

Abstract
Biochemical and biological properties of glycoconjugates are strongly determined by the specific structure of its glycan parts. Glycosylation, the covalent attachment of sugars to proteins and lipids, is very complex and highly-coordinated process involving > 250 gene products. Deficiency of glycosylation enzymes or transporters results in impaired glycosylation, and consequently pathological modulation of many physiological processes. Inborn defects of glycosylation enzymes, caused by the specific mutations, lead to the development of rare, but severe diseases – congenital disorders of glycosylation (CDGs). Up today, there are more than 45 known CDGs. Their clinical manifestations range from very mild to extremely severe (even lethal) and unfortunately, only three of them can be eff ectively treated nowadays. CDG symptoms highly vary, though some are common for several CDG types but also for other unrelated diseases, especially neurological ones, leaving the possibility that many CDGs cases are under- or mis-diagnosed. Glycan analysis of serum transferrin (by isoelectric focusing or more sophisticated methods, such as HPLC (high-performance liquid chromatography) or MALDI (matrix-assisted laser desorption/ionization)) or serum N-glycans (by MS), enzyme activity assays and DNA sequence analysis are the most frequently used methods for CDG screening and identification, since no specific tests are available yet. In this review we summarize the current knowledge on the clinical, biochemical and genetic characteristic of distinct CDGs, as well as existing diagnostic and therapeutic procedures, aiming to contribute to the awareness on the existence of these rare diseases and encourage the eff orts to elucidate its genetic background, improve diagnostics and develop new strategies for their treatment.
Keywords: congenital disorders of glycosylation, CDG, diagnostics, therapy
Sažetak
Šećerni dijelovi glikokonjugata utječu na mnoga biokemijska i biološka svojstva tih molekula. Glikozilacija, kovalentno pripajanje šećera (glikana) proteinima i lipidima vrlo je složen i visokokoordiniran proces koji uključuje više od 250 različitih genskih produkata. Nedostatak enzima i transportera uključenih u glikozilaciju ima za posljedicu nedostatnu ili nepravilnu glikozilaciju te dovodi do patoloških promjena mnogih fizioloških procesa. Specifične mutacije gena čiji su proteinski produkti uključeni u glikozilaciju uzrokuju nastanak rijetkih, ali ozbiljnih bolesti – urođenih poremećaja glikozilacije (engl. congenital disorders of glycosylation, CDGs). Do danas je poznato 45 tipova CDGs. Kliničke manifestacije bolesti širokog su raspona (od vrlo blagih do smrti), a zasad se samo tri tipa CDGs mogu donekle uspješno liječiti. Simptomi CDGs vrlo su raznoliki, a premda su neki uobičajeni za više tipova CDGs, s obzirom na to da se javljaju i kod drugih, nevezanih bolesti (posebice neuroloških), sumnja se da postoje brojni slučajevi CDG koji još nisu dijagnosticirani ili su pogrešno dijagnosticirani. Premda ne postoje specifični testovi za dijagnosticiranje CDGs, u tu se svrhu najčešće koriste glikanske analize serumskog transferina (izoelektričnim fokusiranjem ili sofisticiranijim metodama kao što su HPLC (engl. high-performance liquid chromatography) i MALDI (engl. matrix-assisted laser desorption/ionization)) ili serumskih N-glikana (masenom spektrometrijom), analize enzimske aktivnosti i analize nukleotidnog slijeda DNA. U ovom preglednom članku predočene su trenutne spoznaje o kliničkim, biokemijskim i genetičkim osobinama pojedinih tipova CDGs, kao i postojeći dijagnostički i terapijski postupci, kako bi se proširila svijest o postojanju ovih rijetkih bolesti i potaknula nastojanja da se rasvijetli genetička osnova njihova nastanka, poboljša dijagnostika i razviju novi terapijski pristupi za njihovo liječenje.
Introduction
Glycans (i.e. carbohydrates or sugars) can be covalently linked to the proteins or lipids in the process called glycosylation, thus forming different types of glycoconjugates (glycoproteins, glycolipids, glycosylphosphatidylinositol (GPI) anchors or proteoglycans). The glycan parts of glycoconjugates contribute to diverse physical, biochemical and biological properties of glycoconjugates, ranging from the resistance to protease degradation and localization in the cell, to the regulation of immune response and metastasis spreading (1–3). Therefore it is not surprising that changes of glycosylation are related to many medical problems. Glycosylation of both proteins and lipids occurs in endoplasmic reticulum (ER) and/or Golgi apparatus (GA), depending on the specific type of glycosylation. Depending on the linkage type, through which glycans are bound to the protein part, there are two kinds of protein glycosylaton: N- and O-glycosylation. The first steps of N-glycosylation process involve assembling of a lipid-linked oligosaccharide (LLO) precursor. That process starts at the cytoplasmic side of the ER and continues on the luminal side of the ER after the flipping of the growing precursor across the membrane bilayer by the mechanism which is not yet fully understood. The transfer of completed LLO precursor onto aminogroup of asparagine residue in Asn-X-Ser/Thr sequon of the nascently translated polypeptide is catalyzed by oligosaccharyltransferase. Additional remodeling of the glycans by assembly and processing reactions catalyzed by numerous glycosyltransferases and glycosidases continues in the lumen of ER and GA (4). O-glycosylation, attachment of glycans to the hydroxyl group of threonine or serine moieties of the proteins, does not comprise processing, only assembly, which mainly occurs in the Golgi apparatus. Diverse and complex glycan structures of glycoconjugates are therefore the outcome of the coordinated reactions of glycosyltransferases, glycosidases, nucleotide-sugar-specific transporters, altogether, more than 250 gene products. Gene mutations can cause defects of these proteins resulting in inborn errors of metabolism, characterized by deficient or reduced glycosylation and known as congenital disorders of glycosylation (CDGs).
Congenital disorders of glycosylation
Since 1980, when the first N-glycosylation defect was described and characterized (5) over 45 types of CDGs have been recognized. Although very rare, CDGs are the most rapidly expanding group among the currently known 3000 monogenetic inherited diseases. All known CDGs have a recessive inheritance except EXT1/EXT2-CDG which is dominantly inherited and MGAT1-CDG which is X-linked (6).
CDGs were first classified as type I (CDG-I) related to the disrupted synthesis of the lipid-linked oligosaccharide precursor and type II (CDG-II) involving malfunctioning processing/assembly of the protein-bound oligosaccharide chain (7). However, since 2009, most of the researchers use a novel nomenclature based on the name of the affected gene (e.g. CDG-Ia = PMM2-CDG, CDG-Ib = MPICDG) (8). According to the novel classification, CDGs are divided into 4 categories as defects of:
protein N-glycosylation (16 types of CDGs),
protein O-glycosylation (8 types of CDGs),
lipid glycosylation and glycosylphosphatidylinositol anchor glycosylation (3 types of CDGs), and
defects in multiple glycosylation pathways and in other pathways (17 types of CDGs) (6).
In addition, several CDGs of so far unknown etiology (CDG-x) have been recognized. CDG symptoms highly vary, but some are common for several CDG types, such as psychomotor retardation, failure to thrive, coagulopathies, dysmorphic features, seizures and stroke-like episodes (9,10). Clinical manifestation of CDGs ranges from very mild to extremely severe (literately, all organs can be affected). The resemblance to some other diseases, especially neurological ones and the physicians’ unawareness of the existence of these diseases, are the main reasons why CDGs still remain under-or misdiagnosed. In addition, the population studies on the frequency of the mutations causing CDGs are still scarce. Based on the determined frequency of heterozygotes, the estimated incidence of homozygotes for certain mutations are as high as 1:20,000, suggesting the existence of much higher number of cases than documented (10). In this review, we will provide an overview of the characteristics of currently known CDGs, classified according to novel nomenclature (8) (with the old one given in italics in brackets), as well as biochemical and genetic diagnostic methods and therapeutic approaches for the treatment of these rare diseases. Due to the space limitation, this review describes only CDGs’ types present in more than few patients or characterized by unique phenotype.
