

Abstract
The uterine endometrium is exquisitely sensitive to steroid hormones that act through well-described nuclear receptors. Estrogen drives epithelial proliferation, and progesterone inhibits growth and causes cell differentiation. The importance of progesterone as a key inhibitor of carcinogenesis is reflected by the observation that women who ovulate and produce progesterone almost never get endometrial cancer. In this review we describe seminal research findings that define progesterone as the major endometrial tumor suppressor. We discuss the genes and diverse signaling pathways that are controlled by progesterone through progesterone receptors (PRs) and also the multiple factors that regulate progesterone/PR activity. By defining these progesterone-regulated factors and pathways we identify the principal therapeutic opportunities to control the growth of endometrial cancer.
Hormone-dependency in endometrial cancer
Endometrial cancer is the fourth most common malignancy in women and the most common gynecologic cancer. Rates of endometrial cancer are on the rise, and surprisingly there has been no increase in survival over the past four decades (American Cancer Society). Improving outcomes for women with endometrial cancer depends upon a more complete understanding of endometrial biology. In the normal endometrium, steroid hormones control progression through the menstrual cycle. Estrogen drives proliferation of the endometrial glandular epithelium (the cells most commonly involved in endometrial cancer), whereas progesterone counteracts the effects of estrogen. This balance of estrogen and progesterone activity during the menstrual cycle must be precisely maintained, and a shift in either direction – a gain in estrogen or a loss in the antagonistic activity of progesterone – stimulates oncogenesis. Estrogens act upon the endometrium through estrogen receptors (ERs) resulting in the induction of growth factors such as the epidermal growth factor (EGF) [1], its receptor (EGFR) [2], insulin-like growth factor-1 (IGF-1) [3], and growth-enhancing proto-oncogenes such as c-fos and c-myc [4]. Besides these genomic effects of estrogen in the endometrium, estrogen exerts nongenomic effects via activation of the PI3K/Akt prosurvival signaling pathway [5].
Progesterone acts by binding to PRs, regulating multiple signaling pathways through PR-dependent transcriptional activity. In addition to ligand-mediated regulation, PR activity is also modulated by a variety of factors including micro (mi) RNAs and epigenetic factors. Progesterone has long been explored therapeutically in hormone-based therapies for endometrial carcinomas, and prolongs overall survival in a subset of patients, particularly those that maintain expression of PR. Although hyperplasia, atypical hyperplasia, and some early endometrial cancers are reversed by progestin therapy, response rates in advanced disease are modest and recurrence is common (Box 1). Failure is linked to the loss of PR with prolonged progester-one/progestin treatment. Thus, gradual loss of PR begets resistance to progesterone-based therapy, signifying the need for alternative approaches to pharmacologically modulate the downstream differentiating pathways controlled by progesterone when PR is robustly expressed. By identifying these pathways we can use progesterone/PR as a roadmap to identify the most important differentiating and growth-limiting mechanisms in the endometrium. Even in the absence of PR, new molecular therapies can be targeted to these pathways in future clinical trials. Therefore, a more complete understanding of the effects of progesterone is vital to improving survival in this disease.
Box 1. Types of endometrial carcinomas and response to progestin therapy.
Endometrial hyperplasia: overexposure to estrogen or unopposed estrogen promotes hyperplasia. Hyperplasia without atypia rarely progresses to cancer, whereas 30% of untreated cases of atypical hyperplasia will progress to cancer [79].
Type I endometrial carcinoma: this type represents 70–80% of all endometrial cancers. Type I tumors are endometrioid in origin, hormone-responsive, and prognosis is generally good. Mutations in genes encoding PTEN, PI3KCA, KRAS, and β-catenin and also microsatellite instability are common [79].
Type II endometrial carcinoma: type II is an aggressive non-endometrioid cancer with papillary serous or clear-cell morphology. Tumors are not hormone-responsive, outcome is generally poor, and recurrence is frequent. Loss of p53 and chromosomal instability characterize this tumor type [79].
Response to progestin therapy
Endometrial hyperplasia (both typical and atypical): treatment with progestin promotes regression in 80–90% of cases [73].
Primary endometrial adenocarcinoma (predominantly type I): overall response rate to progestin therapy is 50–70% [73].
Advanced and recurrent endometrial cancer (predominantly type II): objective response rates to progestin therapy are 15–20% [80], and combination therapy of progestin and tamoxifen results in complete or partial response in 33% of patients [77].
PR structure
PR is a nuclear steroid receptor that binds progesterone with high specificity. In humans, the PGR gene located at 11q22–q23 is transcribed to generate mRNAs encoding the two principal isoforms, PRA and PRB. These rise from transcription initiation at alternative promoters on the same gene. PRA is a 90 kD protein whereas PRB is 120 kD; the proteins can form both homo-(A/A, B/B) and hetero-(A/B) dimers. The isoforms are identical except that PRB has a longer N-terminus comprising 164 amino acids that are not present in PRA (Figure 1). This region possesses a third activation function (AF-3). The sequences downstream from the N-terminus of PRB are identical between the A and B isoforms, including a DNA-binding domain (DBD), a hinge region that contains the nuclear localization sequence (NLS) and a region to which co-modulators bind (Co-R), and the ligand-(progesterone) binding domain (LBD) including activation function-2 (AF-2).
Figure 1.
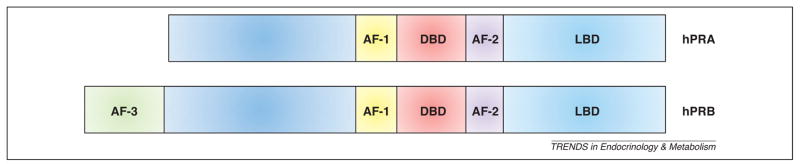
Progesterone receptor structural elements. Diagrammed are the two primary human isoforms of progesterone receptor, PRA and PRB. PRB contains an extra 164 amino acids at the N-terminus. Within this region is AF-3, which is unique to PRB. AF, activation function; DBD, DNA-binding domain; LBD, ligand-binding domain.
