

Abstract
The scintillation proximity assay (SPA) is a powerful technique for measuring radioligand binding to membrane transporters and has become an integral part of high-throughput drug discovery screening efforts. Here we adapt the method for use with purified LeuT, a prokaryotic secondary transporter, reconstituted into phospholipid bilayer nanodiscs. This application surmounts potential challenges with background interference from endogenously-expressed proteins; aggregation and loss of binding activity often accompanying detergent solubilization from native cell membranes; and heterogeneity in size and transporter orientation, where at least some ligand binding sites are inaccessible, associated with reconstitution into lipid vesicles.
Transporters are ubiquitous integral membrane proteins that catalyze the movement of a wide variety of solutes, including neurotransmitters, ions, and amino acids, across the lipid bilayer. Notably, their dysfunction has been implicated in multiple debilitating diseases,1, 2, 3 and they are the target of both therapeutic and deleterious compounds.1, 3 Hence, they are of considerable clinical and pharmacological interest.
Radioligand binding is a fundamental component of evaluating transporter activity. These experiments are usually performed with protein present in intact membranes (cells or vesicles) or in detergent-solubilized form (crude or purified). However, binding studies with unpurified material are often complicated by interference from endogenously-expressed transporters4,5 and/or other cellular components and detergent micelles are frequently inadequate membrane mimics.6, 7
Nanodiscs, on the other hand, provide unique lipid bilayer replicas that enable biochemical and biophysical characterization of membrane proteins in a more physiologically-relevant medium.8 They have been successfully used to study a wide range of membrane proteins such as ion channels,9 G-protein coupled receptors,10,11 chemoreceptors,12 cytochrome oxidases,13 ATP-binding cassette transporters,8, 14–16 and the SecYEG translocase.17
A nanodisc is composed of a nanometer-sized phospholipid bilayer encircled by two α helical, amphipathic membrane scaffold proteins (MSPs).8 These nanoscale models do not suffer from the propensity toward aggregation and geometric distortion typical of micelles6, 7 and some bicelle compositions18 or the heterogeneity in size and transporter orientation,19 where at least some ligand binding sites are inaccessible, often inherent in proteoliposomes. Incorporation of the transmembrane (TM) regions of membrane proteins into the nanometer-sized phospholipid bilayer also makes the protein water-soluble without the need for detergents,8 greatly simplifying downstream applications.
Once the protein-nanodisc complex has been assembled, activity must be assessed, but the traditional radioligand binding studies mentioned above require tedious, time-consuming, and error-prone filtration and washing steps to separate bound from free radioligand.5 Moreover, continuous washing of the sample means that the dissociation constant of low-affinity ligands is extremely difficult to measure20 without the use of centrifugation20 or complex, indirect assays that involve much more planning and development.4 A further complication we have observed with nanodiscs is their tendency to penetrate the relatively large pores present in commonly-used glass fiber and nitrocellulose filters.
An alternative technique is the scintillation proximity assay (SPA). It utilizes fluoromicrospheres or beads filled with scintillant that emit light when excited by a radioligand bound either directly to the bead or to an attached target protein21 Although SPA has been extensively applied to both soluble22, 24 and membrane-bound receptors,23 it has only recently been adapted to transporters.4, 5
Here we describe a method to directly monitor ligand binding to transporters incorporated into nanodiscs by SPA. To our knowledge, this is the first example of such an application despite the many advantages of both nanodiscs and SPA. For this study, we used as an example, LeuT,25 a stable, thoroughly investigated nonpolar amino acid transporter26 and member of the SLC6 (solute carrier 6) family of sodium-coupled symporters.25, 3 Eukaryotic counterparts include the pharmacologically and clinically-significant neurotransmitter transporters for serotonin, dopamine, norepinephrine, γ-aminobutyric acid, and glycine, all of which play crucial roles in terminating synaptic transmission and in shaping the duration and magnitude of synaptic signaling.3 Importantly, their dysfunction has been implicated in multiple neurological and neuropsychiatric diseases and they are the target of a broad array of psychoactive agents such as antidepressants, anticonvulsants, some antipsychotics, amphetamine derivatives, and cocaine.3
Incorporation of purified, his-tagged LeuT (LeuT from now on) into nanodiscs was optimized by varying the molar ratios of the MSP variant MSP1E3D1, LeuT, and lipids. A mixture containing a 3:2 molar ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG), respectively, was employed for the reconstitution. Lipids and LeuT were solubilized in sodium cholate and N-dodecyl-β-D-maltoside (DDM) respectively. A 0.1:1:50:180 molar ratio of LeuT:MSP:lipids:detergent was used to generate the nanodiscs, and assembly was initiated by gradual detergent removal. The resulting nanodiscs were purified via metal affinity chromatography to remove empty nanodiscs followed by size exclusion chromatography (Figure 1A). Purity was confirmed with SDS-PAGE (Figure 1B). Quantitation of LeuT (59.3 kDa) and MSP1E3D1 (62.8 kDa for 2 copies) band intensities yielded a molar ratio of approximately one transporter per nanodisc.