Defects of protein N-glycosylation
Sixteen defects of protein N-glycosylation have been detected so far: 14 assembly defects and 2 processing defects, classified according to the old nomenclature as CDG type I (CDG-I) and CDG type II (CDG-II), respectively.
PMM2-CDG (CDG-Ia)
PMM2 gene (NG-009209.1), mapped on chromosome 16p13, encodes phosphomannomutase (NP-000294.1), an enzyme that transforms mannose-6-phosphate into mannose-1-phosphate (Figure 1) (11). Some of the mutations in PMM2 gene cause PMM2-CDG (CDG-Ia, OMIM≠212065), the most prevalent type of CDGs detected in more than 700 patients worldwide (12). Molecular analysis revealed a large number (> 100) of mutations all over 8 PMM2 exons, but the mutational hot spots were detected in exons 5 and 8 (13,14). The most frequent mutations known by now, p.R141H and p. F119L, both caused by a single base mutation, c.422G>A (rs28936415) and c.357C>A (rs80338701), respectively (15), are located in exon 5. p.R141H has never been observed in the homozygous form, thus being considered lethal (15). Interestingly, PMM2-CDG patients (heterozygotes for p.R141H) have 0–10% of normal PMM2 activity in fibroblasts and leukocytes while their asymptomatic parents, who are also heterozygotes have approximately 50% residual activity of PMM2 enzyme. It seems that compound heterozygosity for p.R141H and some other mutations in PMM2 gene (e.g. p.F119L) is responsible for the decreased PMM2 activity, inadequate for normal glycosylation. Mutation p. F119L itself is very mild, but mentioned compound heterozygosity for p.R141H and p.F119L mutations is the most frequent genotype of CDG-Ia, accounting for 27% of 249 patients from 23 countries (16). Numerous other mutations all over the PMM2 gene also come in different combinations, and the resulting compound heterozygosity leads to more or less severe manifestations of PMM2-CDG.
Figure 1.

N-linked glycan biosynthetic pathway and defects of protein N-glycosylation.
Mannose-6-phosphate (Man-6-P) is generated from the fructose-6-phosphate (Fru-6-P) and converted to mannose-1-phosphate (Man-1-P). Activated mannose (Man) and activated N-acetylglucosamine (GlcNAc) are used to build the lipid-linked oligosaccharide (LLO) precursor, which is assembled on the activated dolichol on the endoplasmic reticulum (ER) membrane and flipped into the ER lumen. Further extension yields oligosaccharide consisting of 2 GlcNAc, 9 Man and 3 glucoses (Glc) which is transferred onto asparagines residues of the polypeptide. Additional trimming and processing of oligosaccharide continues in the Golgi apparatus (GA). CDGs are named according to novel nomenclature, with the abbreviated old names give in brackets. Fru – fructose; Man – mannose; GDP – guanidine diphosphate; UDP – uridine diphosphate; OST – oligosaccharyltransferase.
Clinical presentation and course of PMM2-CDG are highly variable, and range from death in the early childhood due to severe infections, liver insufficiency, or cardiomyopathy (10,17) to mildly affected adults (18). When manifested at birth PMM2-CDG is usually severe multi-organ disease characterized by strong dysfunction of central and peripheral nervous system (19,20). Common features, although not necessarily present in neonates with PMM2-CDG, are inverted nipples, unusual subcutaneous fat, thin upper lip and often muscular hypotonia (21).
MPI-CDG (CDG-Ib)
MPI-CDG (OMIM≠602579) arises due to the deficiency in mannosephosphate isomerase (MPI, NP-002426.1), also known as phosphomannose isomerase (PMI), an enzyme which catalyzes the formation of mannose-6-phosphate from fructose-6-phosphate (Figure 1) (22). It is encoded by MPI gene (NG_008921.1), in which eighteen mutations have been detected by now; 15 point mutations, one splicing defect and two frameshift causing mutations.
Clinical presentation of this disease is unique among CDGs due to the fact that neurological symptoms are usually absent. Gastrointestinal tract and the liver are mainly affected and symptoms usually comprise vomiting, diarrhea, gastrointestinal bleeding, protein-losing enteropathy and hepatomegaly. Additionally, in more severe cases, hypoglycemia, coagulopathy, as well as thrombotic events (protein C and S deficiency, low antithrombin III levels) can be observed. Interestingly, MPI-CDG is one of a few CDGs which can be effectively treated by oral administration of mannose. Since it is potentially lethal, it is of utmost importance to examine patients with inexplicable coagulopathies, intestinal protein loss or hypoglycemia for this disorder.
ALG6-CDG (CDG-Ic)
The second most frequent type of CDGs is ALG6-CDG (OMIM #603147), clinically characterized by psychomotor retardation, dysmorphic features, muscular hypotonia, seizures and epilepsy (23–26). Psychomotor development is less retarded than in PMM2-CDG and ALG6-CDG could be in general considered as a milder type of CDG. It is caused by mutations in ALG6 gene (NG_008925) that encodes dolichyl-P-glucose: mannose9-N-acetyl glucosamine2-pyrophosphate-dolichyl (Dol-P-Glc:Man9GlcNAc2-P-P-Dol) α-1,3 glucosyltransferase (NP_037471.2), an enzyme that participates in the formation of the lipid-linked oligosaccharide precursor of N-linked glycosylation (Figure 1) (27–29). At least 30 patients were identified worldwide (12), which were affected by one of twenty one mutations described so far; sixteen point mutations, one insertion and four deletions (including a large deletion enclosing the complete ALG6 gene). The most common mutation in ALG6 gene causing ALG6-CDG is c.998C>T resulting in p.A333V substitution (23,27). However, recent case of 11-year old Saudi Arabian boy with dilated cardiomyopathy which revealed a novel c.482A>G (p.Y161C) mutation, suggests that CDG should be considered in the differential diagnosis of unexplained cardiomyopathy (30).
Two point mutations c.391T>C in exon 5 and c.911T>C in exon 10 resulting in p.Y131H and p. F304S, respectively, although assumed being single nucleotide polymorphisms (rs35383149 and rs17856039) (31,32) are particularly interesting. It was shown that p.F304S polymorphism may exacerbate the clinical outcome of other CDGs, especially in severely affected patients (33). For the p. Y131H substitution it is still unclear whether the homozygous form c.391C/C is sufficient to cause the disease or some additional genetic alteration has to be present (31). We have recently shown that the frequency of heterozygotes in Croatian population is 3 times higher (6.7%) than in US population (2.1%), the only one screened for p.Y131H until now (31,34). The obtained results suggest the incidence of homozygotes for p.Y131H 1 in 1000 in Croatian population, but up today no patient with ALG6-CDG has been detected in Croatia (34).
ALG3-CDG (CDG-Id)
ALG3-CDG (OMIM#601110) is caused by a defect of mannosyltransferase VI, an enzyme involved in completing of the LLO structure Glc3Man9GlcNAc2-P-P-Dol (NP_001006942.1) (Figure 1). All six patients described so far have displayed a slowly progressive encephalopathy with microcephaly, severe psychomotor retardation and epileptic seizures (35).
ALG12-CDG (CDG-Ig)
ALG12-CDG (OMIM #607143) is caused by a defect of another mannosyltransferase participating in finalizing of LLO - mannosyltransferase VIII (NP_077010.1) (Figure 1) (36). All six patients described so far, exhibited various combination of common CDG symptoms.
ALG8-CDG (CDG-Ih)
ALG8-CDG (OMIM#608104), caused by the deficiency of glucosyltransferase II (NP_001007028.1), participating in one of the last steps of LLO formation (Figure 1), was recognized in five patients. Three of them exhibited severe clinical picture with multiorgan failure, while in two others milder symptoms were observed (37).