Physiology and pathology of progesterone receptors
Progesterone diffuses through the plasma membrane to bind its cognate receptor. When cytoplasmic, PR translocation to the nucleus follows, DNA binding occurs, and transcriptional activity is stimulated. Progesterone also diffuses through the nuclear membrane to bind directly to PRs located in the nucleus. Ligand-dependent dimerization between two individual PRA and/or PRB molecules is required to form the functional transcription factor unit on DNA. The unique N-terminus of PRB, which contains AF-3, confers different functional characteristics on the isoforms: PRB is a stronger transcriptional activator of many genes compared to PRA [6–8], but PRA apparently counters estrogen action directly by inhibiting ER function in a dominant-negative manner [9]. Whereas both isoforms are present in the glands and the stroma, PRA is thought to be the primary form present in the stroma, and PRB predominates in the glandular epithelium [10]. Both isoforms are likely to be required for endometrial differentiation and homeostasis. In endometrial cancer cell lines, they promote differentiation through distinct mechanisms: PRA induces cell senescence whereas PRB induces a secretory phenotype. However, both PRA and PRB sensitize endometrial cancer cells to apoptosis and inhibit the cell cycle at the G1 to S transition [11]. With respect to growth inhibition, PRB appears to be the principal effector in human endometrial cancer cell growth in vitro [12]. PRB protein expression is lost in poorly differentiated endometrial cancer cell lines such as Hec50 and KLE, suggesting that this isoform is important for maintaining endometrial differentiation [6,7]. Data from human endometrial cancer samples suggest either downregulation of PRA and PRB [13] or only PRA [14].
Several rodent models of endometrial cancer have been developed (Box 2), although studies in these models do not always replicate PR biology and pathology in humans. Female knockout mice deficient in both isoforms of PR, and also those deficient in only the A isoform, demonstrate significant endometrial hyperplasia [15–17]. In the PRA knockout model, progesterone treatment causes endometrial proliferation through PRB [17]. Whereas these studies are of interest and require further consideration, differences between rodents and humans must be taken into account. The proliferative effects of progestin through PRB do not appear to be malignant in the PRA knockout model [17], indicating differences between the potential of endometrial cells to respond to hormone stimulation between species. Furthermore, both the production of progesterone in the uterus and placenta [18] and PR expression in the ovary differ between species, indicating that progesterone/PR function is also likely to be different [19]. Nevertheless, the functions of PRA compared to PRB are still under investigation, and the possibility that PRB can be a proliferative signal for the endometrial epithelium in both humans and rodents has not been ruled out. For example, PRB is present in the cytoplasm and interacts with signaling molecules in the absence of ligand that could promote rather than inhibit proliferation.
Box 2. Animal models of endometrial cancer.
Transgenic mice
Pgr−/−: the global (PRKO) and isoform-specific PR knockout animals are valuable tools to understand the normal biology of the endometrium and the tissue-specific actions of the two isoforms [15–17], although these animals develop endometrial hyperplasia but not cancer. Estrogen-induced endometriosis has been documented using PRKO mice and cannot be overcome by addition of progesterone [81].
Pten+/−: these mice develop multiple types of neoplasia, including endometrial tumors, which are similar to the human disease [82,83]. However, tumor formation is not hormone-dependent, and treatment of ovariectomized Pten+/− mice with progestin does not prevent tumor formation [84]. Deletion of Akt1 in the context of Pten+/− prevents endometrial cancer [85].
Pten−/− conditional endometrial knockout (Ptenpr−/−): mice develop in situ endometrial carcinoma in 3–4 weeks [86]. Combined conditional deletion of p53 and PTEN (p53pr−/− Ptenpr−/−) accelerates disease, although p53 is not typically lost in type I endometrial cancers in humans.
Pten+/− Mlh1 Mlh1−/−: deficiency of DNA mismatch repair (Mlh1−/ −) accelerates endometrial cancer formation compared to Pten+/− mice [87]. Tumors also have microsatellite instability, consistent with type I endometrial adenocarcinoma.
Mig-6 conditional endometrial knockout (Mig-6d/d): this conditional knockout was generated similar to the Ptenpr−/ − mice [88]. Treatment of ovariectomized Mig-6d/d mice with estrogen induces invasive endometrioid adenocarcinoma, similar to type I. Combined estrogen and progesterone treatment prevents development of cancer, although these mice do have endometrial hyperplasia.
Xenograft models
Use of either tumor specimens from patients or existing endometrial cancer cell lines in mice or rats.
Spontaneous tumorigenesis in inbred rats
DBII/Han: endometrial cancer is hormone-dependent: >90% of animals die of primary or metastatic disease, and a majority of tumors are adenocarcinomas. Lifelong treatment of DBII/Han rats with progestin prevents carcinogenesis at doses as low as 0.1 mg/ kg/day [83]. Gene amplification of Cdk6, Met, and N-myc has been documented, and overexpression of N-myc is thought to contribute to hormone dependency.
Donryu: endometrial carcinomas develop in 35% of rats, and tumors frequently have mutations in Kras. Tumors are thought to be hormone-dependent because the animals have an imbalance in estrogen/progesterone [83].
DA/Han: 60% of rats die from hormone-dependent endometrial carcinoma. Tumors are well-differentiated histologically and 63% metastasize, particularly to the lung [83].
Han:Wistar: approximately 14% of animals die from endometrial adenocarcinoma. This model is not well-studied [83].
Chemical-induced cancer
Estrogens: neonatal exposure of mice to estrogens, such as estradiol or diethylstilbestrol, causes aggressive endometrial adenocarcinoma in a dose-dependent manner [83].
Carcinogens: N-methyl-N-nitrosourea promotes the development of endometrial cancer from hyperplastic lesions within 30 weeks. Addition of estradiol accelerates tumorigenesis, whereas treatment with progestin prevents tumor formation entirely [83].
Further investigation is warranted to clarify the role of receptor expression in cancer recurrence, particularly the expression of receptor isoforms such as PRA relative to PRB and other hormone-dependent genes that could be potential biomarkers. Studies now indicate that commercially available antibodies do not recognize PRA and PRB with equal affinity by immunohistochemistry (IHC) despite findings to the contrary using immunoblotting [20]. Furthermore, PRB is not recognized by many commonly used antibodies [20], and this raises the possibility that PRB expression might have been under-reported in the past [13]. Therefore, studies to evaluate differential expression of receptor isoforms and their association with clinical outcome will be important for the future understanding of hormone action in endometrial cancer.