Figure 1.

Characterization of LeuT in nanodiscs. (A) Size-exclusion chromatography (SEC) of purified LeuT (red) and LeuT-containing nanodiscs (dark blue). (B) Coomassie-stained SDS-PAGE confirming LeuT incorporation into nanodiscs. Lane I: purified LeuT-containing nanodiscs; Lane II: purified LeuT alone. Note that LeuT runs faster than predicted on SDS-PAGE. (C) Electron microscopy images of negatively stained LeuT-containing nanodiscs. The stain-filled vestibules/openings appear as dark spots within the nanodiscs. Magnified images of individual nanodiscs with 0, 1, or 2 dark spots are displayed (right).
We further validated our nanodisc preparation via electron microscopy (EM). Images of negatively-stained samples (Figure 1C) revealed nanodiscs with a size of ~13 nm, a diameter predicted from nanodiscs made with MSP1E3D1 and sufficient to contain two LeuT monomers, as estimated from the crystal structure (PDB ID: 2A65).25 EM analysis indicated that the vast majority of nanodiscs have one dark spot, which likely represents the stain-filled vestibule/opening of LeuT. A minor fraction (approximately 5%) has 2 dark spots (presumably two LeuT molecules per disc). However, this proportion was markedly reduced (to <1%) when we simply halved the LeuT-to-MSP molar ratio (0.05:1:50:180 LeuT:MSP:lipids:detergent) (Supplementary Figure S1A,C [top]). To determine if we could completely eliminate the chance of incorporating more than one LeuT molecule per nanodisc, we tried the MSP variant MSP1D1 to generate nanodiscs with a smaller diameter (approximately 9.7 nm) using a molar ratio of 0.05:1:30:180 LeuT:MSP:lipids:detergent (Supplementary Figure S1B,C [bottom]). However, we still noticed a minor fraction (<1%) with two dark spots. We therefore carried out binding experiments employing LeuT-nanodiscs made with MSP1E3D1 at the lower LeuT-MSP molar ratio since this larger nanodisc should theoretically provide a more extensive ring of lipids around LeuT, thereby minimizing any potential spatial constraints.
To examine the binding activity of the LeuT-nanodisc complex as well as appraise the scope of our assay, we analyzed two nonpolar amino acid substrates with vastly different affinities and turnover rates: leucine and alanine. Specifically, Leu binds 10–100-fold more tightly but is transported at least 10-fold more slowly than Ala.26, 27 Both substrates were labeled with tritium, which is ideal for SPA because its low-energy β-particles have short path lengths in aqueous solution; indeed, light is emitted only if the isotope is within 1–8 μm of a bead.21 We first immobilized LeuT-nanodiscs onto the surface of copper chelate Yttrium silicate (Cu2+-Ysi) SPA beads (PerkinElmer) (Figure 2A). In preliminary experiments, the bead-nanodisc suspension was added to binding buffer such that the final concentrations of [3H]Leu, [3H]Ala, and LeuT were 50, 1000, and 20 nM, respectively. To account for [3H]substrate binding to nanodisc phospholipids, we performed a parallel assay with empty nanodiscs, and in this case, immobilization was facilitated with his-tagged MSP1E3D1. As shown in Figure 2B, LeuT nanodiscs bind both [3H]Leu and [3H]Ala, whereas empty nanodiscs do not. Although the counts for [3H]Leu binding to empty nanodiscs are slightly above background, we suspect that this is related to the more hydrophobic nature of Leu compared to Ala and thus perhaps a greater propensity to intercalate into the nanodisc phospholipid array.