ALG1-CDG (CDG-Ik)
ALG1-CDG (OMIM #608540) is caused by the deficiency of mannosyltransferase (NP_061982.3) due to the mutations in ALG1 gene (NG_009202.1). This enzyme catalyses addition of the first mannose during formation of the pyrophosphate-linked seven sugar glycan structure (Man5GlcNAc2-P-P-Dol) (Figure 1). Up today, it was diagnosed in four patients with epilepsy, cardiomyopathy, hypogonadism and severe psychomotor retardation (38).
RFT1-CDG (CDG-In)
RFT1-CDG (OMIM#612015), the first CDG recognized to be associated with sensorineural deafness, was described in six patients so far. RFT1 (NP_443091.1), a flippase encoded by the RFT1 gene (NG_009203.1), is involved in the transfer of Man5GlcNAc2-P-P-Dol from the cytoplasmic to the luminal side of the endoplasmic reticulum (Figure 1) (39).
In addition, there are three other CDGs related to the defects of protein N-glycosylation: TUSC3-CDG, MGAT1-CDG and MGAT2-CDG (CDG-IIa) (Fig. 1). TUSC3-CDG and MGAT1-CDG are related to the defects in the subunits of the oligosaccharyltransferase complex, and both are manifested as nonsyndromic mental retardation. Unlike the other known CDGs, MGAT1-CDG is X-linked disorder. MGAT2-CDG reflects impaired N-acetylglucosaminyltransferase II, an enzyme involved in N-glycan processing.
Defects of protein O-glycosylation
CDGs related to the defects of protein O-glycosylation comprise six disorders: EXT1/EXT2-CDG and B4GALT/-CDG (defects in the synthesis of O-xylosylglycans), GALNT3-CDG (defect in the synthesis of O-N-acetylgalactosaminylglycans), SLC35D1-CDG (defect in the O-xylosyl/N-acetylgalactosaminyl/glycans), POMT1/ POMT2-CDG and POMGNT-CDG (defects in the synthesis of O-mannosylglycan core previously known as Walker-Warburg syndrome and muscle-eye-brain disorder, respectively) and SCDO3-CDG and B3GALTL-CDG (defects in the synthesis of O-fucosylglycans). EXT1/EXT2-CDG will be briefly discussed because of its characteristic features distinguished from other CDG types and its prevalence.
EXT1/EXT2-CDG
EXT1/EXT2-CDG (OMIM#133700/133701), also called hereditary multiple osteochondroma (MO) is the only CDG with autosomal dominant inheritance. It is a monosystemic CDG characterized by formation of benign osteochondromas at the ends of long bones in childhood, which, in some cases, can become malignant lesions resulting in osteosarcomas and chondrosarcomas in adult age. Hereditary multiple osteochondromas are usually caused by various mutations in EXT1 or EXT2 genes or in other homologous EXTL genes, while large genomic deletions in EXT1 or EXT2 are responsible for up to 8% of the cases (40). All members of this multigene family encode gylcosyltransferases involved in the adhesion and/or polymerization of heparin sulfate (HS) chains at HS proteoglycans. EXT1 and EXT2 form heterooligomeric protein complex, which in GA catalyses addition of N-acetylglucosamine and glucuronic acid, thus elon-gating HS-chains. With more than 900 affected patients (41), EXT1/EXT2-CDG represents one of the most frequent CDGs, and according to the novel nomenclature it significantly increased the total number of CDGs previously estimated to be about 1000. In addition, the estimated incidence of 1/50 000 in Western population (42,43), is expected to be an underestimation because of many patients with very mild symptoms that are often not identified.
Defects of glycosphingolipid and GPI-anchor glycosylation
This group of defects comprises tree disorders: PIGM-CDG, PIGMV-CDG (caused by the defects of the first and the second mannosyl-transferases in the GPI-anchor biosynthesis pathway, respectively) and SIA9-CDG (Amish infantile epilepsy) caused by the defect of GM3 synthase, a sialyltransferase, which catalyses the initial step in the biosynthesis of complex gangliosides.
Defects of multiple glycosylation and other pathways
Many of the defects which were previously included in CDG nomenclature are in fact not limited to the N- or O-glycosylation pathways, and their manifestations are due to the defects in other pathways involved in carbohydrate metabolism. CMP-sialic acid transporter (SLC35A1) deficiency of which was related to CDG-IIf (OMIM#603585) and the GDP-fucose transporter (SLC35C1) (previously being related to CDG-IIc, OMIM#266265) are examples of the proteins affecting multiple glycosylation pathways. DK1-CDG (CDG-Im) (OMIM#610768) is also a good example, because dolichol kinase 1 provides a substrate for N- and O-linked glycans, GPI anchors and C- and O-mannosylation and in that way impairs multiple pathways (44). Up today, defects of multiple glycosylation and other pathways comprise 17 defects (8), and some of them are briefly described in the next sessions.
COG-CDG
Conserved oligomeric Golgi (COG) complex is a large, eight-subunits (Cog1-Cog8) complex which plays a key role in protein transport between ER and Golgi and within Golgi (10). Complex is organized into two substructures; lobe A contains Cog 2–4 and lobe B Cog 5–7 (45,46), while Cog1 and Cog8 bridge those lobes. Six COG-CDG types in 10 patients have been identified so far (47,48). In addition, cutis laxa syndrome, a rare skin disorder characterized by wrinkled skin due to defective synthesis of elastic fibers and other proteins of the extracellular matrix, has been described as COG7-CDG (OMIM#608779) (49). Common features of COG-CDGs are dysmorphy, hypotonia, feeding problems, growth retardation and cerebral atrophy. COG complex is probably not only involved in glycosylation but also in other cellular functions, so it was proposed the COG defects to be called the “CDG-plus” (12).
ATP6V0A2-CDG (OMIM#611716)
This disorder could be considered as another “CDG-plus” defect. Although laboratory findings of type 2 pattern on transferrin isoelectrofocusing (Tf-IEF), abnormal isoelectrofocusing of apolipoprotein C-III (apoC-III), and abnormal mass spectrometry of glycans of total serum proteins could be ascribed to the classical CDG type II caused by defects of enzymes involved in glycan processing (50,51), the cause of glycosylation disturbance lies in defects of α-subunit of the vesicular ATPase H+-pump. It seems that disturbed H+ concentration in vesicules affects proper processing of glycans. All patients have generalized cutis laxa at birth, but ophtalmological abnormalities and delayed motor development that improves with age were also described (49).
SLC35C1-CDG (CDG-IIc)
This disease (OMIM#266265), also known as Leukocyte Adhesion Deficiency type II (LAD II) syndrome (CDG-IIc), is caused by defective GDP-fucose transporter (SLC35C1; NG_009875.1; NP_060859.4), which leads to the lack of fucosylated selectin ligands and, consequently, to the developmental defects and immunodeficiency (52). Patients with SLC35C1-CDG also exhibit strong mental and growth retardation.
B4GALT1-CDG (CDG-IId)
B4GALT1-CDG (OMIM 607091) is delineated to a deficiency of β1,4-galactosyltransferase (E.C.2.4.1.38) (53). Typical clinical presentations are Dandy-Walker malformation and myopathy. MALDI analysis of transferrin N-linked glycans revealed the presence of species lacking both the terminal sialic acids and galactose residues (54).