Crosstalk and steroid receptor expression in endometrial glands and stroma
The endometrium is composed of two cell types, the glandular epithelium and the stroma. Endometrial cancers arise most commonly in the glandular epithelium. However, the glands and stroma communicate directly, and it is likely that abnormal interactions between the two tissues could occur in the process of endometrial carcinogenesis [21]. The epithelial glands and the stroma both express ER and PR, and expression in both tissue types is likely to be necessary to induce normal growth and differentiation of the glandular epithelium. For example, reconstitution experiments using stromal and epithelial cells from estrogen receptor knockout (ERKO) and normal mice demonstrate that epithelial cell proliferation in the mouse is mediated via ER action from the stroma [21–24]. Interestingly, ER expression in epithelial cells is necessary to prevent apoptosis before proliferation, but is not required for estrogen-dependent PR downregulation [24]. In addition, stromal PR levels are much higher than epithelial levels at the time of implantation, making it clear that the effects of progesterone at crucial stages in endometrial function are mediated through the stroma [25]. It is predicted that some aspects of epithelial cell differentiation in response to progesterone could be mediated through stromal PR [26] in which PRA is the predominantly expressed isoform [10]. Therefore, evaluation of receptor expression in both the stroma and the glands could provide insight into the process of carcinogenesis and the responsiveness of the tumor to progestin therapy.
Genomic effects of steroid receptors
Estrogens and progesterone/progestins act via genomic and non-genomic pathways to control cell function and proliferation. The estrogen and progesterone receptors are best characterized as nuclear transcription factors that bind to DNA, recruit a protein complex of co-modulators as well as the basal transcriptional unit, and initiate the expression of hormone-dependent proteins [27–29]. The genomic pathway leads directly to the transcription of hormone-dependent genes and is the principal and best-studied mode of action. Gene expression profiling in response to steroid hormones through their cognate receptors has provided new information on the large number of pathways controlled by these factors. Cytokines, cell-adhesion molecules, growth factors and their receptors, signaling molecules, pro- and anti-apoptotic factors, DNA-binding proteins, enzymes, other classes of transcription factors and cell-cycle regulators are now recognized to be under hormonal control [11,12,30–41]. Whereas progestins promote proliferation in breast cancer cells, this has been less evident in endometrial cells where luteal promotion and differentiation are the primary effects [11,42–45]. Nevertheless, it is possible that the PR isoforms modulate both growth-promoting and growth-inhibitory pathways, as has been demonstrated in PR-knockout mouse models [15,17,46,47]. It is clear that the receptor isoforms have distinct functions and activate unique as well as overlapping gene sets [11,45]. This is reminiscent of the findings with the glucocorticoid receptor (GR) where different GR species activate both different and overlapping sets of genes [48–50]. Therefore, a complete understanding of tumor response to progestins will depend upon the PR isoform composition.
Progesterone-regulated genes and pathways
The importance of progesterone in endometrial cancer had fuelled research to understand how progesterone governs expression of a vast array of signaling pathways through transcriptional regulation to prevent tumor development (Figure 2). Many progesterone target genes contain progesterone response elements (PRE) within the basal promoter regions: these include genes encoding pepsinogen C, c-myc, insulin-like growth factor binding protein-1, mucin (MUC)-1, cyclin D1, Ets-1, Forkhead-box protein O1 (FOXO1), glycodelin, and matrix metalloproteases (MMP)-1, -2, and -9. However, progesterone-mediated gene expression is not limited to genes with a PRE in the promoter; in this case, Sp1 is the crucial element in the basal promoter of these genes. Examples of progesterone-controlled genes lacking a PRE include the genes encoding p21, p27, and folate receptor type α. Details of the mechanisms for progesterone regulation of these genes are lacking. Also, these findings are preclinical observations and should be validated in translational endpoints from future clinical studies of women with endometrial cancer. To this end, the Gynecologic Oncology Group (GOG) has undertaken a non-therapeutic trial, GOG211, which examines the effect of Depo-Provera, a progestin, given in the neoadjuvant setting before surgery, on endometrial cancer pathologic characteristics and on the expression of key factors at the time of hysterectomy. Results of this trial are still under analysis but are anticipated to be published in the coming year.
Figure 2.

Progesterone web of influence. Through genomic and non-genomic regulation, progesterone acts as a tumor suppressor to promote (➞) apoptosis, differentiation, and cell cycle arrest, and to inhibit (
 ) inflammation and invasion.
) inflammation and invasion.
Our group has extensively investigated progesterone transcriptional regulation to understand the mechanisms by which progesterone exerts its tumor-suppressor effects. In one study we found that ligand-dependent PR transcriptional suppression of activating protein (AP-1) family members, including c-jun, serves to decrease proliferation of endometrial cancer cells [43]. In a separate study, progesterone was found to abrogate the transcriptional activity of NFκB in poorly differentiated endometrial cancer cells (Hec50). This modulation of NFκB prevents transcription of pro-inflammatory genes associated with tumor development [44,45]. Several cell-adhesion molecules are also downregulated by progesterone, including fibronectin, integrins α3, β1, and β3, and cadherin-6 [11,42], which could be important to metastatic disease.
In relation to differentiation, progesterone also regulates the expression of key components of the Wnt signaling pathway. Specifically, progesterone induces the expression of the Wnt inhibitory proteins dickkopf homolog 1 (DKK1) and FOXO1 [51]. These data are consistent with a report by another group that FOXO1 was upregulated at the protein level by progestin in well-differentiated endometrial cancer cell lines (Ishikawa), specifically through PRB [52]. This report not only provides a link between progesterone and the Akt pro-survival pathway, but also reveals a novel isoform-specific effect.
Progesterone/progestins regulate a variety of cell-cycle proteins to promote G1/S arrest and upregulation of the cyclin-dependent kinase inhibitors (CDKIs) including p21, p27, and p53. As mentioned, progesterone blocks transcription of the gene encoding c-jun [43]. However, recruitment of c-jun to the cyclin D1 promoter is also inhibited by progesterone by a mechanism that is not fully understood [43]. Furthermore, p21 and p27 expression increases upon progestin treatment in a PRB-dependent manner [53]; mechanistically, this upregulation occurs through enhanced binding of AP-1 to the p21 promoter in response to progesterone [43]. In addition to progesterone regulation of AP-1 binding to the p21 promoter, levels of p21 and other CDKIs can be increased by another progesterone-controlled factor, glycodelin. Overexpression of glycodelin results in G1/S arrest through upregulation of CDKIs in endometrial cancer cells [54]. Finally, p53 expression is also regulated by progesterone, although indirectly through AP-1 binding to the p53 promoter [43].