Figure 2.
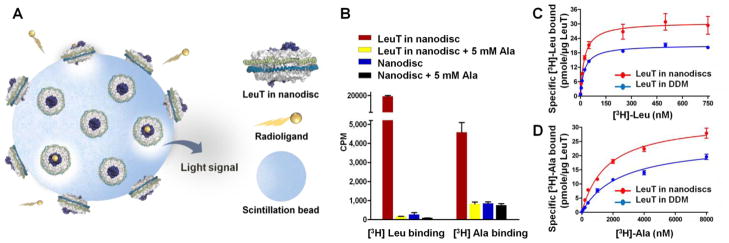
[3H]Leu and [3H]Ala binding to LeuT as measured by SPA. (A) Schematic of the nanodisc-SPA assay. (B) Comparison of [3H]Leu (50 nM) and [3H]Ala (1000 nM) binding to empty and LeuT-containing nanodiscs. Nonspecific binding was assessed in the presence of 5 mM cold Ala. (C) [3H]Leu binding to LeuT in nanodiscs (red) versus DDM (blue). (D) [3H]Ala binding to LeuT in nanodiscs (red) versus DDM (blue). Data represent the mean ± SD from three (B, D) or two (C) independent experiments, each performed in duplicate. [3H]Leu and [3H]Ala specific activities were 21.2 and 12.0 Ci/mmol respectively.
To quantitate binding affinity, we next conducted a series of experiments in which [3H]Leu and [3H]Ala concentrations were varied from 1–750 nM and 10–8000 nM, respectively. The LeuT-nanodisc complex binds to [3H]Leu with an affinity 70 times greater than it does to [3H]Ala (dissociation constant [Kd] of 23.4 versus 1620 nM) but with a similar number of maximum binding sites (Bmaxs of 30.7 versus 32.7 pmol [3H]substrate/μg LeuT) (Figures 2C,D, Supplementary Table S1 [top row]). When compared with DDM-solubilized material, [3H]Leu affinity is similar, but [3H]Ala affinity and both substrate Bmax values are all almost 1.5-fold higher (Figures 2C,D, Supplementary Table S1 [bottom]), indicating that LeuT is more active in the presence of a lipid bilayer, as would be expected for a membrane protein. (Refer to Supplementary Figure S2 for additional graphs and associated discussion).
In conclusion, we have demonstrated the suitability and ease of combining SPA with nanodisc technology to probe radioligand binding of a prokaryotic secondary transporter in a more native-like environment. Importantly, the assay preserves the unique ability of SPA to permit Kd determination for exceedingly weak interactions. Such preliminary scrutiny is an essential prelude to more extensive and intricate downstream experiments such as electron paramagnetic resonance (EPR), single-molecule Förster resonance energy transfer, and hydrogen-deuterium exchange mass spectrometry, techniques designed to interrogate ligand-induced conformational changes critical for assembling a truly mechanistic understanding of membrane transporter function. This is particularly crucial for eukaryotic membrane proteins, many of which are labile when extracted from the lipid bilayer or with prolonged exposure to detergent.28 Furthermore, their activity and conformational dynamics are heavily influenced by lipid composition, especially cholesterol.29, 30, 31 Thus, the ability to conduct a rapid assay to first assess the impact of distinct lipids on activity is indispensable. Although proteoliposomes also offer the ability to control lipid content and provide the vectorial gradient required for substrate transport impossible with nanodiscs, there is precedent for liposomes decreasing access to ligand binding sites and imposing conformational constraints on transporter movement,15 rendering them ill-suited to SPA and even EPR in some cases.15 Nanodiscs, by contrast, suffer from neither of these deficiencies, making such preparations an ideal part of comprehensive biophysical characterization of membrane transporter activity and regulation.
Supplementary Material
Acknowledgments
We thank Kaifeng Zhou for assistance in acquiring the TEM images. We also thank Bruce Davis, Silvia Ravera, Sotiria Tavoulari, Sita Ram Meena and Suraj Adhikary for helpful discussion.
Funding Sources
This work was supported by grants to S.K.S. from the Goodman Gilman Yale Scholar Award, the Alfred P. Sloan Foundation, and the NIH (R00MH083050 and R21MH098180).
Footnotes
The authors declare no competing financial interests.