Laboratory diagnostics
Because of high variety of CDG symptoms and the resemblance to other diseases, diagnosis of CDG is very demanding. Thus, a suspicion on CDG should arise in a case of any unexplained neurological syndrome, particularly when associated with other organ disease, as well as in any unexplained syndrome without obvious neurological symptoms. Therefore, clinical features such as syndromic fat pads and/or inverted nipples, chronic diarrhea, liver fibrosis, ichtiosis syndrome, different neurosyndomatic conditions (e.g. cataract, sensorineural deafness, radio-ulnar synostosis, recurrent infections), syndromic cardiomyopathy are for certain the conditions in which suspicion on CDG should arise (6). In that sense, laboratory analysis could be very helpful, although no specific tests are available yet. In general, four approaches can be applied: glycans analyses, measurements of enzyme activities and metabolites, classical biochemical analyses, and molecular diagnostics. At the moment, there is no algorithm recommended by any professional organization, and majority of the reported studies and case reports are outcomes of personal and scientific approach of physicians, usually performed in collaboration with the scientific groups focused on CDGs. However, according to Jaeken (12), some general guidelines can be proposed (Figure 2). The first step in laboratory diagnostics should unquestionably be isoelectric focusing (IEF) of serum transferrin (Tf) which provides valuable data for diagnostic of CDGs caused by impaired N-glycosylation. If the obtained pattern is abnormal, possible other causes e.g. transferrin mutants, galactosemia or hereditary fructose intolerance should be excluded. Depending on the obtained pattern (type 1 – CDG-Ix or type 2 – CDG-IIx) further analyses should be performed. In the case of Tf IEF type 1 pattern, enzymatic activity of PMM2 and MIP in fibroblasts or leukocytes should be determined. If their activities are normal, the analysis of dolichol-linked oligosaccharide (LLO) should be the next step. Higher quantity of glycan intermediate(s) implies measurement of particular enzyme activity or mutation analyses. If Tf IEF has showed type 2 pattern, direct analysis of transferrin glycans and IEF of apolipoprotein C-III should be performed. The finding of specific transferrin glycans profiles could give a clue on impaired enzyme, but it usually results reveal just a reduced sialylation or galactosylation. In these cases, further steps in laboratory diagnostics should be chosen in accordance with specific clinical manifestations. However, in some CDGs in which Tf IEF pattern is normal, as well as Apo C-III IEF pattern, mutation analyses remain the only choice.
Figure 2.

Schematic illustration of the strategy for CDG laboratory diagnostics (for N-glycosylation, core 1 mucin-type O-glycosylation and combined N- and O-glycosylation disorders). IEF – isolectric focusing; Tf – transferrin, ApoC-III apolipoprotein C-III).
Glycan analysis
All CDGs are associated with at least in some extent by changes of glycosylation. This is why the analyses of glycans emerge as the most promising screening approach. Yet, lack of a reliable, user friendly screening method, which could detect specific glycosylation changes of particular CDG, implies the use of some less specific, but still useful approaches.
The simplest and still wildly used methods for CDG screening are isoelectric focusing (IEF) of transferrin (Tf) and apolipoprotein C-III (ApoC-III) for N-and O-glycan synthesis defects, respectively. Plasma transferrin usually carries 2 complex-type biantennary N-glycans with terminal sialic acids (usually 4) (Figure 3). Since terminal residues and the branching of N-glycans are variable, several isoforms of transferrin can be distinguished in healthy individuals: tetrasialo-transferrin as a dominant fraction and small amounts of mono-, di-, tri-, penta- and occasionally hexasialotransferrins. ApoC-III is a plasma protein containing one core 1 mucin-type O-glycan. Three isoforms of ApoC-III can be distinguished, ApoC-III0, ApoC-III1, and ApoC-III2, for which the isoform number, as in transferrin, is the number of sialic acid residues attached to the core 1 mucin-type O-glycan. Because sialic acid has a negative charge, the IEF patterns of hyposialylated transferrin and ApoC-III show a cathodal shift that can be caused either by structural glycan changes or by a different number of glycans. Thus, CDG type I is associated with an increase of asialoand disialotransferrin bands, while CDG type II shows an increase of mono and/or trisialotransferrin. However, some other conditions, such as uncontrolled galactosaemia, fructose intolerance and alcoholism, can also be associated with altered transferrin glycosylation. Additionally, several transferrin gene polymorphisms result in a shifted IEF pattern, caused by the differences in the transferrin aminoacid composition (55,56). Furthermore, some CDG types cannot be identified by transferrin IEF analysis because in some of them transferrin sialylation is not altered e.g. deficiency of GLS1 (CDG-IIb), SLC35C1 (CDG-IIc), one of the two Golgi GDP-fucose transporters, and TUSC3-CDG. Finally, IEF is laborious, time-consuming technique, and not suitable for automation or accurate quantification, so during last two decades high-performance liquid chromatography (HPLC) (57) and capillary zone electrophoresis (CZE) (58) have been applied for diagnostics of CDGs. They enabled detection of altered and even abnormal patterns, which might be missed by transferrin IEF (59), though some disadvantages such as column cost and frequent and complex column regeneration are still present. Additional improvement of CDG diagnostics was achieved by the employment of mass spectrometric (MS) analyses (54,60,61). Because of high specificity and sensitivity of MS, and possibility to be fully automated, different kinds of MS found the application in CDG diagnostics, e.g. ESIMS (electrospray ionization MS) of transferrin (62), MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight MS) of transferrin and α-1-antitrypsin (63) as well as isolated serum N-linked (64) and O-linked glycans (65) and its variation -SELDI-TOF-MS (surface-enhanced laser desorption/ionization TOF MS) (66). Next generation of glycan analysis techniques will provide new opportunities for identification of novel glycosylation defects, especially those which could not be identified by classical methods.
Figure 3.

Schematic illustration of transferrin isoforms. Oligosacharrides of normal transferrin (Tf) have most usually four sialic acids (tetrasialotransferrin) (Ctrl - control, line 1), but in CDG patients the IEF of serum transferrin reveals additional bands as results of different structural alterations. In CDG type I patients shows abnormal pattern of disialo- and asialotransferrin (CDG-I, line 2). In CDG-II patients there are combinations of elevated asialo-, disialo, trisialo and monosialo-Tf fractions (CDG-type II, line 3). Structural illustrations of the major typical isoforms are given on the right side of the picture.
Measurements of enzyme activities and metabolites
Impaired glycosylation is a result of deficient or absent enzyme or transporter activity, thus direct measurement of these parameters represents the most accurate parameters for CDGs diagnostics. Unfortunately, availability of these methods is limited, and the analyses can be performed in only few laboratories in the world. Yet, measurement of phosphomannomutase 2 (PMM2) activity in fibroblasts or leukocytes should be the first choice when CDG is suspected, since PMM2-CDG is the most frequent CDG. In the case of solely hepato-intestinal clinical symptoms, mannosephosphate isomerase (MPI) measurements should be performed. Normal activities of the mentioned enzymes indicate further analysis of the lipid-linked oligosaccharides (LLO) in fibroblasts or other assays to identify known or unknown CDG defect. Many CDGs are characterized by accumulation of the specific intermediate structures, mainly substrates of the deficient enzyme in fibroblasts (e.g. Glc1Man9GlcNAc2-P-P-Dol) in ALG8-CDG (37), Man5GlcNAc2-P-P-Dol in RFT1-CDG (39), Glc1Man9−GlcNAc2-P-P-Dol in ALG1-CDG (67), or serum (e.g. lactosylceramide in SIAT9-CDG (68).
Biochemical analysis
There are also some standard biochemical tests which can contribute to the diagnostics of particular CDGs. Thus, low plasma cholesterol concentration and cholinesterase activity with proteinuria are common characteristics of PMM2-CDG. MPICDG is usually associated with high aminotransferases activities (24), while CDGs previously classified as type II are characterized by increased aspartate aminotransferase (AST) and normal alanine aminotransferase (ALT) activity but CDGs type-I are not (69). PIGV is characterized by hyper-posphatasia (70), while high plasma transferrin concentration and thrombocytopenia have been also seen in various cases (20). All these finding are not surprising since many proteins, especially membrane and secreted ones are glycosylated and glycans highly affect their structure and consequently function, as well as localization and secretion.
