

Abstract
Purpose of review
The phospholipase A2 (PLA2) family of proteins includes lipolytic enzymes that liberate the sn-2 fatty acyl chains from phospholipids to yield non-esterified fatty acids and lysophospholipids. The purpose of this review is to discuss recent findings showing distinct roles of several of these PLA2 enzymes in inflammatory metabolic diseases such as diabetes and atherosclerosis.
Recent findings
The Group 1B PLA2 (PLA2G1B) digestion of phospholipids in the intestinal lumen facilitates postprandial lysophospholipid absorption, which suppresses hepatic fatty acid oxidation leading to increased VLDL synthesis, decreased glucose tolerance, and promotion of tissue lipid deposition to accentuate diet-induced obesity, diabetes, and hyperlipidemia. Other secretory PLA2s promote inflammatory metabolic diseases by generating bioactive lipid metabolites to induce inflammatory cytokine production, whereas the major intracellular PLA2s, cPLA2α and iPLA2, generate arachidonic acid and lysophosphatic acid in response to extracellular stimuli to stimulate leukocyte chemotactic response.
Summary
Each member of the phospholipase A2 family of enzymes serves a distinct role in generating active lipid metabolites that promote inflammatory metabolic diseases including atherosclerosis, hyperlipidemia, obesity, and diabetes. The development of specific drugs that target one or more of these PLA2 enzymes may be novel strategies for treatment of these chronic inflammatory metabolic disorders.
Keywords: Obesity, diabetes, hyperlipidemia, atherosclerosis, inflammation, lysophospholipid
Introduction
The phospholipase A2 (PLA2) family of proteins includes lipolytic enzymes that catalyze the hydrolysis of the ester bond at the sn-2 position of phospholipids to yield free fatty acids and lysophospholipids. Currently, these enzymes are classified into 5 major groups with many subgroups depending on their functions and cellular locations. The secreted PLA2 enzymes are small secreted proteins of 14–18 kDa. The lipoprotein-associated PLA2 is also a secreted enzyme but it has platelet-activating factor acetylhydrolase activity and is associated with plasma lipoproteins. The other 3 groups of PLA2 enzymes are located intracellularly: the cytosolic PLA2s (molecular sizes of 61–114 kDa) that are calcium-dependent, the calcium-independent PLA2s (85 to 88-kDa) that are also present in the cytosol, and the lysosomal PLA2 that functions exclusively in the lysosomes with optimal activity at lower pH. Research over the past decade using genetically-modified mouse models and association studies in humans have indicated distinct roles of each PLA2 in biological processes such as digestion, tissue injury/repair, host defense, inflammation, atherosclerosis, and obesity/diabetes. This review will focus primarily on the recently discovered role of the group 1B phospholipase A2 (PLA2G1B) in obesity/diabetes, hyperlipidemia, and atherosclerosis. The influence of other PLA2s in these metabolic disorders will be discussed briefly for comparation. Readers interested in these PLA2 enzymes are referred to several excellent recent review articles for more details [1–4].
Group 1B Phospholipase A2
The PLA2G1B is a secreted phospholipase expressed primarily in the acinar cells of the pancreas and to lesser extent also in the lung and islet β-cells. In pancreatic acinar cells, PLA2G1B is found within zymogen granules along with other digestive enzymes. Upon meal feeding, contents within the zymogen granules are released into pancreatic juice and transported to the intestinal lumen where the enzymes can mediate nutrient digestion and absorption. Based on its enzymatic activity in hydrolyzing phospholipids, the role of PLA2G1B in the digestive tract was presumed to be the hydrolysis of phospholipids, thereby contributing to the necessary remodeling of lipid particles in the intestinal lumen prior to lipid nutrient absorption. However, knockout mouse studies revealed that PLA2G1B is not the only phospholipid hydrolytic enzyme in the digestive tract, and other phospholipases in the intestine can compensate for the lack of this enzyme in catalyzing phospholipid digestion and intestinal lipid absorption in Pla2g1b-null mice.