The relative contribution of coregulators to PR function is important. For example, the well-studied steroid receptor coactivator (SRC)-1 enhances gene transcription by PR through multiple mechanisms. For a more in-depth summary of the coregulators that impinge on PR function the reader is referred to a review by Lonard et al. [55].
Immunologic effects of progesterone
There are two unanswered questions regarding progestin-based hormone therapy: (i) why do patients who express PR not respond to progestin therapy, and (ii) why do patients with an apparent lack of PR respond to the therapy? This second issue has been addressed by investigating the immune response after progestin treatment [56]. Using endometrial carcinoma tumor samples pre-and post-progestin treatment it was found that regulatory T cells are decreased in patients who received progestin. By contrast, progestin treatment dramatically increases the recruitment of natural killer (NK) cells to the tumor. These data signify that progestin possesses immunomodulatory activity, although the effect is unlikely to be direct because NK cells do not express PR mRNA [57].
Other factors regulate PR activity
On one hand, progesterone and PR regulate many genes and signaling pathways to function as a tumor suppressor in endometrial cancer; however, on the other hand, in endometrial cancer cells PR itself can be downregulated by multiple factors to lose the tumor-suppressor feature of progesterone, and thereby facilitate tumor growth. PR expression, a general marker for a differentiated endometrial cell, is commonly lost in cultured endometrial cell lines, although the mechanism remains poorly understood. Despite the long history of hormonal therapy through progesterone/progestin in endometrial cancer, the outcome is generally modest, most likely due to the gradual loss of PR. The loss of PR, particularly in poorly differentiated tumors, has been validated in a number of studies including those from our own group [58,59]. It is now known that PR activity can be suppressed at least at four different levels. First, PR activity can be inhibited at the transcription level by epigenetic regulation, such as methylation of its promoter and within exons [60–62]. Second, miRNAs downregulate PR activity at the post-transcriptional level [63], although this has not yet been tested in endometrial cancer. Third, small ubiquitin-like modifier (SUMO) proteins regulate PR at the post-translational level [64,65]. Finally, PR stability is regulated in a negative-feedback loop involving ligand-induced degradation [66,67].
The mechanism by which the gene encoding PR, PGR, is epigenetically silenced can be exploited to restore the tumor-suppressor function of progesterone in endometrial cancer. Towards this goal it has been shown that the PGR gene is silenced by aberrant DNA methylation on its promoter and the first exon [60]. Furthermore, methylation is exclusive to the PRB promoter both in endometrial cancer cell lines and in patient samples [68]. In 87% of endometrial cancer specimens, PRB promoter hypermethylation was observed and correlated with loss of PRB protein expression [68], suggesting that hypermethylation significantly contributes to the loss of PR and corresponding progesterone insensitivity.
PR also potentially loses its expression through down-regulation by miRNAs. Although miRNAs that target progesterone/PR have not been studied in endometrial cancer, two independent groups have described miRNAs that correlate with PR status in breast cancer [69,70]. For example, overexpression of miR-26a and miR-181a blocked estradiol-dependent PRA and PRB expression, both at the mRNA and protein levels, in MCF7 breast cancer cells [71]. It is unclear whether miR-26a and miR-181a target PGR in endometrial cancer and thus requires further investigation.
In addition to the epigenetic and post-transcriptional silencing of PR expression, post-translational modifications of PR have also been shown to regulate activity. PR possesses a single consensus Ψ KXE SUMO-conjugation motif (where Ψ is a large hydrophobic residue and X is any residue) in its N-terminus that is conserved in both isoforms [64,72]. SUMOylation of PR stringently represses progesterone-mediated transcriptional activity [64]. Strikingly, however, overexpression of SUMO can also promote PR transcriptional activity indirectly by enhancing binding of the SRC-1 coactivator to SUMOylated PR [64]. Ligand-induced SUMOylation of PR is also inhibited via cAMP signaling pathways in endometrial stromal cells [65]. Because these studies were conducted using breast cancer cells or normal endometrial stromal cells, it will be of interest to determine both the level of PR SUMOylation in endometrial cancer cells and how this post-translational modification affects its activity.
A fourth mechanism by which progestins alter PR expression through a negative-feedback loop has also been described. Ligand-dependent activation of PR activates a signaling cascade that includes MAPK phosphorylation of PR; this in turn induces ubiquitination of PR and targeting to the proteosome for degradation [66,67]. However, phosphorylation of PR has not been documented in endometrial cancer patient samples to date. To prove this mechanism of downregulation, use of MAPK or proteosome inhibitors could be of therapeutic value to restore PR expression. For example, treatment of breast cancer cells with proteosome inhibitors (lactacystin and calpain inhibitor I), an ERK inhibitor (PD98059), and p38 MAPK inhibitor (SB202190), but not another p38 inhibitor (SB203580), prevents ligand-dependent degradation of PR [66]. Ligand-independent degradation of immature PR also involves the proteosome, because expression can be restored with proteasome inhibitors [66]. These experiments have not yet been replicated using endometrial cancer cells to determine if use of these inhibitors might be clinically relevant; however, this reasoning provides a direction which can be tested in future clinical trials.
Progestin therapy in endometrial cancer
Progestin therapy has been addressed in detail in a recently published review [73]. Progestins cause differentiation of susceptible tumor cells and could produce an anti-tumor effect either by permitting apoptosis or by removing them from the pool of actively dividing cells. Expression of PR has been positively correlated with a good prognosis and response to progestin treatment – with 72% overall response in patients with PR-rich tumors but only 12% response in patients with PR-poor lesions [74]. However, in responders the overall clinical benefit is typically of short duration. Progestins downregulate both estrogen receptor and PR, and this could limit the duration of efficacy of progestin therapy [75,76].