Molecular diagnostics
Molecular basis of most of all known CDGs has been elucidated. In many cases, not only one, two or several, but over 100 mutations are known to be related to particular CDG (e.g. in PMM2 gene). Mutations can be identified by simple single-strand conformational polymorphism analysis (SSCP) (71,72), restriction fragment length polymorphism (RFLP) (32), real time-polymerase chain reaction (PCR) (34), and DNA sequencing (73,74). Compound heterozygous or homozygous mutations in the coding sequence, but also in promoters, introns and splice junctions, as well as deletions of a whole region, can be confirmed at the genomic level. In addition, in some cases sequencing of the entire gene including all exons, introns and promoters is required. Thus, it is obvious that molecular diagnostic is valuable approach for a confirmation of the presence of the mutation(s) in the particular gene(s), but primarily when suspicion on CDG is established based on clinical symptoms and other laboratory analyses. Even then, several suspected genes have to be analyzed to identify and characterize mutation(s) which cause the impaired protein/enzyme functionality and consequently the disease. Genetic testing approach can be also applied for screening for the mutation carriers in the population studies. This is why the development of novel, cheap, fast and user friendly screening methods, which can be applied on small quantity of sample, is of utmost importance (75,76).
Prenatal diagnosis
Prenatal diagnostics of CDGs is also available and it can be performed in few genetic centers in Europe and USA, especially for the individuals coming from the families with higher risk for CDGs. However, until today, prenatal diagnostics has principally been applied in the cases of the suspicion on PPM2-CDG, although it can be performed for some other CDGs on the request, but only in the research laboratories.
If both parents have already been identified as carriers of a recessive gene involved in CDG pathway, chorionic villous sampling or amniocentesis should be offered. Prenatal diagnostics can be performed by analyzing the activity of the particular enzyme in fibroblasts, leukocytes or lymphocytes of the proband or in the leukocytes of the parents, or screening for mutations in the specific gene (77). It is worth to point out that prenatal diagnoses are also made according to CDG subtype. PMM2-CDG, deficiency of phosphomannomutase 2, can also be detected in cultured trophoblasts and amniocytes from the affected fetus. However, prenatal diagnosis by enzyme assay can give false positive results because of the low values of the enzyme in poorly growing amniocytes when the fetus is not affected. Therefore, prenatal testing should only be offered to the families where mutations in the PMM2 gene are known, since DNA analysis is the most reliable method. It is important to know that prenatal diagnostics for PMM2-CDG cannot be performed by Tf IEF or any other assay that would determine the glycosylation of proteins in the fetus (78).
Due to the severity of the disease, prenatal diagnostic services based on combined mutation and linkage analysis with polymorphic markers are often required (79,80).
Therapy
The therapy for only three (MPI-CDG (CDG-Ib), SLC35C1-CDG (CDG-IIc) and PIGM-CDG) of almost 50 known CDG defects is available so far. Yet, lot of efforts is putting in mouse models which were shown to be very useful not only in the studies of molecular basis of these diseases (21,81), but also in the therapeutic studies (82).
MPI-CDG (CDG-Ib)
Defect of mannosephosphate isomerase which blocks the endogenous mannose formation from glucose (Figure 2), can be overcome by oral administration of mannose (83). Oral mannose supply bypasses the enzymatic block using alternative way catalysed by hexokinase (Figure 4) and leads to the significant metabolic normalization and disappearance of symptoms. With such therapy patients usually can live normally. If oral intake of mannose is not possible, mannose should be applied intravenously, yet life-threatening central nervous system disturbances can occur, probably because of intracellular energy failure. Clinical symptoms of such energy deficiency should be treated early and aggressively (84).
Figure 4.
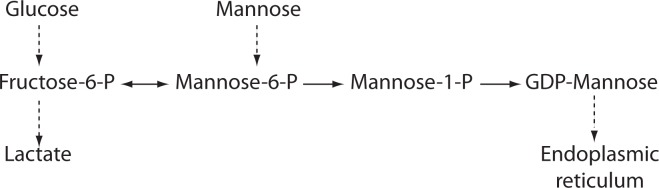
Mannose metabolic pathway. Mannose-6-phosphate (Man-6-P) is mainly generated from fructose-6-phosphate (Fru-6-P) by the action of mannosephosphate isomerase (MPI), or alternatively from mannose by the action of hexokinase (HK). Thus, decreased Man-6-P production due to the MPI deficiency can be restored by the mannose supplementation therapy. Phosphomannomutase (PMM2) converts Man-6-P to Man-1 -P, but since MPI can also catabolizes Man-6-P, supplementary mannose treatment does not improve PMM2-CDG symptoms. Inhibition of MPI activity might provide more Man-6-P and thus overcome low residual activity of PMM2.
SLC35C1-CDG (CDG-IIc)
Clinical symptoms of SLC35C1-CDG are the consequences of the defect of GDP-fucose transporter and include immunodeficiency caused by the absence of carbohydrate-based selectin ligands on the surface of neutrophils resulting in recurrent infections with hyperleukocytosis as well as severe psychomotor and mental retardation. In some patients, increased level of fucose achieved by oral supplementation might overcome low affinity of the fucose transporters and in that way result in clinical improvements (85). However, the observations that fucose treatment did not have the effect in some other cases, suggest that the effectiveness of fucose therapy depends on the nature of the mutation (12).
PIGM-CDG
Constitutional mutation in the promoter of a housekeeping gene PIGM causes histone hypoacetylation and disruption of binding of SP1 transcription factor, resulting in deficiency of the first mannosyltransferase in the GPI-anchor biosynthesis pathway and consequently low glycosylphosphatidylinositol content (86). PIGM-CDG is characterized by splanchnic vein thrombosis and epilepsy. Treatment with a histone deacetylase inhibitor, butyrate, was proposed as an effective therapy for PIGM-CDG in vitro as well as in vivo, since it was shown to increase PIGM transcription and GPI expression, and is able to cause complete cessation of intractable seizures on one PIGM-CDG patient (87). It may be an effective therapeutic option for other diseases caused by Sp1-dependent hypoacetylation, so further investigations are needed.
Unfortunately, the successful therapy for PMM2-CDG, the most prevalent CDG is not yet available, although many attempts to design the effective therapeutic approach have been undertaken. One of the examples is the application of cell permeable mannose-1-phosphate derivatives that succeeded to restore glycosylation to normal levels, but the half-life of these derivatives was too short (88). In addition, zaragozic acid A, a squalene synthase inhibitor, was shown to be able to improve protein N-glycosylation, by redirecting the flow of the polyisoprene pathway toward dolichol by lowering cholesterol biosynthesis (89). As mentioned before, deficiency of phosphomannomutase 2 (PMM2) and mannosephospho isomerase (MPICDG) reduces the metabolic flux of mannose-6-phosphate (Man-6-P) (Figure 2), which results in impaired N-glycosylation. Both enzymes compete for the same substrate, Man-6-P. Mannose supplementation reverses most of the symptoms of MPICDG patients, but has no effect on PMM2-CDG patients because Man-6-P is catabolized by MPI. It was recently proposed that inhibition of MPI activity might provide more Man-6-P for glycosylation and possibly help PMM2-CDG patients with residual PMM2 activity (90). Application of a potent MPI inhibitor from the benzoisothiazolone series successfully diverted Man-6-P towards glycosylation in various cell lines including fibroblasts from PMM2-CDG patients and improved N-glycosylation. Hopefully, this novel therapeutic approach will be also effective in clinical trials and beneficial for at least a subset of PMM2-CDG patients.
Conclusions and remarks
Glycobiology is an emerging and one of the most promising fields of biomedicine. Many glycosylation pathways are not yet elucidated and numerous glycan roles wait to be identified. 250 gene products are recognized to be involved in glycosylation pathways, thus it is reasonable to believe that in addition to 45 recognized CDGs many others will be discovered. The biggest breakthrough is expected to be achieved in recognition of defects of lipid glycosylation, dolichol synthesis, proteoglycans, and organ-specific glycosylation pathways, but also in the field of complex genetic disorders where glycosylation is not the main focus of interest yet. Hopefully, enormous efforts put in mouse models of CDGs will also provide new insights and knowledge in the field. Although belonging to the orphan (i.e. rare) diseases group, CDGs also represents a huge challenge for both pharmaceutical industry and scientists who are trying to understand complex synthetic pathways of glycosylation, the most complex modifications of proteins and lipids. In addition, the estimation of thousands undiscovered or misdiagnosed CDG patients alerts on the necessity of the improvement of clinical awareness of CDGs and development of better biochemical and molecular diagnostics.
