitutions represented obligatory anchor residues for pocket 1 and general classes of amino acids for pockets 4, 6, 7, and 9 (5). Each peptide was bound at various concentrations to recombinant DR0901 protein in competition with the biotinylated TT reference peptide. The sequences of these 48 peptides and their RBA are summarized in Table I.
Table I.
Binding of substituted TT 503–515 peptides
| Sequence | Substitution | RBA | KD (nM) |
|---|---|---|---|
| LNFNYSLDKIIVD | Unmodified | 1.0 ± 0.24 | 73 ± 16 |
| LNYNYSLDKIIVD | P1 F→Y | 0.65 ± 0.35 | 113 ± 40 |
| LNLNYSLDKIIVD | P1 F→L | 0.16 ± 0.03 | 450 ± 80 |
| LNMNYSLDKIIVD | P1 F→M | 0.12 ± 0.04 | 600 ± 150 |
| LNVNYSLDKIIVD | P1 F→V | 0.16 ± 0.07 | 440 ± 130 |
| LNFNYYLDKIIVD | P4 S→Y | 0.02 ± 0.01 | 3,400 ± 1,100 |
| LNFNYLLDKIIVD | P4 S→L | 0.02 ± 0.02 | 3,600 ± 1,800 |
| LNFNYMLDKIIVD | P4 S→M | 0.50 ± 0.03 | 146 ± 9 |
| LNFNYPLDKIIVD | P4 S→P | 0.25 ± 0.07 | 290 ± 70 |
| LNFNYALDKIIVD | P4 S→A | 1.3 ± 0.28 | 55 ± 10 |
| LNFNYGLDKIIVD | P4 S→G | 2.3 ± 0.34 | 38 ± 5 |
| LNFNYRLDKIIVD | P4 S→R | 0.06 ± 0.02 | 1,230 ± 340 |
| LNFNYQLDKIIVD | P4 S→Q | 0.06 ± 0.02 | 1,500 ± 450 |
| LNFNYHLDKIIVD | P4 S→H | 0.87 ± 0.19 | 80 ± 15 |
| LNFNYDLDKIIVD | P4 S→D | 0.04 ± 0.01 | 1,040 ± 240 |
| LNFNYNLDKIIVD | P4 S→N | 0.15 ± 0.06 | 290 ± 90 |
| LNFNYELDKIIVD | P4 S→E | 0.001 ± 0.001 | 116,000 ± 60,000 |
| LNFNYSLYKIIVD | P6 D→Y | 0.65 ± 0.13 | 67 ± 11 |
| LNFNYSLLKIIVD | P6 D→L | 0.65 ± 0.09 | 67 ± 8 |
| LNFNYSLMKIIVD | P6 D→M | 0.94 ± 0.05 | 46 ± 2 |
| LNFNYSLPKIIVD | P6 D→P | 1.39 ± 0.08 | 31 ± 2 |
| LNFNYSLSKIIVD | P6 D→S | 1.41 ± 0.16 | 31 ± 3 |
| LNFNYSLAKIIVD | P6 D→A | 1.6 ± 0.86 | 51 ± 18 |
| LNFNYSLRKIIVD | P6 D→R | 0.61 ± 0.10 | 130 ± 19 |
| LNFNYSLHKIIVD | P6 D→H | 0.94 ± 0.10 | 87 ± 8 |
| LNFNYSLNKIIVD | P6 D→N | 0.88 ± 0.23 | 93 ± 19 |
| LNFNYSLDRIIVD | P7 K→R | 1.7 ± 0.40 | 53 ± 10 |
| LNFNYSLDHIIVD | P7 K→H | 3.0 ± 1.2 | 29 ± 9 |
| LNFNYSLDDIIVD | P7 K→D | 0.50 ± 0.29 | 176 ± 65 |
| LNFNYSLDYIIVD | P7 K→Y | 2.1 ± 0.69 | 41 ± 10 |
| LNFNYSLDNIIVD | P7 K→N | 3.3 ± 0.11 | 27 ± 1 |
| LNFNYSLDSIIVD | P7 K→S | 0.77 ± 0.52 | 110 ± 45 |
| LNFNYSLDFIIVD | P7 K→F | 3.1 ± 0.76 | 28 ± 6 |
| LNFNYSLDPIIVD | P7 K→P | 2.5 ± 1.2 | 36 ± 12 |
| LNFNYSLDMIIVD | P7 K→M | 3.7 ± 0.88 | 24 ± 5 |
| LNFNYSLDAIIVD | P7 K→A | 1.4 ± 0.60 | 65 ± 20 |
| LNFNYSLDKIYVD | P9 I→Y | 0.34 ± 0.05 | 240 ± 33 |
| LNFNYSLDKILVD | P9 I→L | 0.25 ± 0.06 | 330 ± 68 |
| LNFNYSLDKIMVD | P9 I→M | 0.29 ± 0.08 | 280 ± 61 |
| LNFNYSLDKIPVD | P9 I→P | 0.05 ± 0.008 | 1,700 ± 250 |
| LNFNYSLDKISVD | P9 I→S | 0.11 ± 0.02 | 770 ± 100 |
| LNFNYSLDKIAVD | P9 I→A | 0.09 ± 0.03 | 940 ± 250 |
| LNFNYSLDKIRVD | P9 I→R | 0.01 ± 0.02 | 6,700 ± 1,200 |
| LNFNYSLDKIHVD | P9 I→H | 0.15 ± 0.04 | 740 ± 170 |
| LNFNYSLDKIDVD | P9 I→D | 0.14 ± 0.02 | 790 ± 120 |
| LNFNYSLDKINVD | P9 I→N | 0.04 ± 0.02 | 2,600 ± 740 |
| LNFNYSLDKIEVD | P9 I→E | 0.10 ± 0.05 | 890 ± 290 |
| LNFNYSLDKIWVD | P9 I→W | 0.16 ± 0.02 | 530 ± 50 |
Corresponding to these observed binding affinities, the preferences for each pocket are summarized in Fig. 3. For this analysis, values within 2.5-fold of the reference peptide (RBA ≥ 0.4) were preferred, values within 10-fold of the reference peptide (0.4 > RBA ≥ 0.1) were tolerated, and values <10-fold of the reference peptide (RBA ≤ 0.1) were not tolerated. As expected, pocket 1 preferred aromatic anchor residues. Consistent with previous reports, pocket 4 preferred small anchor residues but also accepted Met and His and tolerated Pro and Asn. Pocket 6 appeared to be completely permissive, accepting residues of varied size, hydrophobicity, and charge. Pocket 7 was nearly as permissive, accommodating Asp somewhat less than other residues, but accepting every residue tested. In contrast to previous reports, pocket 9 was not completely permissive, barely tolerating Ser and Asn while not allowing Arg, Ala, Pro, and Glu.