Our group has demonstrated that progestin control of cell growth can be re-established by expression of PRB in PR-negative endometrial cancer cell lines [42], and in GOG study 119, tamoxifen (acting as an estrogen surrogate to induce PR) was added to enhance the clinical activity of progestin [59,77]. In the GOG119 phase II trial, patients with advanced or recurrent endometrial cancer received daily tamoxifen and alternating weekly doses of progestin (medroxyprogesterone acetate) [59,77]. The overall response rate was 33% [77]. ER expression positively correlated with response [59]. For ER-positive tumors the clinical response rate was 44% compared to 25% for ER-negative tumors. The correlation between ER expression and clinical response was statistically significant as a continuous variable in a logistic model ( p = 0.02). The presence of the other receptors was not clinically significant due to the relatively small study n value; however, trends were evident. Fifty-eight percent of patients with PRA and 38% of those with PRB responded [59]. Tumors expressing both PRA and PRB had a surprisingly high response rate of 67%. However, these observations should be confirmed in larger future trials.
One additional mechanism of PR downregulation is promoter methylation, and two groups have explored how DNA methyltransferase (DNMT) inhibitors can restore PR expression with the hopes of developing an alternative treatment strategy [60,62]. Although one group demonstrated a decrease in proliferation in response to DNMT inhibition [60], the clinical efficacy of these agents in restoring progestin sensitivity has not yet been tested.
Conclusions and future directions
Among hormone-sensitive malignancies, the impact of progesterone as a tumor suppressor is most obvious in cancers of the uterine endometrium. Progestins used alone are clearly efficacious in reversing pre-malignant endometrial hyperplasia, preventing the development of frank carcinomas, and even treating early-grade endometrial neoplasia in women seeking to preserve fertility. However, hormonal therapy for advanced endometrial cancer results in lower response rates than chemotherapy in the frontline setting, and does not often cure patients with recurrent disease. Hence, hormonal therapy is used less commonly in the US in recent decades compared to previous practice trends. Nevertheless, the study of biologic effects of progesterone in the endometrium is crucial. Such studies provide an unprecedented type of roadmap for clinical investigators, who, with the growth of individualized molecular therapeutics, can more easily identify the specific pathways which must be blocked to prevent tumor growth. The GOG is addressing this issue in the selection of molecular agents in trials within the 229 and the 211 queue at the present time; it is anticipated that this knowledge will result in identifying molecular therapies with clinical activity. Hence, future progress in preventing and treating endometrial cancer depends upon understanding the differentiating actions of progesterone in the endometrium.
Progesterone controls many pathways that culminate in growth inhibition and tissue homeostasis. To accomplish this regulation, progesterone signals through two predominant PR isoforms, PRA and PRB, which allows progesterone to weave a vast web of biological influence in the endometrium. Endometrial cancer is an understudied disease with more questions than answers (Box 3), and no significant therapeutic breakthroughs have occurred in the past decade. Although the effects of progesterone on differentiation in PR-positive endometrial cancer cells are undeniable, questions remain regarding how to maintain sensitivity to progestin therapy in PR-positive tumors, and how to regain PR expression in cells that have lost this receptor. Even though progesterone hormonal therapy is a traditional alternative, response is generally modest, particularly for recurrent endometrial carcinoma in which the objective response rate to progestin is ~15–20% [73]. However, new strategies to enhance progestin sensitivity, including adding an estrogen to induce PR or using a DNMT inhibitor to reverse the suppressed PR expression, could hold promise for the future.
Box 3. Barriers and unanswered questions.
How can PR expression be maintained or reinstated in endometrial cancer, thereby ensuring higher response rates to progestin therapy?
What are the best agents to combine with progestins in the treatment of recurrent endometrial cancer? For example, does resistance to hormonal therapy result in the simultaneous activation of multiple oncogene-initiated signaling pathways (EGFR, VEGF, mTOR), and can resistance be overcome with combinatorial therapies?
Novel biomarker identification to screen for predicted responders and monitor response. For example, is PR expression status sufficient, or is a functional measurement required?
One major area for progesterone therapy improvement is efficacy, in particular through combinations with chemotherapy and molecular targeted therapies. For example, a trend to enhance the therapeutic benefit of progesterone is the use of combined hormone and chemotherapy regimens [78]. Of course, the issue of chemoresistance remains a significant roadblock for all cancers, including endometrial cancer. The emerging hormone-combination therapies seek to demonstrate how clinical response can be improved by combining progestin with other targeted therapeutics (e.g. kinase inhibitors). For example, because many cancers rely on multiple pathways for oncogenic signaling, we must first determine how progesterone regulates growth-factor signaling and vice versa. This knowledge will guide future clinical directions for combinatorial therapies. Towards that goal, the GOG is now testing both single-agent targeted molecules and combinations aimed at inhibiting the same proliferative pathways controlled by progesterone. Thus, knowledge of progesterone action delineates the roadmap to endometrial differentiation, and improving clinical outcomes in the future will depend upon a more complete understanding of the biologic effects of progesterone which identify new therapeutic targets.
Acknowledgments
Funding for this work was provided by National Institutes of Health (NIH) grant CA99908 to K.L., the Department of Obstetrics and Gynecology Academic Enrichment Fund, the GOG Core Laboratory for Receptors and Targets funded by NIH grant CA27469, and the Cory Beach Family in honor of Mrs. Barbara Beach. We also thank Drs Donghai Dai and Xiangbing Meng for their comments on the manuscript.