Footnotes
Potential conflict of interest
None declared.
References
- 1.Roth J, Zuber C, Park S, Jang I, Lee Y, Kysela KG, et al. Protein N-glycosylation, protein folding, and protein quality control. Mol Cells. 2010;30:497–506. doi: 10.1007/s10059-010-0159-z. [DOI] [PubMed] [Google Scholar]
- 2.Moran AP, Gupta A, Joshi L. Sweet-talk: role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut. 2011;60:1412–25. doi: 10.1136/gut.2010.212704. [DOI] [PubMed] [Google Scholar]
- 3.Larkin A, Imperiali B. The expanding horizons of asparagine-linked glycosylation. Biochemistry. 2011;50:4411–26. doi: 10.1021/bi200346n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol. 2011;21:576–82. doi: 10.1016/j.sbi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Jaeken J, Vanderschueren-Lodewyckx M, Snoeck L, Corbeel L, Wggermont E, Eeckels R. Familiar psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG deficiency, increased serum arylsulphatase A and increased CSF protein: a new syndrome? Pediar Res. 1980;14:179. [Google Scholar]
- 6.Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J Inherit Metab Dis. 2011;34:853–8. doi: 10.1007/s10545-011-9299-3. [DOI] [PubMed] [Google Scholar]
- 7.Aebi M, Helenius A, Schenk B, Barone R, Fiumara A, Berger EG, et al. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG. First International Workshop on CDGS. Glycoconj J. 1999;16:669–71. doi: 10.1023/a:1017249723165. [DOI] [PubMed] [Google Scholar]
- 8.Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: time for a change! Biochim Biophys Acta. 2009;1792:825–6. doi: 10.1016/j.bbadis.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eklund EA, Freeze HH. The congenital disorders of glycosylation: a multifaceted group of syndromes. NeuroRx. 2006;3:254–63. doi: 10.1016/j.nurx.2006.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006;7:537–51. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 11.Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ, et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome) Nat Genet. 1997;16:88–92. doi: 10.1038/ng0597-88. [DOI] [PubMed] [Google Scholar]
- 12.Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–8. doi: 10.1111/j.1749-6632.2010.05840.x. [DOI] [PubMed] [Google Scholar]
- 13.Kjaergaard S. Congenital disorders of glycosylation type Ia and Ib. Genetic, biochemical and clinical studies. Dan Med Bull. 2004;51:350–63. [PubMed] [Google Scholar]
- 14.Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30:1628–41. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- 15.Matthijs G, Schollen E, Van Schaftingen E, Cassiman JJ, Jaeken J. Lack of homozygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type 1A. Am J Hum Genet. 1998;62:542–50. doi: 10.1086/301763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matthijs G, Schollen E, Bjursell C, Erlandson A, Freeze H, Imtiaz F, et al. Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia) Hum Mutat. 2000;16:386–94. doi: 10.1002/1098-1004(200011)16:5<386::AID-HUMU2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 17.Kjaergaard S. Congenital disorders of glycosylation type Ia and Ib. Dan Med Bull. 2004;51:350–63. [PubMed] [Google Scholar]
- 18.Coman D, McGill J, MacDonald R, Morris D, Klingberg S, Jaeken J, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci. 2007;14:668–72. doi: 10.1016/j.jocn.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Jaeken J, Stibler H, Hagberg B. The carbohydrate-deficient glycoprotein syndrome. A new inherited multisystemic disease with severe nervous system involvement. Acta Paediatr Scand Suppl. 1991;375:1–71. [PubMed] [Google Scholar]
- 20.Jaeken J. Komrower Lecture. Congenital disorders of glycosylation (CDG): it’s all in it! J Inherit Metab Dis. 2003;26:99–118. doi: 10.1023/a:1024431131208. [DOI] [PubMed] [Google Scholar]
- 21.Grunewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia) Biochim Biophys Acta. 2009;1792:827–34. doi: 10.1016/j.bbadis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Niehues R, Hasilik M, Alton G, Korner C, Schiebe-Sukumar M, Koch HG, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998;101:1414–20. doi: 10.1172/JCI2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grunewald S, Imbach T, Huijben K, Rubio-Gozalbo ME, Verrips A, de Klerk JB, et al. Clinical and biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N-glycan synthesis. Ann Neurol. 2000;47:776–81. [PubMed] [Google Scholar]
- 24.Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr. 2003;162:359–79. doi: 10.1007/s00431-002-1136-0. [DOI] [PubMed] [Google Scholar]
- 25.Damen G, de Klerk H, Huijmans J, den Hollander J, Sinaasappel M. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J Pediatr Gastroenterol Nutr. 2004;38:282–7. doi: 10.1097/00005176-200403000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Sun L, Eklund EA, Van Hove JL, Freeze HH, Thomas JA. Clinical and molecular characterization of the first adult congenital disorder of glycosylation (CDG) type Ic patient. Am J Med Genet A. 2005;137:22–6. doi: 10.1002/ajmg.a.30831. [DOI] [PubMed] [Google Scholar]
- 27.Newell JW, Seo NS, Enns GM, McCraken M, Mantovani JF, Freeze HH. Congenital disorder of glycosylation Ic in patients of Indian origin. Mol Genet Metab. 2003;79:221–8. doi: 10.1016/s1096-7192(03)00089-1. [DOI] [PubMed] [Google Scholar]
- 28.Burda P, Borsig L, de Rijk-van Andel J, Wevers R, Jaeken J, Carchon H, et al. A novel carbohydrate-deficient glycoprotein syndrome characterized by a deficiency in glucosylation of the dolichol-linked oligosaccharide. J Clin Invest. 1998;102:647–52. doi: 10.1172/JCI2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imbach T, Burda P, Kuhnert P, Wevers RA, Aebi M, Berger EG, et al. A mutation in the human ortholog of the Saccharomyces cerevisiae ALG6 gene causes carbohydrate-deficient glycoprotein syndrome type-Ic. Proc Natl Acad Sci U S A. 1999;96:6982–7. doi: 10.1073/pnas.96.12.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Owain M, Mohamed S, Kaya N, Zagal A, Matthijs G, Jaeken J. A novel mutation and first report of dilated cardiomyopathy in ALG6-CDG (CDG-Ic): a case report. Orphanet J Rare Dis. 2010;5:7. doi: 10.1186/1750-1172-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westphal V, Xiao M, Kwok PY, Freeze HH. Identification of a frequent variant in ALG6, the cause of Congenital Disorder of Glycosylation-Ic. Hum Mutat. 2003;22:420–1. doi: 10.1002/humu.9195. [DOI] [PubMed] [Google Scholar]
- 32.Vuillaumier-Barrot S, Le Bizec C, Durand G, Seta N. The T911C (F304S) substitution in the human ALG6 gene is a common polymorphism and not a causal mutation of CDGIc. J Hum Genet. 2001;46:547–8. doi: 10.1007/s100380170038. [DOI] [PubMed] [Google Scholar]
- 33.Westphal V, Kjaergaard S, Schollen E, Martens K, Grunewald S, Schwartz M, et al. A frequent mild mutation in ALG6 may exacerbate the clinical severity of patients with congenital disorder of glycosylation Ia (CDG-Ia) caused by phosphomannomutase deficiency. Hum Mol Genet. 2002;11:599–604. doi: 10.1093/hmg/11.5.599. [DOI] [PubMed] [Google Scholar]