FIGURE 3.
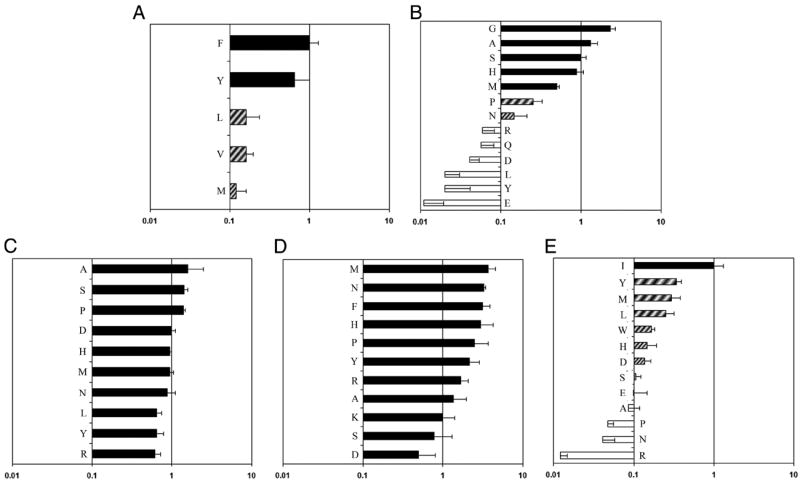
Pocket Amino acid preferences. Binding preferences for pocket 1 (A), pocket 4 (B), pocket 6 (C), pocket 7 (D), and pocket 9 (E) of the DR0901 motif. ■ represents values within 2.5-fold of the reference peptide (RBA ≥ 0.4, “preferred”), ▨ represents values within 10-fold of the reference peptide (0.4 > RBA ≥ 0.1, “tolerated”), and □ represents values within <10-fold of the reference peptide (RBA ≤ 0.1, “excluded”).
Modeling analysis of the DR0901 peptide binding motif
Models of the TT 503–515/DR0901 complex were created based on the crystal structure of DR0101 in complex with the HLA-A2 peptide (2) after energy minimization at pH 5.4, as described in Materials and Methods. The modeling results suggested that TT 503–515 binds with geometry consistent with a high-affinity interaction (Fig. 4A). This view highlights several DR0901 residues that appear to interact with the antigenic peptide (shown in space-filling form). This modeling indicated that the peptide binds with 505F in pocket 1, 508S in pocket 4, 510D in pocket 6, 511K in pocket 7, and 513I in pocket 9.
FIGURE 4.

Molecular modeling of DRB1*0901 in complex with peptide. A, TCR view of the antigenic peptide (LNFNYSLDKIIVD, anchors in bold) in the groove of DRB1*0901 (DR0901) after energy minimization at pH 5.4 based on the crystal structure of HLA-DRB1*0101 in complex with the HLA-A2 peptide (2). The α1β1 domain of the DR0901 molecule is depicted in van der Waals surface representation, with the surface atomic charges color-coded (blue, positive; gray, neutral; red, negative), and the antigenic peptide is shown in space-filling form (color code: carbon, green; oxygen, red; nitrogen, blue; hydrogen, white; sulfur, yellow). P1Phe is in pocket 1, p6Asp in pocket 6, and p9Ile in pocket 9 (all three pointing into the plane of the paper) as expected of a peptide bound to DR0901 with high affinity. Several DR0901 residues that have interactions with the antigenic peptide (α54Phe, α58Gly, α65Val, α72Ile, α73Met, β57Val, β60Ser, β61Trp, β67Phe, β70Arg, β71Arg, β74Glu, and β78Val) are shown in stick form with the same color-code as the antigenic peptide with the exception of carbon (orange). B, TCR view of pocket 4 of the complex of the peptide LNFNYSLDKIIVD with HLA-DR0901. The arrangement of the various residues in this pocket promotes the anchoring of either small aliphatic/polar amino acids at p4. The figure has been rotated by 45° with respect to the y-axis (left side into the plane of the paper, right side above the plane of the paper) so that the interactions between various residues could be fully exposed. Because of this, the aromatic ring of β13Phe is totally covered by p4Ser. This view highlights the overall compactness of this pocket, which limits the size of residues that may anchor. Color and depiction conventions are identical to those in A. C, Side view of pocket 6 of the complex of the peptide LNFNYSLDKIIVD with HLA-DR0901. The β9Lys residue (typically a pocket 9 residue) swings into this pocket and remains there, promoting the anchoring of acidic residues (as in the case of p6Asp shown here). This pocket is depicted from the Cα of β57Val at the level of the β-sheet floor (similar to Fig. 4 of Ref. 28). For orientation purposes, part of the α1 helix (residues 62–69), the β-sheet floor from where residues originate, and the peptide backbone are shown. Color and depiction conventions are identical to those in A. In addition, α-helix is in red, β-sheet is in turquoise, and all other secondary structure forms (random coil, polyproline type II helix of the antigenic peptide) are in gray. D, Side view of pocket 6 of the complex of the peptide LNFNYSLKKIIVD with HLA-DR0901. This residue can be accommodated, albeit not with the same ease as p6D. As in the other panels, pocket 9 is occupied by isoleucine. The orientation of the HLA-DRB1*0901 chains and the antigenic peptide chain is identical to the previous figure of pocket 6 with Asp. In contrast to C, the β9Lys residue assumes a slightly different position within pocket 6, whereas P6Lys is diverted and points toward the base of pocket 9. The orientation, color, and depiction conventions are identical to those in C. E, TCR view of pocket 9 of the complex of the peptide LNFNYSLDKIYVD with HLA-DR0901. The Tyr anchor residue is depicted in pocket 9 to illustrate the ample space within this pocket. Color and depiction conventions are identical to those in A.
Fig. 4B shows a TCR view of pocket 4 of the bound study peptide, tilted by 45° with respect to the y-axis, to present a better perspective of the residues that comprise this pocket. These combine to form a compact pocket, promoting the anchoring of either small aliphatic or small polar amino acids. The presence of β70Arg/β74Glu hinders the binding of acidic and basic residues in this pocket. Likewise, the presence of β13Phe and β26Tyr (and to some degree α9Gln) leaves insufficient space for bulky aliphatic or aromatic residues in this pocket. Although histidine is positively charged at pH 5.4, its imidazole side chain is planar and hence can squeeze through the aforementioned residues constituting the base of pocket 4, in close contact with β13Phe and β26Tyr (via pi-pi aromatic interactions).