References
- 1.Huet-Hudson YM, et al. Estrogen regulates the synthesis of epidermal growth factor in mouse uterine epithelial cells. Mol Endocrinol. 1990;4:510–523. doi: 10.1210/mend-4-3-510. [DOI] [PubMed] [Google Scholar]
- 2.Lingham RB, et al. Estrogen regulation of epidermal growth factor receptor messenger ribonucleic acid. Mol Endocrinol. 1988;2:230–235. doi: 10.1210/mend-2-3-230. [DOI] [PubMed] [Google Scholar]
- 3.Murphy LJ, et al. Estrogen induces insulin-like growth factor-I expression in the rat uterus. Mol Endocrinol. 1987;1:445–450. doi: 10.1210/mend-1-7-445. [DOI] [PubMed] [Google Scholar]
- 4.Weisz A, Bresciani F. Estrogen induces expression of c-fos and c-myc protooncogenes in rat uterus. Mol Endocrinol. 1988;2:816–824. doi: 10.1210/mend-2-9-816. [DOI] [PubMed] [Google Scholar]
- 5.Guo RX, et al. 17 beta-estradiol activates PI3K/Akt signaling pathway by estrogen receptor (ER)-dependent and ER-independent mechanisms in endometrial cancer cells. J Steroid Biochem Mol Biol. 2006;99:9–18. doi: 10.1016/j.jsbmb.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 6.Kumar NS, et al. Selective down-regulation of progesterone receptor isoform B in poorly differentiated human endometrial cancer cells: implications for unopposed estrogen action. Cancer Res. 1998;58:1860–1865. [PubMed] [Google Scholar]
- 7.Leslie KK, et al. Differential expression of the A and B isoforms of progesterone receptor in human endometrial cancer cells Only progesterone receptor B is induced by estrogen and associated with strong transcriptional activation. Ann N Y Acad Sci. 1997;828:17–26. doi: 10.1111/j.1749-6632.1997.tb48520.x. [DOI] [PubMed] [Google Scholar]
- 8.Jacobsen BM, et al. New human breast cancer cells to study progesterone receptor isoform ratio effects and ligand-independent gene regulation. J Biol Chem. 2002;277:27793–27800. doi: 10.1074/jbc.M202584200. [DOI] [PubMed] [Google Scholar]
- 9.Vegeto E, et al. Human progesterone receptor A form is a cell-and promoter-specific repressor of human progesterone receptor B function. Mol Endocrinol. 1993;7:1244–1255. doi: 10.1210/mend.7.10.8264658. [DOI] [PubMed] [Google Scholar]
- 10.Mote PA, et al. Colocalization of progesterone receptors A and B by dual immunofluorescent histochemistry in human endometrium during the menstrual cycle. J Clin Endocrinol Metab. 1999;84:2963–2971. doi: 10.1210/jcem.84.8.5928. [DOI] [PubMed] [Google Scholar]
- 11.Dai D, et al. Progesterone inhibits human endometrial cancer cell growth and invasiveness: down-regulation of cellular adhesion molecules through progesterone B receptors. Cancer Res. 2002;62:881–886. [PubMed] [Google Scholar]
- 12.Smid-Koopman E, et al. Distinct functional differences of human progesterone receptors A and B on gene expression and growth regulation in two endometrial carcinoma cell lines. J Soc Gynecol Investig. 2003;10:49–57. [PubMed] [Google Scholar]
- 13.Arnett-Mansfield RL, et al. Relative expression of progesterone receptors A and B in endometrioid cancers of the endometrium. Cancer Res. 2001;61:4576–4582. [PubMed] [Google Scholar]
- 14.Fujimoto J, et al. Expression of progesterone receptor form A and B mRNAs in gynecologic malignant tumors. Tumour Biol. 1995;16:254–260. doi: 10.1159/000217942. [DOI] [PubMed] [Google Scholar]
- 15.Lydon JP, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9:2266–2278. doi: 10.1101/gad.9.18.2266. [DOI] [PubMed] [Google Scholar]
- 16.Mulac-Jericevic B, et al. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci USA. 2003;100:9744–9749. doi: 10.1073/pnas.1732707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulac-Jericevic B, et al. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science. 2000;289:1751–1754. doi: 10.1126/science.289.5485.1751. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Zimra M, et al. Uterine and placental expression of steroidogenic genes during rodent pregnancy. Mol Cell Endocrinol. 2002;187:223–231. doi: 10.1016/s0303-7207(01)00713-4. [DOI] [PubMed] [Google Scholar]
- 19.Stocco C, et al. The molecular control of corpus luteum formation, function, and regression. Endocr Rev. 2007;28:117–149. doi: 10.1210/er.2006-0022. [DOI] [PubMed] [Google Scholar]
- 20.Mote PA, et al. Detection of progesterone receptor forms A and B by immunohistochemical analysis. J Clin Pathol. 2001;54:624–630. doi: 10.1136/jcp.54.8.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooke PS, et al. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc Natl Acad Sci USA. 1997;94:6535–6540. doi: 10.1073/pnas.94.12.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooke PS, et al. Mechanism of estrogen action: lessons from the estrogen receptor-alpha knockout mouse. Biol Reprod. 1998;59:470–475. doi: 10.1095/biolreprod59.3.470. [DOI] [PubMed] [Google Scholar]
- 23.Buchanan DL, et al. Role of stromal and epithelial estrogen receptors in vaginal epithelial proliferation, stratification, and cornification. Endocrinology. 1998;139:4345–4352. doi: 10.1210/endo.139.10.6241. [DOI] [PubMed] [Google Scholar]
- 24.Winuthayanon W, et al. Uterine epithelial estrogen receptor alpha is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci USA. 2010;107:19272–19277. doi: 10.1073/pnas.1013226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wathes DC, et al. Regulation of oxytocin, oestradiol and progesterone receptor concentrations in different uterine regions by oestradiol, progesterone and oxytocin in ovariectomized ewes. J Endocrinol. 1996;151:375–393. doi: 10.1677/joe.0.1510375. [DOI] [PubMed] [Google Scholar]
- 26.Rider V, et al. Progesterone–growth factor interactions in uterine stromal cells. Biol Reprod. 1998;59:464–469. doi: 10.1095/biolreprod59.3.464. [DOI] [PubMed] [Google Scholar]
- 27.Onate SA, et al. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 28.Jensen EV, DeSombre ER. Estrogen–receptor interaction. Science. 1973;182:126–134. doi: 10.1126/science.182.4108.126. [DOI] [PubMed] [Google Scholar]
- 29.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mutter GL, et al. Global expression changes of constitutive and hormonally regulated genes during endometrial neoplastic transformation. Gynecol Oncol. 2001;83:177–185. doi: 10.1006/gyno.2001.6352. [DOI] [PubMed] [Google Scholar]