- 34.Goreta SS, Dabelic S, Pavlinic D, Lauc G, Dumic J. Frequency Determination of alpha-1,3 Glucosyltransferase p.Y131H and p.F304S Polymorphisms in the Croatian Population Revealed Five Novel Single Nucleotide Polymorphisms in the hALG6 Gene. Genet Test Mol Biomarkers. 2012;16:50–3. doi: 10.1089/gtmb.2011.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rimella-Le-Huu A, Henry H, Kern I, Hanquinet S, Roulet-Perez E, Newman CJ, et al. Congenital disorder of glycosylation type Id (CDG Id): phenotypic, biochemical and molecular characterization of a new patient. J Inherit Metab Dis. 2008;31(Suppl2):S381–6. doi: 10.1007/s10545-008-0959-x. [DOI] [PubMed] [Google Scholar]
- 36.Kranz C, Basinger AA, Gucsavas-Calikoglu M, Sun L, Powell CM, Henderson FW, et al. Expanding spectrum of congenital disorder of glycosylation Ig (CDG-Ig): sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am J Med Genet A. 2007;143A:1371–8. doi: 10.1002/ajmg.a.31791. [DOI] [PubMed] [Google Scholar]
- 37.Stolting T, Omran H, Erlekotte A, Denecke J, Reunert J, Marquardt T. Novel ALG8 mutations expand the clinical spectrum of congenital disorder of glycosylation type Ih. Mol Genet Metab. 2009;98:305–9. doi: 10.1016/j.ymgme.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 38.Kranz C, Denecke J, Lehle L, Sohlbach K, Jeske S, Meinhardt F, et al. Congenital disorder of glycosylation type Ik (CDGIk): a defect of mannosyltransferase I. Am J Hum Genet. 2004;74:545–51. doi: 10.1086/382493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaeken J, Vleugels W, Regal L, Corchia C, Goemans N, Haeuptle MA, et al. RFT1-CDG: Deafness as a novel feature of congenital disorders of glycosylation. J Inherit Metab Dis. 2009 doi: 10.1007/s10545-009-1297-3. [DOI] [PubMed] [Google Scholar]
- 40.Jennes I, de Jong D, Mees K, Hogendoorn PC, Szuhai K, Wuyts W. Breakpoint characterization of large deletions in EXT1 or EXT2 in 10 multiple osteochondromas families. BMC Med Genet. 2011;12:85. doi: 10.1186/1471-2350-12-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jennes I, Pedrini E, Zuntini M, Mordenti M, Balkassmi S, Asteggiano CG, et al. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb) Hum Mutat. 2009;30:1620–7. doi: 10.1002/humu.21123. [DOI] [PubMed] [Google Scholar]
- 42.Schmale GA, Conrad EU, 3rd, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg Am. 1994;76:986–92. doi: 10.2106/00004623-199407000-00005. [DOI] [PubMed] [Google Scholar]
- 43.Stancheva-Ivanova MK, Wuyts W, van Hul E, Radeva BI, Vazharova RV, Sokolov TP, et al. Clinical and molecular studies of EXT1/EXT2 in Bulgaria. J Inherit Metab Dis. 2011;34:917–21. doi: 10.1007/s10545-011-9314-8. [DOI] [PubMed] [Google Scholar]
- 44.Kranz C, Jungeblut C, Denecke J, Erlekotte A, Sohlbach C, Debus V, et al. A defect in dolichol phosphate biosynthesis causes a new inherited disorder with death in early infancy. Am J Hum Genet. 2007;80:433–40. doi: 10.1086/512130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ungar D, Oka T, Vasile E, Krieger M, Hughson FM. Subunit architecture of the conserved oligomeric Golgi complex. J Biol Chem. 2005;280:32729–35. doi: 10.1074/jbc.M504590200. [DOI] [PubMed] [Google Scholar]
- 46.Ungar D, Oka T, Brittle EE, Vasile E, Lupashin VV, Chatterton JE, et al. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J Cell Biol. 2002;157:405–15. doi: 10.1083/jcb.200202016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foulquier F. COG defects, birth and rise! Biochim Biophys Acta. 2009;1792:896–902. doi: 10.1016/j.bbadis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 48.Ng BG, Sharma V, Sun L, Loh E, Hong W, Tay SK, et al. Identification of the first COG-CDG patient of Indian origin. Mol Genet Metab. 2011;102:364–7. doi: 10.1016/j.ymgme.2010.11.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohamed M, Kouwenberg D, Gardeitchik T, Kornak U, Wevers RA, Morava E. Metabolic cutis laxa syndromes. J Inherit Metab Dis. 2011;34:907–16. doi: 10.1007/s10545-011-9305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morava E, Lefeber DJ, Urban Z, de Meirleir L, Meinecke P, Gillessen Kaesbach G, et al. Defining the phenotype in an autosomal recessive cutis laxa syndrome with a combined congenital defect of glycosylation. Eur J Hum Genet. 2008;16:28–35. doi: 10.1038/sj.ejhg.5201947. [DOI] [PubMed] [Google Scholar]
- 51.Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32–4. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- 52.Wild MK, Luhn K, Marquardt T, Vestweber D. Leukocyte adhesion deficiency II: therapy and genetic defect. Cells Tissues Organs. 2002;172:161–73. doi: 10.1159/000066968. [DOI] [PubMed] [Google Scholar]
- 53.Peters V, Penzien JM, Reiter G, Korner C, Hackler R, Assmann B, et al. Congenital disorder of glycosylation IId (CDGIId) - a new entity: clinical presentation with Dandy-Walker malformation and myopathy. Neuropediatrics. 2002;33:27–32. doi: 10.1055/s-2002-23597. [DOI] [PubMed] [Google Scholar]
- 54.Sturiale L, Barone R, Garozzo D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis. 2011;34:891–9. doi: 10.1007/s10545-011-9306-8. [DOI] [PubMed] [Google Scholar]
- 55.Fujita M, Satoh C, Asakawa J, Nagahata Y, Tanaka Y, Hazama R, et al. Electrophoretic variants of blood proteins in Japanese. VI. Transferrin. Jinrui Idengaku Zasshi. 1985;30:191–200. doi: 10.1007/BF01876469. [DOI] [PubMed] [Google Scholar]
- 56.Kamboh MI, Ferrell RE. Human transferrin polymorphism. Hum Hered. 1987;37:65–81. doi: 10.1159/000153680. [DOI] [PubMed] [Google Scholar]
- 57.Helander A, Eriksson G, Stibler H, Jeppsson JO. Interference of transferrin isoform types with carbohydrate-deficient transferrin quantification in the identification of alcohol abuse. Clin Chem. 2001;47:1225–33. [PubMed] [Google Scholar]
- 58.Carchon HA, Chevigne R, Falmagne JB, Jaeken J. Diagnosis of congenital disorders of glycosylation by capillary zone electrophoresis of serum transferrin. Clin Chem. 2004;50:101–11. doi: 10.1373/clinchem.2003.021568. [DOI] [PubMed] [Google Scholar]
- 59.Quintana E, Montero R, Casado M, Navarro-Sastre A, Vilaseca MA, Briones P, et al. Comparison between high performance liquid chromatography and capillary zone electrophoresis for the diagnosis of congenital disorders of glycosylation. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:2513–8. doi: 10.1016/j.jchromb.2009.06.031. [DOI] [PubMed] [Google Scholar]
- 60.Barone R, Sturiale L, Garozzo D. Mass spectrometry in the characterization of human genetic N-glycosylation defects. Mass Spectrom Rev. 2009;28:517–42. doi: 10.1002/mas.20201. [DOI] [PubMed] [Google Scholar]
- 61.Wada Y, Nishikawa A, Okamoto N, Inui K, Tsukamoto H, Okada S, et al. Structure of serum transferrin in carbohydrate-deficient glycoprotein syndrome. Biochem Biophys Res Commun. 1992;189:832–6. doi: 10.1016/0006-291x(92)92278-6. [DOI] [PubMed] [Google Scholar]