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rask-Andersen M, Masuram S, Fredriksson R, Schioth HB. Mol Aspects Med. 2013;34:702–710. doi: 10.1016/j.mam.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 2.Gadsby DC, Vergani P, Csanady L. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kristensen AS, Andersen J, Jorgensen TN, Sorensen L, Eriksen J, Loland CJ, Stromgaard K, Gether U. Pharmacol Rev. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- 4.Quick M, Javitch JA. Proc Natl Acad Sci U S A. 2007;104:3603–3608. doi: 10.1073/pnas.0609573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harder D, Fotiadis D. Nat Protoc. 2012;7:1569–1578. doi: 10.1038/nprot.2012.090. [DOI] [PubMed] [Google Scholar]
- 6.Chou JJ, Kaufman JD, Stahl SJ, Wingfield PT, Bax A. J Am Chem Soc. 2002;124:2450–2451. doi: 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- 7.Garavito RM, Ferguson-Miller S. J Biol Chem. 2001;276:32403–32406. doi: 10.1074/jbc.R100031200. [DOI] [PubMed] [Google Scholar]
- 8.Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG. Methods Enzymol. 2009;464:211–231. doi: 10.1016/S0076-6879(09)64011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raschle T, Hiller S, Yu TY, Rice AJ, Walz T, Wagner G. J Am Chem Soc. 2009;131:17777–17779. doi: 10.1021/ja907918r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. Proc Natl Acad Sci U S A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inagaki S, Ghirlando R, White JF, Gvozdenovic-Jeremic J, Northup JK, Grisshammer R. J Mol Biol. 2012;417:95–111. doi: 10.1016/j.jmb.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boldog T, Grimme S, Li M, Sligar SG, Hazelbauer GL. Proc Natl Acad Sci U S A. 2006;103:11509–11514. doi: 10.1073/pnas.0604988103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nasvik Ojemyr L, von Ballmoos C, Gennis RB, Sligar SG, Brzezinski P. FEBS Lett. 2012;586:640–645. doi: 10.1016/j.febslet.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawai T, Caaveiro JM, Abe R, Katagiri T, Tsumoto K. FEBS Lett. 2011;585:3533–3537. doi: 10.1016/j.febslet.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 15.Alvarez FJ, Orelle C, Davidson AL. J Am Chem Soc. 2010;132:9513–9515. doi: 10.1021/ja104047c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou P, McHaourab HS. Biophys J. 2010;98:L18–20. doi: 10.1016/j.bpj.2009.12.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alami M, Dalal K, Lelj-Garolla B, Sligar SG, Duong F. EMBO J. 2007;26:1995–2004. doi: 10.1038/sj.emboj.7601661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Czerski L, Sanders CR. Anal Biochem. 2000;284:327–333. doi: 10.1006/abio.2000.4720. [DOI] [PubMed] [Google Scholar]
- 19.Grote M, Polyhach Y, Jeschke G, Steinhoff HJ, Schneider E, Bordignon E. J Biol Chem. 2009;284:17521–17526. doi: 10.1074/jbc.M109.006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keen M. Meth Mol Biol. 1995;41:1–16. doi: 10.1385/0-89603-298-1:1. [DOI] [PubMed] [Google Scholar]
- 21.Khawaja X, Dunlop J, Kowal D. Expert Opin Drug Discov. 2008;3:1267–1280. doi: 10.1517/17460441.3.11.1267. [DOI] [PubMed] [Google Scholar]
- 22.Sun S, Almaden J, Carlson TJ, Barker J, Gehring MR. Metab Eng. 2005;7:38–44. doi: 10.1016/j.ymben.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr, Civelli O. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- 24.Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ. Proc Natl Acad Sci U S A. 1999;96:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 26.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. Mol Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott DJ, Kummer L, Tremmel D, Pluckthun A. Curr Opin Chem Biol. 2013;17:427–435. doi: 10.1016/j.cbpa.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Divito CB, Amara SG. Mol Interv. 2009;9:252–262. doi: 10.1124/mi.9.5.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong WC, Amara SG. J Biol Chem. 2010;285:32616–32626. doi: 10.1074/jbc.M110.150565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orlowski S, Martin S, Escargueil A. Cell Mol Life Sci. 2006;63:1038–1059. doi: 10.1007/s00018-005-5554-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.