- 31.Richer JK, et al. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277:5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- 32.Salleh MN, et al. A comparison of gene expression changes in response to diethylstilbestrol treatment in wild-type and p53+/− hemizygous knockout mice using focussed arrays. Toxicology. 2003;185:49–57. doi: 10.1016/s0300-483x(02)00587-5. [DOI] [PubMed] [Google Scholar]
- 33.Levenson AS, et al. Molecular classification of selective oestrogen receptor modulators on the basis of gene expression profiles of breast cancer cells expressing oestrogen receptor alpha. Br J Cancer. 2002;87:449–456. doi: 10.1038/sj.bjc.6600477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levenson AS, et al. Gene expression profiles with activation of the estrogen receptor alpha-selective estrogen receptor modulator complex in breast cancer cells expressing wild-type estrogen receptor. Cancer Res. 2002;62:4419–4426. [PubMed] [Google Scholar]
- 35.Kramer PR, Wray S. 17-Beta-estradiol regulates expression of genes that function in macrophage activation and cholesterol homeostasis. J Steroid Biochem Mol Biol. 2002;81:203–216. doi: 10.1016/s0960-0760(02)00065-1. [DOI] [PubMed] [Google Scholar]
- 36.Wan Y, Nordeen SK. Overlapping but distinct gene regulation profiles by glucocorticoids and progestins in human breast cancer cells. Mol Endocrinol. 2002;16:1204–1214. doi: 10.1210/mend.16.6.0848. [DOI] [PubMed] [Google Scholar]
- 37.Wan Y, Nordeen SK. Identification of genes differentially regulated by glucocorticoids and progestins using a Cre/loxP-mediated retroviral promoter-trapping strategy. J Mol Endocrinol. 2002;28:177–192. doi: 10.1677/jme.0.0280177. [DOI] [PubMed] [Google Scholar]
- 38.Kao LC, et al. Global gene profiling in human endometrium during the window of implantation. Endocrinology. 2002;143:2119–2138. doi: 10.1210/endo.143.6.8885. [DOI] [PubMed] [Google Scholar]
- 39.Naciff JM, et al. Gene expression profile induced by 17alpha-ethynyl estradiol, bisphenol A, and genistein in the developing female reproductive system of the rat. Toxicol Sci. 2002;68:184–199. doi: 10.1093/toxsci/68.1.184. [DOI] [PubMed] [Google Scholar]
- 40.Rockett JC, et al. DNA arrays to monitor gene expression in rat blood and uterus following 17beta-estradiol exposure: biomonitoring environmental effects using surrogate tissues. Toxicol Sci. 2002;69:49–59. doi: 10.1093/toxsci/69.1.49. [DOI] [PubMed] [Google Scholar]
- 41.Weston GC, et al. Microarray analysis of VEGF-responsive genes in myometrial endothelial cells. Mol Hum Reprod. 2002;8:855–863. doi: 10.1093/molehr/8.9.855. [DOI] [PubMed] [Google Scholar]
- 42.Dai D, et al. Molecular tools to reestablish progestin control of endometrial cancer cell proliferation. Am J Obstet Gynecol. 2001;184:790–797. doi: 10.1067/mob.2001.113844. [DOI] [PubMed] [Google Scholar]
- 43.Dai D, et al. Progesterone regulation of activating protein-1 transcriptional activity: a possible mechanism of progesterone inhibition of endometrial cancer cell growth. J Steroid Biochem Mol Biol. 2003;87:123–131. doi: 10.1016/j.jsbmb.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Davies S, et al. Identification of a novel mechanism of NF-kappaB inactivation by progesterone through progesterone receptors in Hec50co poorly differentiated endometrial cancer cells: induction of A20 and ABIN-2. Gynecol Oncol. 2004;94:463–470. doi: 10.1016/j.ygyno.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 45.Davies S, et al. Immunomodulatory and transcriptional effects of progesterone through progesterone A and B receptors in Hec50co poorly differentiated endometrial cancer cells. J Soc Gynecol Investig. 2004;11:494–499. doi: 10.1016/j.jsgi.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Conneely OM, Lydon JP. Progesterone receptors in reproduction: functional impact of the A and B isoforms. Steroids. 2000;65:571–577. doi: 10.1016/s0039-128x(00)00115-x. [DOI] [PubMed] [Google Scholar]
- 47.Conneely OM, et al. Reproductive functions of the progesterone receptor. J Soc Gynecol Investig. 2000;7:S25–32. doi: 10.1016/s1071-5576(99)00062-3. [DOI] [PubMed] [Google Scholar]
- 48.Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009;1179:167–178. doi: 10.1111/j.1749-6632.2009.04986.x. [DOI] [PubMed] [Google Scholar]
- 49.Lewis-Tuffin LJ, et al. Human glucocorticoid receptor {beta} binds RU-486 and is transcriptionally active. Mol Cell Biol. 2007;27:2266–2282. doi: 10.1128/MCB.01439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu NZ, Cidlowski JA. Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. 2006;16:301–307. doi: 10.1016/j.tcb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, et al. Progesterone inhibition of Wnt/beta-catenin signaling in normal endometrium and endometrial cancer. Clin Cancer Res. 2009;15:5784–5793. doi: 10.1158/1078-0432.CCR-09-0814. [DOI] [PubMed] [Google Scholar]
- 52.Ward EC, et al. The regulation and function of the Forkhead transcription factor Forkhead Box O1, is dependent on the progesterone receptor in endometrial carcinoma. Endocrinology. 2008;149:1942–1950. doi: 10.1210/en.2007-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watanabe J, et al. Significance of p27 as a predicting marker for medroxyprogesterone acetate therapy against endometrial endometrioid adenocarcinoma. Int J Gynecol Cancer. 2006;16:452–457. doi: 10.1111/j.1525-1438.2006.00418.x. [DOI] [PubMed] [Google Scholar]
- 54.Ohta K, et al. Glycodelin blocks progression to S phase and inhibits cell growth: a possible progesterone-induced regulator for endometrial epithelial cell growth. Mol Hum Reprod. 2008;14:17–22. doi: 10.1093/molehr/gam081. [DOI] [PubMed] [Google Scholar]
- 55.Lonard DM, et al. Nuclear receptor coregulators and human disease. Endocr Rev. 2007;28:575–587. doi: 10.1210/er.2007-0012. [DOI] [PubMed] [Google Scholar]