- 62.Lacey JM, Bergen HR, Magera MJ, Naylor S, O’Brien JF. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem. 2001;47:513–8. [PubMed] [Google Scholar]
- 63.Sturiale L, Barone R, Palmigiano A, Ndosimao CN, Briones P, Adamowicz M, et al. Multiplexed glycoproteomic analysis of glycosylation disorders by sequential yolk immunoglobulins immunoseparation and MALDI-TOF MS. Proteomics. 2008;8:3822–32. doi: 10.1002/pmic.200700496. [DOI] [PubMed] [Google Scholar]
- 64.Guillard M, Morava E, van Delft FL, Hague R, Korner C, Adamowicz M, et al. Plasma N-glycan profiling by mass spectrometry for congenital disorders of glycosylation type II. Clin Chem. 2011;57:593–602. doi: 10.1373/clinchem.2010.153635. [DOI] [PubMed] [Google Scholar]
- 65.Faid V, Chirat F, Seta N, Foulquier F, Morelle W. A rapid mass spectrometric strategy for the characterization of N- and O-glycan chains in the diagnosis of defects in glycan biosynthesis. Proteomics. 2007;7:1800–13. doi: 10.1002/pmic.200600977. [DOI] [PubMed] [Google Scholar]
- 66.Mills K, Mills P, Jackson M, Worthington V, Beesley C, Mann A, et al. Diagnosis of congenital disorders of glycosylation type-I using protein chip technology. Proteomics. 2006;6:2295–304. doi: 10.1002/pmic.200500682. [DOI] [PubMed] [Google Scholar]
- 67.Schwarz M, Thiel C, Lubbehusen J, Dorland B, de Koning T, von Figura K, et al. Deficiency of GDP-Man:GlcNAc2-PP-dolichol mannosyltransferase causes congenital disorder of glycosylation type Ik. Am J Hum Genet. 2004;74:472–81. doi: 10.1086/382492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36:1225–9. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- 69.Jaeken J. Congenital disorders of glycosylation (CDG): update and new developments. J Inherit Metab Dis. 2004;27:423–6. doi: 10.1023/b:boli.0000031221.44647.9e. [DOI] [PubMed] [Google Scholar]
- 70.Krawitz PM, Schweiger MR, Rodelsperger C, Marcelis C, Kolsch U, Meisel C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42:827–9. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 71.Goreta SS, Dabelic S, Dumic J. Employment of single-strand conformation polymorphism analysis in screening for alpha-1,3 glucosyltransferase gene mutation A333V in Croatian population. J Clin Lab Anal. 2011;25:65–70. doi: 10.1002/jcla.20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bjursell C, Erlandson A, Nordling M, Nilsson S, Wahlstrom J, Stibler H, et al. PMM2 mutation spectrum, including 10 novel mutations, in a large CDG type 1A family material with a focus on Scandinavian families. Hum Mutat. 2000;16:395–400. doi: 10.1002/1098-1004(200011)16:5<395::AID-HUMU3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 73.Imbach T, Grunewald S, Schenk B, Burda P, Schollen E, Wevers RA, et al. Multi-allelic origin of congenital disorder of glycosylation (CDG)-Ic. Hum Genet. 2000;106:538–45. doi: 10.1007/s004390000293. [DOI] [PubMed] [Google Scholar]
- 74.Mizugishi K, Yamanaka K, Kuwajima K, Yuasa I, Shigemoto K, Kondo I. Missense mutations in the phosphomannomutase 2 gene of two Japanese siblings with carbohydrate-deficient glycoprotein syndrome type I. Brain Dev. 1999;21:223–8. doi: 10.1016/s0387-7604(99)00004-2. [DOI] [PubMed] [Google Scholar]
- 75.Lefeber DJ, Morava E, Jaeken J. How to find and diagnose a CDG due to defective N-glycosylation. J Inherit Metab Dis. 2011;34:849–52. doi: 10.1007/s10545-011-9370-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jones MA, Bhide S, Chin E, Ng BG, Rhodenizer D, Zhang VW, et al. Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation. Genet Med. 2011;13:921–32. doi: 10.1097/GIM.0b013e318226fbf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matthijs G, Schollen E, Van Schaftingen E. The prenatal diagnosis of congenital disorders of glycosylation (CDG) Prenat Diagn. 2004;24:114–6. doi: 10.1002/pd.815. [DOI] [PubMed] [Google Scholar]
- 78.Clayton P, Winchester B, Di Tomaso E, Young E, Keir G, Rodeck C. Carbohydrate-deficient glycoprotein syndrome: normal glycosylation in the fetus. Lancet. 1993;341:956. doi: 10.1016/0140-6736(93)91244-g. [DOI] [PubMed] [Google Scholar]
- 79.Nogueira C, Quelhas D, Vilarinho L. Prenatal diagnosis for CDG Ia based on post-mortem molecular study of Guthrie card. Mol Genet Metab. 2006;87:379. doi: 10.1016/j.ymgme.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 80.Charlwood J, Clayton P, Keir G, Mian N, Young E, Winchester B. Prenatal diagnosis of the carbohydrate-deficient glycoprotein syndrome type 1A (CDG1A) by a combination of enzymology and genetic linkage analysis after amniocentesis or chorionic villus sampling. Prenat Diagn. 1998;18:693–9. [PubMed] [Google Scholar]
- 81.Thiel C, Korner C. Mouse models for congenital disorders of glycosylation. J Inherit Metab Dis. 2011;34:879–89. doi: 10.1007/s10545-011-9295-7. [DOI] [PubMed] [Google Scholar]
- 82.Schneider A, Thiel C, Rindermann J, Derossi C, Popovici D, Hoffmann GF, et al. Successful prenatal mannose treatment for congenital disorder of glycosylation-Ia in mice. Nat Med. 2011;18:71–3. doi: 10.1038/nm.2548. [DOI] [PubMed] [Google Scholar]
- 83.Panneerselvam K, Freeze HH. Mannose corrects altered N-glycosylation in carbohydrate-deficient glycoprotein syndrome fibroblasts. J Clin Invest. 1996;97:1478–87. doi: 10.1172/JCI118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schroeder AS, Kappler M, Bonfert M, Borggraefe I, Schoen C, Reiter K. Seizures and stupor during intravenous mannose therapy in a patient with CDG syndrome type 1b (MPICDG) J Inherit Metab Dis. 2011 doi: 10.1007/s10545-010-9252-x. [DOI] [PubMed] [Google Scholar]
- 85.Marquardt T, Luhn K, Srikrishna G, Freeze HH, Harms E, Vestweber D. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood. 1999;94:3976–85. [PubMed] [Google Scholar]
- 86.Georgiou E, Layton M, Karadimitris A. Inherited GPI deficiency: a disorder of histone hypoacetylation. Birth Defects Res C Embryo Today. 2009;87:327–34. doi: 10.1002/bdrc.20166. [DOI] [PubMed] [Google Scholar]
- 87.Almeida AM, Murakami Y, Baker A, Maeda Y, Roberts IA, Kinoshita T, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med. 2007;356:1641–7. doi: 10.1056/NEJMoa063369. [DOI] [PubMed] [Google Scholar]
- 88.Eklund EA, Merbouh N, Ichikawa M, Nishikawa A, Clima JM, Dorman JA, et al. Hydrophobic Man-1-P derivatives correct abnormal glycosylation in Type I congenital disorder of glycosylation fibroblasts. Glycobiology. 2005;15:1084–93. doi: 10.1093/glycob/cwj006. [DOI] [PubMed] [Google Scholar]
- 89.Haeuptle MA, Welti M, Troxler H, Hulsmeier AJ, Imbach T, Hennet T. Improvement of dolichol-linked oligosaccharide biosynthesis by the squalene synthase inhibitor zaragozic acid. J Biol Chem. 2011;286:6085–91. doi: 10.1074/jbc.M110.165795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sharma V, Ichikawa M, He P, Bravo Y, Dahl R, Ng BG, et al. Phosphomannose Isomerase Inhibitors Improve N-Glycosylation in Selected Phosphomannomutase-deficient Fibroblasts. J Biol Chem. 2011;286:39431–8. doi: 10.1074/jbc.M111.285502. [DOI] [PMC free article] [PubMed] [Google Scholar]