Fig. 4C depicts a side view (from the Cα of β57Val at the level of the β-sheet floor) of pocket 6 of the peptide-MHC complex. The flexible arrangement of the residues in this pocket and the ability of β9Lys to participate in this pocket (seen clearly in the case of Asp shown in pocket 6 here) promote the anchoring of a wide variety of residues. The presence of β30Gly provides ample space for bulky residues, whereas the presence of three acidic residues (α11Glu, α66Asp, and β11Asp) and two basic ones (β28His and β9Lys, the latter of which is not considered a classical pocket 6 residue but can be drawn toward acidic p6 anchor residues) provides a suitable environment for the binding of positively and negative charged residues in this pocket. In contrast, the binding of the basic p6Lys is in an unconventional manner, because β9Lys is too close to the various negative charges of pocket 6 to be completely displaced (Fig. 4D). Thus, β9Lys still participates in pocket 6, whereas the incoming p6Lys residue assumes a position pointing toward the bottom of pocket 9. In this position, p6Lys might well be expected to influence the preferences of pocket 9.
Fig. 4E shows a TCR view of pocket 9 of the complex. The arrangement of the residues in this pocket promotes the anchoring of either large aliphatic or aromatic amino acids at this position. In particular, the presence of β30Gly leaves ample space for bulky residues. In a manner that is analogous to pocket 9 of HLA-DQ0604 (20), the combination α72Ile/α73Met/β37Asn/β57Val appears to disfavor the anchoring of basic (and to some degree acidic) amino acids within this pocket. The β9Lys residue, typically regarded as belonging to pocket 9, has been diverted away by electrostatic attraction to α11Glu, α66Asp, β11Asp, and p6Asp, creating additional space within pocket 9.
Motif analysis of peptides containing DR0901 epitopes
As summarized in Table II, a total of 31 peptides were shown by tetramer-guided epitope mapping to contain putative DR0901-restricted epitopes. These peptides were derived from the NP, M1, Flu B HA, HA Pan, HA NC, TT, and TTL proteins. For each of these peptides, RBA were calculated using the standard prediction method as described in Materials and Methods. The predicted score of the best nonameric core epitope for each peptide is shown in the RBA column of Table II. For the majority of these peptides, single nonameric core epitopes consistent with the DR0901 binding motif were identified (boldface). A few peptides contained two distinct registers that could be expected to bind DR0901. For these, the second motif is underlined.
Table II.
Motif analysis for DR0901 epitopes
| Protein | Residues | Epitopea | RBAb | CRBAc |
|---|---|---|---|---|
| NP | NP 161–180d | PRMCSLMQGSTLPRRSGAAG | 0.01 | 0.57 |
| NP 217–236 | IAYERMCNILKGKFQTAAQK | 0.07 | 0.49 | |
| NP 225–244 | ILKGKFQTAAQKAMMDQVRE | 0.27 | 0.29 | |
| NP 233–252d | AAQKAMMDQVRESRNPGNAE | 0.01 | 0.21 | |
| NP 449–468d | ESARPEDVSFQGRGVFELSD | 0.04 | 0.10 | |
| NP 457–476 | SFQGRGVFELSDEKAASPIV | 0.08 | 0.10 | |
| NP 465–484 | ELSDEKAASPIVPSFDMSNE | 0.03 | 0.03 | |
| NP 473–492 | SPIVPSFDMSNEGSYFFGDN | 0.15 | 0.15 | |
| M1 | MP 97–116 | VKLYRKLKREITFHGAKEIS | 0.28 | 0.28 |
| MP 105–124 | REITFHGAKEISLSYSAGAL | 0.72 | 0.72 | |
| Flu B HA | HA 145–164 | NVTNGNGFFATMAWAVPKNE | 0.62 | 1.6 |
| HA Pan | HA 105–124 | YASLRSLVASSGTLEFNNES | 0.10 | 0.35 |
| HA 185–204 | PSTDSDQISLYAQASGRVTV | 0.15 | 0.68 | |
| HA 193–212 | SLYAQASGRVTVSTKRSQQT | 0.15 | 0.68 | |
| HA 321–340 | RNVPEKQTRGIFGAIAGFIE | 0.21 | 0.21 | |
| HA 329–348 | RGIFGAIAGFIENGWEGMVD | 0.21 | 0.21 | |
| HA 331–350 | IFGAIAGFIENGWEGMVDGW | 0.21 | 0.21 | |
| HA NC | HA 249–268 | LLEPGDTIIFEANGNLIAPW | 0.69 | 0.92 |
| HA 257–276 | IFEANGNLIAPWYAFALSRG | 0.69 | 0.69 | |
| HA 265–284 | IAPWYAFALSRGFGSGIITS | 0.49 | 1.7 | |
| TT | TT 498–517 | TKNKPLNFNYSLDKIIVDYN | 1.0 | 1.0 |
| TT 722–741 | VKAKWLGTVNTQFQKRSYQM | 0.08 | 0.28 | |
| TT 730–749 | VNTQFQKRSYQMYRSLEYQV | 0.08 | 0.08 | |
| TT 738–757 | SYQMYRSLEYQVDAIKKIID | 0.08 | 0.08 | |
| TT 810–829 | INEAKKQLLEFDTQSKNILM | 0.11 | 0.10 | |
| TT 850–869d | KINKVFSTPIPFSYSKNLDC | 0.36 | 0.96 | |
| TT 1178–1197 | KRYTPNNEIDSFVKSGDFIK | 0.06 | 0.60 | |
| TTL | TT 185–204 | PCRDGFGSIMQMAFCPEYVP | 0.07 | 0.18 |
| TT 193–212 | IMQMAFCPEYVPTFDNVIEN | 0.84 | 0.84 | |
| TT 249–268 | SHEIIPSKQEIYMQHTYPIS | 0.10 | 0.10 | |
| TT 257–276 | QEIYMQHTYPISAEELFTFG | 0.10 | 0.10 |
For each peptide, the most likely binding motif (based in predicted binding affinity) is boldface. Plausible secondary motifs are underlined.
RBA of most likely motif predicted based on binding of singly substituted peptides.
CRBA of most likely motif predicted based on pocket 6/9 interactions observed for doubly substituted peptides.
Motifs that were clarified based on CRBA values are double underlined.