- 56.Witkiewicz AK, et al. Increased natural killer cells and decreased regulatory T cells are seen in complex atypical endometrial hyperplasia and well-differentiated carcinoma treated with progestins. Hum Pathol. 2010;41:26–32. doi: 10.1016/j.humpath.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 57.van den Heuvel MJ, et al. A review of trafficking and activation of uterine natural killer cells. Am J Reprod Immunol. 2005;54:322–331. doi: 10.1111/j.1600-0897.2005.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leslie KK, et al. Progesterone receptor isoform identification and subcellular localization in endometrial cancer. Gynecol Oncol. 2005;96:32–41. doi: 10.1016/j.ygyno.2004.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singh M, et al. Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: a Gynecologic Oncology Group Study. Gynecol Oncol. 2007;106:325–333. doi: 10.1016/j.ygyno.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 60.Ren Y, et al. Down-regulation of the progesterone receptor by the methylation of progesterone receptor gene in endometrial cancer cells. Cancer Genet Cytogenet. 2007;175:107–116. doi: 10.1016/j.cancergencyto.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 61.Zhou XC, et al. Epigenetic considerations for endometrial cancer prevention, diagnosis and treatment. Gynecol Oncol. 2007;107:143–153. doi: 10.1016/j.ygyno.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 62.Xiong Y, et al. Epigenetic-mediated upregulation of progesterone receptor B gene in endometrial cancer cell lines. Gynecol Oncol. 2005;99:135–141. doi: 10.1016/j.ygyno.2005.05.035. [DOI] [PubMed] [Google Scholar]
- 63.Tessel MA, et al. Steroid receptor and microRNA regulation in cancer. Curr Opin Oncol. 2010;22:592–597. doi: 10.1097/CCO.0b013e32833ea80c. [DOI] [PubMed] [Google Scholar]
- 64.Abdel-Hafiz H, et al. Mechanisms underlying the control of progesterone receptor transcriptional activity by SUMOylation. J Biol Chem. 2009;284:9099–9108. doi: 10.1074/jbc.M805226200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones MC, et al. Regulation of the SUMO pathway sensitizes differentiating human endometrial stromal cells to progesterone. Proc Natl Acad Sci USA. 2006;103:16272–16277. doi: 10.1073/pnas.0603002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lange CA, et al. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiu M, et al. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol Endocrinol. 2003;17:628–642. doi: 10.1210/me.2002-0378. [DOI] [PubMed] [Google Scholar]
- 68.Sasaki M, et al. Progesterone receptor B gene inactivation and CpG hypermethylation in human uterine endometrial cancer. Cancer Res. 2001;61:97–102. [PubMed] [Google Scholar]
- 69.Iorio MV, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 70.Lowery A, et al. MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res. 2009;11:R27. doi: 10.1186/bcr2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maillot G, et al. Widespread estrogen-dependent repression of microRNAs involved in breast tumor cell growth. Cancer Res. 2009;69:8332–8340. doi: 10.1158/0008-5472.CAN-09-2206. [DOI] [PubMed] [Google Scholar]
- 72.Abdel-Hafiz H, et al. The inhibitory function in human progesterone receptor N termini binds SUMO-1 protein to regulate autoinhibition and transrepression. J Biol Chem. 2002;277:33950–33956. doi: 10.1074/jbc.M204573200. [DOI] [PubMed] [Google Scholar]
- 73.Kim JJ, Chapman-Davis E. Role of progesterone in endometrial cancer. Semin Reprod Med. 2010;28:081, 090. doi: 10.1055/s-0029-1242998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ehrlich CE, et al. Steroid receptors and clinical outcome in patients with adenocarcinoma of the endometrium. Am J Obstet Gynecol. 1988;158:796–807. doi: 10.1016/0002-9378(88)90075-0. [DOI] [PubMed] [Google Scholar]
- 75.Satyaswaroop PG, et al. Apparent resistance in human endometrial carcinoma during combination treatment with tamoxifen and progestin may result from desensitization following downregulation of tumor progesterone receptor. Cancer Lett. 1992;62:107–114. doi: 10.1016/0304-3835(92)90180-4. [DOI] [PubMed] [Google Scholar]
- 76.Mortel R, et al. Designing a schedule of progestin administration in the control of endometrial carcinoma growth in the nude mouse model. Am J Obstet Gynecol. 1990;162:928–934. doi: 10.1016/0002-9378(90)91293-l. discussion 934–926. [DOI] [PubMed] [Google Scholar]
- 77.Whitney CW, et al. Phase II study of medroxyprogesterone acetate plus tamoxifen in advanced endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2004;92:4–9. doi: 10.1016/j.ygyno.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 78.Chaudhry P, Asselin E. Resistance to chemotherapy and hormone therapy in endometrial cancer. Endocr Relat Cancer. 2009;16:363–380. doi: 10.1677/ERC-08-0266. [DOI] [PubMed] [Google Scholar]
- 79.Samarnthai N, et al. Molecular profiling of endometrial malignancies. Obstet Gynecol Int. 2010;2010:162363. doi: 10.1155/2010/162363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thigpen JT, et al. Oral medroxyprogesterone acetate in the treatment of advanced or recurrent endometrial carcinoma: a dose-response study by the Gynecologic Oncology Group. J Clin Oncol. 1999;17:1736–1744. doi: 10.1200/JCO.1999.17.6.1736. [DOI] [PubMed] [Google Scholar]
- 81.Fang Z, et al. Intact progesterone receptors are essential to counteract the proliferative effect of estradiol in a genetically engineered mouse model of endometriosis. Fertil Steril. 2004;82:673–678. doi: 10.1016/j.fertnstert.2004.01.048. [DOI] [PubMed] [Google Scholar]
- 82.Stambolic V, et al. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in Pten+/− mice. Cancer Res. 2000;60:3605–3611. [PubMed] [Google Scholar]
- 83.Vollmer G. Endometrial cancer: experimental models useful for studies on molecular aspects of endometrial cancer and carcinogenesis. Endocr Relat Cancer. 2003;10:23–42. doi: 10.1677/erc.0.0100023. [DOI] [PubMed] [Google Scholar]
- 84.Fyles A, et al. Neither ovariectomy nor progestin treatment prevents endometrial neoplasia in Pten+/− mice. Gynecol Oncol. 2008;108:395–401. doi: 10.1016/j.ygyno.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 85.Chen ML, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006;20:1569–1574. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Daikoku T, et al. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008;68:5619–5627. doi: 10.1158/0008-5472.CAN-08-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang H, et al. DNA mismatch repair deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. Am J Pathol. 2002;160:1481–1486. doi: 10.1016/S0002-9440(10)62573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jeong JW, et al. Mig-6 modulates uterine steroid hormone responsiveness and exhibits altered expression in endometrial disease. Proc Natl Acad Sci USA. 2009;106:8677–8682. doi: 10.1073/pnas.0903632106. [DOI] [PMC free article] [PubMed] [Google Scholar]