For 11 of the 31 peptides, determining the preferred binding register was problematic because the predicted RBA values were low for every possible register. For several, the predicted RBA was depressed because of unfavorable residues at position 9. For example, NP 161–180 was problematic because its most likely nonameric register (LMQGSTLPR) contains a poorly tolerated Arg at position 9. Likewise, NP 233–252 was problematic because its most likely register (VRESRNPGN) contains a suboptimal Asn at position 9. Other peptides, such as TT 722–741 and TT 185–204 were also problematic. However, additional findings (see next section) clarified the binding motifs for many of these peptides.
Binding of peptides with dual substitutions to DR0901
As mentioned above, our findings for the binding preferences of pocket 9 differed significantly from previously published results. In addition, several antigenic peptides, which lacked good DR0901 motifs, were problematic mainly because of poorly tolerated residues at position 9. Our modeling results indicated that for our study peptide, the side chain of β9Lys was diverted toward pocket 6 through electrostatic attraction with Asp anchored in pocket 6 but was diverted with Lys anchored in pocket 6, allowing p6Lys to point toward the base of pocket 9 (Fig. 4, C and D). We hypothesized that the β9Lys side chain can assume various positions depending on electrostatic properties, size, and orientation of the residue anchored in pocket 6, thereby altering the binding preferences of pocket 9. To investigate these possible interactions between pockets 6 and 9 through β9Lys, a panel of TT 503–515-derived peptides was designed with amino acid substitutions at both of these positions. These substitutions consisted of a negatively charged (D), uncharged (S), or positively charged (K) residue at position 6 and several classes of amino acids at position 9. Each peptide was bound at various concentrations to recombinant DR0901 protein in competition with the biotinylated TT reference peptide. To isolate the effect of pocket 9 residues alone for dually substituted peptides, RBAadj values were calculated as the IC50 value of the dually substituted peptide divided by the IC50 value of the singly substituted (or nonsubstituted) peptide with a matching pocket 6 residue. Corresponding to these RBAadj, the preferences for pocket 9 are summarized in Fig. 5A.
FIGURE 5.
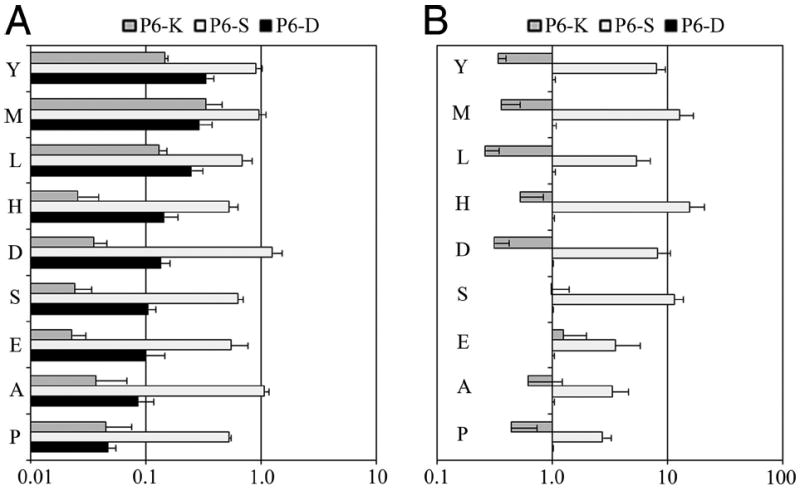
The amino acid preferences of pocket 9 of DR0901 vary cooperatively for distinct substitutions in pocket 6. A, RBAadj values plotted for dually substituted peptides. ■ represents values for various amino acids in pocket 9 with Asp (negatively charged) in pocket 6, □ represents values for various amino acids in pocket 9 with Ser (uncharged) in pocket 6, and
 represents values for various amino acids in pocket 9 with Lys (positively charged) in pocket 6. RBAadj values are normalized for relative p6 binding affinity to reflect the effect for pocket 9 alone on amino acid preference (calculated as described in Materials and Methods). B, Coopertivity values (calculated as described in Materials and Methods) plotted for dually substituted peptides. □ represents cooperativity values for p6Ser in conjunction with various amino acids in pocket 9.
represents cooperativity values for p6Lys in conjunction with various amino acids in pocket 9. By definition, cooperativity values are 1 for the singly substituted p6Asp peptides.
represents values for various amino acids in pocket 9 with Lys (positively charged) in pocket 6. RBAadj values are normalized for relative p6 binding affinity to reflect the effect for pocket 9 alone on amino acid preference (calculated as described in Materials and Methods). B, Coopertivity values (calculated as described in Materials and Methods) plotted for dually substituted peptides. □ represents cooperativity values for p6Ser in conjunction with various amino acids in pocket 9.
represents cooperativity values for p6Lys in conjunction with various amino acids in pocket 9. By definition, cooperativity values are 1 for the singly substituted p6Asp peptides.
Given the marked differences in pocket 9 preferences for different pocket 6 residues, we suspected that cooperative effects may exist. To investigate this directly, the cooperativity for each doubly substituted peptide was calculated as described in Materials and Methods. These values, shown in Fig. 5B, reflect the effect of the double substitution (observed) divided by the expected effect of the two single substitutions (calculated). Cooperativity values were >1 for p6Ser in conjunction with every p9 residue tested, indicating enhanced binding compared with the combined effect of the two single substitutions. Cooperativity values were <1 for p6Lys in conjunction with many of the p9 residues tested, indicating reduced binding compared with the combined effect of the two single substitutions. However, a few were not significantly different than 1, indicating that there was no cooperativity for those residues.
These results indicate that the permissiveness of pocket 9 is dependent on pocket 6 because of cooperative effects, most likely mediated by the positioning of β9Lys. On the basis of the modeling results, the three negatively charged residues of position 6 draw β9Lys away from pocket 9 (and p6Asp further reinforces this arrangement), creating a spacious pocket. In agreement, the binding results indicated that pocket 9 prefers large residues and excludes basic amino acids when Asp was anchored in pocket 6. Conversely, the modeling indicated that a positively charged Lys residue anchored in pocket 6 is oriented pointing toward the base of pocket 9, creating a constrained pocket 9. In agreement, the binding results indicated that pocket 9 accepted only a narrow range of hydrophobic residues when Lys was anchored in pocket 6. On the basis of the modeling, it might be expected that an uncharged Ser residue at position 6 would allow β9Lys some flexibility of movement (while still remaining in close proximity to the negative charges of pocket 6). In agreement with this notion, pocket 9 accommodated a diverse range of residues when Ser was anchored in pocket 6. As such, the peptide binding preferences for pocket 9 can be envisioned as a continuum of submotifs, in which the role of pocket 9 varies from being permissive (with small, uncharged residues such as Ser in pocket 6) to being extremely selective (with positively charged residues such as Lys in pocket 6).
On the basis of these findings, corrected relative binding affinities (CRBA) were calculated using a modified prediction method as described in Materials and Methods. The predicted score of the best nonameric core epitope for each peptide is shown in the CRBA column of Table II. This modified prediction method clarified the binding motif for several peptides. For example, the binding motif for NP 161–180 (LMQGSTLPR) is no longer problematic. Because this peptide contains an uncharged Thr (similar to Ser) at position 6, its pocket 9 can accommodate Arg. Similarly, in the motif for NP 233–252 (VRESRNPGN), Asn should be allowed at position 9 because it has an uncharged Asn at position 6. For 3 of the 31 peptides, determining the preferred binding register remained problematic. One of these, NP 465–484, may bind with Phe in pocket 1 (NP residue 479) and Ser in pocket 4 (NP residue 482) and the remaining pockets empty (bolded residues) as has been previously suggested for DR0901 (14, 15). The remaining two peptides, TT 730–749 and TT 738–757, appear to share a low-affinity motif (MYRSLEYQV) that is antigenic despite being predicted to bind weakly. Binding motifs that were clarified by these findings are double underlined in Table II.
Binding of peptides with dual substitutions to DR0301
The results above indicate that for DR0901, the permissiveness of pocket 9 is dependent on pocket 6, most likely through the positioning of β9Lys. Although β9Lys is unique to DR9 alleles, β9Glu is present in several common alleles, including DR0301. We hypothesized that this side chain may also assume different positions (depending on the residue present in pocket 6), thereby altering the binding preferences of pocket 9. To investigate these possible interactions between pockets 6 and 9 through β9Glu, a panel of 27 sw myoglobin 137–148-derived peptides was designed with amino acid substitutions at both of these positions. These substitutions consisted of a negatively charged (E), uncharged (A), or positively charged (K) residue at position 6 and several classes of amino acids at position 9. Each peptide was bound at various concentrations to recombinant DR0301 protein in competition with the biotinylated sw myoglobin reference peptide. To isolate the effect of pocket 9 residues alone for dually substituted peptides, RBAadj were calculated as the IC50 value of the dually substituted peptide divided by the IC50 value of the singly substituted (or nonsubstituted) peptide with a matching pocket 6 residue. Corresponding to these RBAadj values, the preferences for pocket 9 for DR0301 are summarized in Fig. 6A.
FIGURE 6.
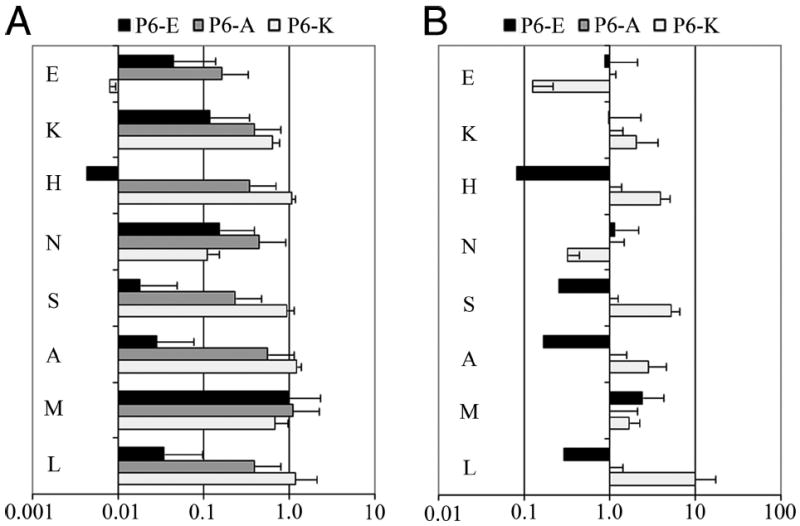
The amino acid preferences of pocket 9 of DR0301 vary cooperatively for distinct substitutions in pocket 6. A, RBAadj values plotted for dually substituted peptides. ■ represents values for various amino acids in pocket 9 with Glu (negatively charged) in pocket 6,
represents values for various amino acids in pocket 9 with Ala (uncharged) in pocket 6, and □ represents values for various amino acids in pocket 9 with Lys (positively charged) in pocket 6. RBAadj values are normalized for relative p6 binding affinity to reflect the effect for pocket 9 alone on amino acid preference (calculated as described in Materials and Methods). B, Coopertivity values (calculated as described in Materials and Methods) plotted for dually substituted peptides. ■ represents cooperativity values for p6Glu in conjunction with various amino acids in pocket 9. □ represents cooperativity values for p6Lys in conjunction with various amino acids in pocket 9. By definition, cooperativity values are 1 for the singly substituted p6Ala peptides.
To directly investigate cooperative effects for pockets 6 and 9 of DR0301, the cooperativity for each doubly substituted peptide was calculated as described in Materials and Methods. These values, shown in Fig. 6B, reflect the effect of the double substitution (observed) divided by the expected effect of the two single substitutions (calculated). Cooperativity values were >1 for p6Lys in conjunction with most of the p9 residues tested, indicating enhanced binding compared with the combined effect of the two single substitutions. However, two (Glu and Asn) were <1, indicating reduced binding compared with the combined effect of the two single substitutions. Cooperativity values were <1 for p6Glu in conjunction with half of the p9 residues tested, indicating reduced binding compared with the combined effect of the two single substitutions. However, the cooperativity value for Met was modestly >1, indicating enhanced binding compared with the combined effect of the two single substitutions. The cooperativity values for the remaining residues were not significantly different than 1, indicating that there was no cooperativity for these residues.
These results indicate that the permissiveness of pocket 9 of DR0301 is also dependent on pocket 6 because of cooperative effects. In this case, the effect is more subtle. When a positively charged Lys residue is anchored at position 6, β9Glu is somewhat free to move away from pocket 9, creating a pocket that accommodates basic and uncharged residues of various sizes. However, acidic amino acids are excluded. A negatively charged Glu residue at position 6 pushes β9Glu toward pocket 9, creating a constrained pocket that accepts Met, Asn, and Lys while excluding aliphatic and acidic residues. A small hydrophobic Ala residue at position 6 allows β9Glu to move freely, creating a pocket 9 that can accommodate diverse residues. On the basis of these findings, it would be expected that DR0301 would prefer different residues at position 9, depending on the character of the residue at position 6. As shown in Table III, the residues seen at position 9 for several novel or previously described (21) tetramer-positive DR0301-restricted epitopes are consistent with these findings. For peptides with basic pocket 6 anchors (K or R), primarily small or basic residues are found within pocket 9 (G, V, T, L, and K). For peptides with acidic pocket 6 anchors, a basic (R) or long aliphatic (L) residue is present. For peptides with an uncharged pocket 6 anchor, all classes of amino acids are accepted (E, T, S, L, Y, K, and R).
Table III.
Motif analysis for DR0301 epitopes
| Protein | Residues | Epitopea |
|---|---|---|
| NP | NP 54–65b | GRLIQNSLTIER |
| NP 107–118b | ELILYDKEEIRR | |
| NP 486–497b | SYFFGDNAEEYD | |
| Flu B HA | HA 110–121b | FPIMHDRTKIRQ |
| HA 217–228b | AKLYGDSKPQKF | |
| HA 253–264b | GGLPQSGRIVVD | |
| HA 259–270b | GRIVVDYMVQKS | |
| HA 265–276b | YMVQKSGKTKTI | |
| TT | TT 510–521c | DKIIVDYNLQSK |
| TT 830–841c | QYIKANSKFIGI | |
| TT 994–1005c | SLKGNNLIWTLK | |
| TT 1062–1073c | GAIREDNNITLK | |
| TT 1120–1131c | NPLRYDTEYYLI | |
| M1 | MP 169–180b | PLIRHENRMVLA |
For each peptide, the most likely binding motif is underlined. Pockets 6 and 9 residues are boldface.
Novel DR0301-restricted epitope identified by tetramer-guided epitope mapping.
Previously identified tetramer-positive DR0301 epitope (22).
Discussion
The simplest view of peptide binding to class II MHC is that the preferences of individual binding pockets are independent of other pockets. However, previous studies have suggested both negative and positive cooperative effects between pocket residues (7). Ignoring such cooperative interactions can hinder efforts to understand and predict the binding and presentation of epitopes by HLA of interest. For example, previous studies of DR0901 were not able to define amino acid preferences for pocket 9 (14, 15). These studies used a peptide sequence (ANDGATGWVASMSSAY, anchor residues underlined) that bound with Ser in pocket 6. Our current findings suggest that a small, uncharged residue at position 6 (such as Ser or Ala) creates a permissive pocket 9 that can accommodate diverse residues. Therefore, these previous results are completely consistent with our present findings. However, the preferences of pocket 9 change completely when a charged residue is anchored within pocket 6. This dramatic change in binding preferences is dependent on the positioning of β9Lys and the influence of the residue anchored within pocket 6. On the basis of these molecular modeling results, this residue has the ability to participate in pocket 6 in a stable electrostatic attraction with three negatively charged residues (α11Glu, α66Asp, and β11Asp); furthermore, the presence of β30Gly leaves ample freedom for the movement for any bulky residues in pocket 6. This is the only MHC class II allele known with such a substantial shift in the position of the β9 residue from pocket 9 into pocket 6. A negatively charged residue at position 6 creates a spacious pocket 9 that prefers large residues and excludes basic amino acids. A positively charged residue at position 6 creates a constrained pocket 9 (as explained previously) that can accept only a narrow range of hydrophobic residues.
Because of this, the binding preferences for pockets 6 and 9 of DR0901 are interdependent. Similarly, our current findings suggest that the binding preferences of pockets 6 and 9 of DR0301 are also interdependent, depending on the relative positioning of β9Glu. Although the β9Glu side chain of DR3 is two carbons shorter than β9Lys of DR9, this amino acid can be drawn toward pocket 6 when a positively charged residue is anchored within this pocket. Note that in DRB1*0301 the presence of α11Glu, α66Asp, and β11Ser would not attract β9Glu toward this pocket. Yet, the presence of a basic anchor in pocket 6 attracts β9Glu toward it, simultaneously shielding the two negative charges in pocket 6 that would otherwise repel β9Glu. This notion of cooperativity between pockets 6 and 9 of DR0301 is supported not only by our experimental observations but also by the prior observation of multiple submotifs for DR0301 (22), one characterized by basic residues at pocket 6 and a small residue at pocket 9 (e.g., TT 830–843 and Lol pollen 171–190) and a second with a nonbasic residue pocket 6 and accommodating larger residues at pocket 9 (e.g., sperm whale myoglobulin 132–151 and heat shock protein 65 3–13). These different submotifs can now be attributed to the relative positioning of β9Glu, leading to more constrained binding preferences in pockets 6 and 9 when β9Glu is diverted toward basic p6 anchors (as observed for Lol pollen 171–190, compared with sperm whale myoglobulin 132–151 (22)). It is possible that similar effects could be observed in other alleles as well. For example, a previous study of DR0101 noted cooperativity when comparing the binding of modified versions of the HA 306–318 peptide with single, double, or triple substitutions (7). In this study, the single P6-A substitution bound almost to the same extent (RBA = 1.03) as the unmodified sequence. The dual substitutions P6-A/P9-A or P6-A/P9-S bound somewhat worse than the P6-A sequence (RBA = 0.69 and 0.39, respectively) but somewhat better than the P9-A and P9-S single substitutions (RBA = 0.37 and 0.33, respectively). These results suggest that DR0101, a β9Trp allele, better accommodates small residues in pocket 9 when a smaller Ala residue is anchored in pocket 6.
The results of this current study indicate that epitope prediction can be substantially improved by accounting for interactions between pockets 6 and 9, at least for certain alleles. When using a simple, noncooperative model, DR0901 minimal binding motifs could only be predicted for 20 of 31 antigenic peptides identified using tetramers. However, 28 of 31 minimal binding motifs could be predicted using a more complex method, which accounted for cooperative interactions between pockets 6 and 9. Given these observations for DR0901 (particularly the improved prediction of epitopes) and DR0301, it is clear that the peptide binding motif of some HLA class II alleles should not be viewed (or modeled) as a simple combination of individual pocket preferences. Rather their binding motifs could be viewed as a combination of submotifs, in which preferences of certain pockets (most notably pocket 9) can vary due to cooperative effects. These current findings are a specific, clear example of the cooperative effects that have been previously noted in other studies of peptide binding to HLA class II (6, 7).
DR0901 is interesting in other subtle ways. This is only the third human MHC class II allele (besides HLA-DQA1*0501/B1*0201 and HLA-DQA1*0102/B1*0604 (20, 23-25)) that has been shown to bind to Trp, which has the bulkiest and most inflexible amino acid side chain, within pocket 9. In addition, HLA-DRB1*0901 is the second MHC class II allele (besides HLA-DQA1*0102/B1*0604) that has been shown to exclude acidic residues in the absence of β57Asp, albeit only when there is an acidic or basic anchor residue at p6. Both of these characteristics may be attributed to β57Val (this residue may be Ala, Asp, Ser, or Val in other HLA-DR alleles), which apparently does not allow optimal interaction between an acidic anchor residue in pocket 9 and the invariant α76Arg residue. Yet, when a Ser/Thr residue is the p6 anchor of DRB1*0901, acidic residues are allowed in pocket 9. Most probably, the presence of β37Asn (instead of the bulky and inflexible Tyr of DQB1*0604) participates in the interaction of p9Asp/Glu with α76Arg when p6 is occupied by Ser/Thr. In addition, it is notable that DR0901 accepts Pro in several anchor positions. This may explain the association of this allele with gelatin sensitivity and may suggest reactivity to other proline-rich proteins. It is true that proline has not been tested as an anchor residue in most MHC class II alleles, with the exception of HLA-DQA1*0501/B1*0201, which is found in >90% of patients with celiac disease, where the target Ags are proline rich (26). This allele has been shown to accept proline at p1 and p6. In addition, a recent detailed analysis of bound peptides to HLA-DQ2 rarely detected proline at anchor positions p4, p7, and p9 (25). Proline anchoring in these positions would cost at least one hydrogen bond in antigenic peptide-polar side chain interaction (27). Preference for proline at pocket 6 has also recently been observed for HLA-DRB1*1401 (28). It is also interesting that in HLA-DRB1*0901 pocket 6 shows almost as wide an acceptance of amino acids as pocket 7, which usually is the pocket most accommodating of bulky aromatic residues.
Whereas the pocket 6–pocket 9 interaction effects observed in this study for DR0901 and DR0301 can be explained to a large extent based on the positioning of their respective β9 residues, other factors such as the presence of water molecules within pockets 6 and 9 may also play a role in defining the binding preferences of these two pockets. For example, it has been previously shown for I-Ek that a water molecule is interposed among β9Glu, α66Asp, α69Asn, and p6Glu, altering the ionization of these carboxylates and facilitating the anchoring of this acidic residue in the middle of these three negative charges (29). Therefore, water molecules could be expected to play an important role for β9Glu alleles, such as DR0301 (and probably others as well, as in HLA-DQ2 (27)). Unfortunately, it is not currently possible to model water molecules within these binding pockets.
In conclusion, this study demonstrated interactions between peptide binding pockets 6 and 9 based on the peptide binding properties of both DR0901 and DR0301. Peptide binding was measured for singly substituted peptides derived from high-affinity binding sequences and evaluated by molecular modeling studies. In particular for DR0901, minimal binding motifs were difficult to predict for antigenic peptides (identified by tetramer guided epitope mapping) using a simple, noncooperative model but predicted well by a more complex method, which accounted for cooperative interactions between pockets 6 and 9. Although the β9Lys residue is unique to DR9 alleles, similar behavior was exhibited by DR0301, a β9Glu allele. This allele had been reported to have multiple submotifs. These findings support the general idea that an amino acid in one pocket of the MHC class II peptide binding groove can influence binding elsewhere in the groove, providing a foundation for improved epitope prediction.
Supplementary Material
Acknowledgments
We thank Laurel Huston for technical assistance and Gerald Nepom for critical review of this manuscript.
Footnotes
This work was supported in part by National Institutes of Health Contract HHSN266200400028C.
Abbreviations used in this paper: DR0901, DRA1/B1*0901; DR0301, DRA1/B1*0301; NP, influenza A/Puerto Rico/8/34 nucleoprotein; M1, influenza A/Puerto Rico/8/34 matrix protein; Flu B HA, influenza B/Hong Kong/330/2001 hemagglutinin; HA Pan, influenza A/Panama/2007/99 hemagglutinin; HA NC, influenza A/New Caledonia/20/99 hemagglutinin; RBA, relative binding affinity; RBAadj, adjusted RBA; CRBA, corrected RBA; TT, tetanus toxin H chain; TTL, tetanus toxin L chain.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Bondinas GP, Moustakas AK, Papadopoulos GK. The spectrum of HLA-DQ and HLA-DR alleles, 2006: a listing correlating sequence and structure with function. Immunogenetics. 2007;59:539–553. doi: 10.1007/s00251-007-0224-8. [DOI] [PubMed] [Google Scholar]
- 2.Murthy VL, Stern LJ. The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implications for the structural basis of the specificity of peptide binding. Structure. 1997;5:1385–1396. doi: 10.1016/s0969-2126(97)00288-8. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh P, Amaya M, Mellins E, Wiley DC. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature. 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 4.Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, Wiley DC. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 5.Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, Braxenthaler M, Gallazzi F, Protti MP, Sinigaglia F, Hammer J. Generation of tissue-specific and promiscuous HLA ligand database using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17:555–561. doi: 10.1038/9858. [DOI] [PubMed] [Google Scholar]
- 6.Anderson MW, Gorski J. Cooperativity during the formation of peptide/MHC class II complexes. Biochemistry. 2005;44:5617–5624. doi: 10.1021/bi048675s. [DOI] [PubMed] [Google Scholar]
- 7.Ferrante A, Gorski J. Cooperativity of hydrophobic anchor interactions: evidence for epitope selection by MHC class II as a folding process. J Immunol. 2007;178:7181–7189. doi: 10.4049/jimmunol.178.11.7181. [DOI] [PubMed] [Google Scholar]
- 8.Itoh A, Shimada A, Kodama K, Morimoto J, Suzuki R, Oikawa Y, Irie J, Nakagawa Y, Shigihara T, Kanazawa Y, et al. GAD-reactive T cells were mainly detected in autoimmune-related type 1 diabetic patients with HLA DR9. Ann NY Acad Sci. 2004;1037:33–40. doi: 10.1196/annals.1337.006. [DOI] [PubMed] [Google Scholar]
- 9.Imagawa A, Hanafusa T, Uchigata Y, Kanatsuka A, Kawasaki E, Kobayashi T, Shimada A, Shimizu I, Maruyama T, Makino H. Different contribution of class II HLA in fulminant and typical autoimmune type 1 diabetes mellitus. Diabetologia. 2005;48:294–300. doi: 10.1007/s00125-004-1626-x. [DOI] [PubMed] [Google Scholar]
- 10.Murao S, Makino H, Kaino Y, Konoue E, Ohashi J, Kida K, Fujii Y, Shimizu I, Kawasaki E, Fujiyama M, et al. Differences in the contribution of HLA-DR and -DQ haplotypes to susceptibility to adult- and childhood-onset type 1 diabetes in Japanese patients. Diabetes. 2004;53:2684–2690. doi: 10.2337/diabetes.53.10.2684. [DOI] [PubMed] [Google Scholar]
- 11.Park YS, Wang CY, Ko KW, Yang SW, Park M, Yang MC, She JX. Combinations of HLA DR and DQ molecules determine the susceptibility to insulin-dependent diabetes mellitus in Koreans. Hum Immunol. 1998;59:794–801. doi: 10.1016/s0198-8859(98)00076-7. [DOI] [PubMed] [Google Scholar]
- 12.Sugihara S, Sakamaki T, Konda S, Murata A, Wataki K, Kobayashi Y, Minamitani K, Miyamoto S, Sasaki N, Niimi H. Association of HLA-DR, DQ genotype with different β-cell functions at IDDM diagnosis in Japanese children. Diabetes. 1997;46:1893–1897. doi: 10.2337/diab.46.11.1893. [DOI] [PubMed] [Google Scholar]
- 13.Kumagai T, Yamanaka T, Wataya Y, Saito A, Okui T, Yano S, Tsutsumi H, Chiba S, Wakisaka A. A strong association between HLA-DR9 and gelatin allergy in the Japanese population. Vaccine. 2001;19:3273–3276. doi: 10.1016/s0264-410x(00)00555-7. [DOI] [PubMed] [Google Scholar]
- 14.Fujisao S, Matsushita S, Nishi T, Nishimura Y. Identification of HLA-DR9 (DRB1*0901)-binding peptide motifs using a phage fUSE5 random peptide library. Hum Immunol. 1996;45:131–136. doi: 10.1016/0198-8859(95)00169-7. [DOI] [PubMed] [Google Scholar]
- 15.Fujisao S, Nishimura Y, Matsushita S. Evaluation of peptide-HLA binding by an enzyme-linked assay and its application to the detailed peptide motifs for HLA-DR9 (DRB1*0901) J Immunol Methods. 1997;201:157–163. doi: 10.1016/s0022-1759(96)00234-7. [DOI] [PubMed] [Google Scholar]
- 16.Novak EJ, Liu AW, Nepom GT, Kwok WW. MHC class II tetramers identify peptide-specific human CD4+ T cells proliferating in response to influenza A antigen. J Clin Invest. 1999;104:R63–R67. doi: 10.1172/JCI8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novak EJ, Liu AW, Gebe JA, Falk B, Nepom GT, Koelle DM, Kwok WW. Tetramer-guided epitope mapping: rapid identification and characterization of immunodominant CD4+ T cell epitopes from complex antigens. J Immunol. 2001;166:6665–6670. doi: 10.4049/jimmunol.166.11.6665. [DOI] [PubMed] [Google Scholar]
- 18.Ettinger RA, Kwok WW. A peptide binding motif for HLA-DQA1*0102/DQB1*0602, the class II MHC molecule associated with dominant protection in insulin-dependent diabetes mellitus. J Immunol. 1998;160:2365–2373. [PubMed] [Google Scholar]
- 19.Reichstetter S, Papadopoulos GK, Moustakas AK, Swanson E, Liu AW, Beheray S, Ettinger RA, Nepom GT, Kwok WW. Mutational analysis of critical residues determining antigen presentation and activation of HLA-DQ0602-restricted T cell clones. Hum Immunol. 2002;63:185–193. doi: 10.1016/s0198-8859(01)00377-9. [DOI] [PubMed] [Google Scholar]
- 20.Ettinger RA, Papadopoulos GK, Moustakas AK, Nepom GT, Kwok WW. Allelic variation in key peptide-binding pockets discriminates between closely related diabetes-protective and diabetes-susceptible HLA-DQB1*06 alleles. J Immunol. 2006;176:1988–1998. doi: 10.4049/jimmunol.176.3.1988. [DOI] [PubMed] [Google Scholar]
- 21.James EA, Bui J, Berger D, Huston L, Roti M, Kwok WW. Tetramer-guided epitope mapping reveals broad, individualized repertoires of tetanus toxin-specific CD4+ T cells and suggests HLA-based differences in epitope recognition. Int Immunol. 2007;19:1291–1301. doi: 10.1093/intimm/dxm099. [DOI] [PubMed] [Google Scholar]
- 22.Geluk A, van Meijgaarden KE, Southwood S, Oseroff C, Drijfhout JW, de Vries RR, Ottenhoff TH, Sette A. HLA-DR3 molecules can bind peptides carrying two alternative specific submotifs. J Immunol. 1994;152:5742–5748. [PubMed] [Google Scholar]
- 23.Vartdal F, Johansen BH, Friede T, Thorpe CJ, Stevanović S, Eriksen JE, Sletten K, Thorsby E, Rammensee H-G, Sollid LM. The peptide binding motif of the disease associated HLA-DQ (α1*0501/β1*0201) molecule. Eur J Immunol. 1996;26:2764–2772. doi: 10.1002/eji.1830261132. [DOI] [PubMed] [Google Scholar]
- 24.Van de Wal Y, Kooy YM, Drijfhout JW, Amons R, Papadopoulos GK, Koning F. Unique peptide binding characteristics of the disease-associated DQ(α1*0501, β1*0201) vs the non-disease-associated DQ(α1*0201, β1*0202) molecule. Immunogenetics. 1997;46:484–492. doi: 10.1007/s002510050309. [DOI] [PubMed] [Google Scholar]
- 25.Koelle DM, Johnson ML, Ekstrom AN, Byers P, Kwok WW. Preferential presentation of herpes simplex virus T cell antigen by HLA DQA1*0501/DQB1*0201 in comparison to HLA DQA1*0201/DQB1*0201. Hum Immunol. 1997;53:195–205. doi: 10.1016/S0198-8859(97)00034-7. [DOI] [PubMed] [Google Scholar]
- 26.Stepniak D, Wiesner M, de Ru AH, Moustakas AK, Papadopoulos GK, van Veelen PA, Koning F. Large scale characterization of natural ligands explains the unique binding properties of HLA-DQ2. J Immunol. 2008;180:3268–3278. doi: 10.4049/jimmunol.180.5.3268. [DOI] [PubMed] [Google Scholar]
- 27.Kim C-Y, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci USA. 2004;101:4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.James EA, Moustakas AK, Berger D, Huston L, Papadopoulos GK, Kwok WW. Definition of the peptide binding motif within novel DRB1*1401 restricted epitopes by peptide competition and structural modeling. Mol Immunol. 2008;45:2651–2659. doi: 10.1016/j.molimm.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fremont DH, Hendrickson WA, Marrack P, Kappler J. Structures of an MHC class II molecule with covalently bound single peptides. Science. 1996;272:1001–1004. doi: 10.1126/science.272.5264.1001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.