Genome-wide association studies have identified polymorphisms in the PLA2G1B gene as a determinant of central obesity in humans [5]. The association between PLA2G1B gene polymorphism and obesity in humans can be recapitulated in genetically-modified mouse models, with Pla2g1b-null mice displaying resistance to diet-induced obesity, hyperglycemia, and hyperinsulinemia whereas transgenic over-expression of the Pla2g1b gene in pancreas accentuates these metabolic diseases [6, 7]. The differences in sensitivity to diet-induced metabolic diseases between wild type Pla2g1b+/+ mice with Pla2g1b−/− and Pla2g1b-transgenic mice are not due to differences in food intake. Although the most straight forward explanation for their metabolic differences is that PLA2G1B may be require for complete phospholipid hydrolysis when lipid input into the intestinal lumen exceeds the capacity of the compensatory mechanism, similar amount of fat was found to be absorbed between these animals when they were fed the high fat diet. Additionally, the fat-fed Pla2g1b−/− mice absorbed more fat than chow-fed Pla2g1b+/+ mice yet their body weight gains were similar [6]. The Pla2g1b+/+ and Pla2g1b−/− mice also displayed similar fat absorption efficiency when maintained on a low-fat chow diet, yet the Pla2g1b−/− mice displayed increased glucose tolerance and insulin sensitivity compared to their wild type counterparts under these conditions [6]. These results indicated that PLA2G1B has a direct role in regulating adiposity, glucose metabolism, and insulin sensitivity, and this unique property of PLA2G1B is not shared by other phospholipases in the intestinal tract.
One difference in intestinal phospholipid digestion between Pla2g1b+/+ and Pla2g1b−/− mice is the lipolytic products formed and absorbed after meal consumption. The Pla2g1b+/+ mice absorbed significantly more lysophospholipids into the liver and circulation compared with Pla2g1b−/− mice, but the absorption of phospholipid-derived fatty acids and phosphatidylcholine- associated choline was found to be similar between Pla2g1b+/+ and Pla2g1b−/− mice [8]. These results suggested that the compensatory enzyme for phospholipid digestion in the intestinal lumen in the absence of PLA2G1B is not specific for the ester bond at the sn-2 position of phospholipids, but can release both sn-1 and sn-2 fatty acyl chains from phospholipids to yield 2 nonesterified fatty acids and glycerophosphocholine for absorption by enterocytes. In contrast, luminal digestion of diet- and/or biliary-derived phospholipids by PLA2G1B yields nonesterified fatty acids and lysophospholipids that can be rapidly absorbed by intestinal cells.
Differences in lysophospholipid absorption accounted for the differences between Pla2g1b+/+, Pla2g1b−/−, and Pla2g1b-transgenic mice in their sensitivity to diet-induced metabolic diseases. Postprandial lysophospholipid levels were significantly lower in Pla2g1b−/− mice and higher in Pla2g1b-transgenic mice compared to those observed in wild type Pla2g1b+/+ mice. The infusion of lysophospholipids into Pla2g1b−/− mice to levels similar to the levels observed during the postprandial state of Pla2g1b+/+ mice abolished the protective effects of Pla2g1b gene deletion against exaggerated postprandial insulin resistance and hyperglycemia [8]. Lysophospholipids promote postprandial hyperglycemia by reducing glucose uptake by the liver, heart, and muscle tissues, as well as suppressing insulin-stimulated glycogen synthesis in the liver [8]. Lysophospholipids administration also ameliorated the enhanced hepatic fatty acid oxidation observed in Pla2g1b−/− mice, thereby increasing substrate availability for deposition into adipose tissues to promote obesity [9]. Lysophospholipids reduce fatty acid oxidation by inhibiting the expression of peroxisome proliferator-activated receptor (PPAR)-α, -δ, and –γ genes as well as genes for proteins responsible for fatty acid transport and utilization such as CD36 and UCP2 [9].
The reduced hepatic fatty acid oxidation due to PLA2G1B-mediated lysophospholipid absorption also increases fatty acid substrates available for triglyceride biosynthesis [10]. Additionally, lysophospholipids are also efficiently re-acylated to phospholipids as well as promote triglyceride synthesis [11]. Lysophospholipids also prevent premature intracellular degradation of apoB in the liver [12]. Therefore, PLA2G1B-mediated lysophospholipid absorption promotes hyperlipidemia by accentuating hepatic VLDL synthesis and secretion after fat feeding [10]. Moreover, PLA2G1B gene inactivation has also been reported to improve plasma clearance of triglyceride-rich lipoproteins in high fat-fed mice [10], suggesting that lysophospholipids derived from PLA2G1B-mediated phospholipid hydrolysis in the digestive tract may also suppress the expression of LDL receptor and/or LDL receptor-related proteins in the liver. Taken together, these results indicate that the underlying mechanism by which PLA2G1B gene inactivation protects against diet-induced hyperlipidemia is also mediated via reducing lysophospholipid absorption after fat feeding, thereby ameliorating diet-induced postprandial VLDL synthesis and increasing plasma clearance of triglyceride-rich lipoproteins. The lower plasma lipid levels in Pla2g1b−/− mice are likely to reduce lipid deposition to adipose tissues, skeletal muscle, and the vessel wall, thereby may also indirectly suppress diet-induced obesity, insulin resistance, and atherosclerosis through this mechanism. A schematic diagram depicting the model of how PLA2G1B influences lipid metabolism and related metabolic diseases is shown in Figure 1.
Figure 1. Schematic diagram depicting pathway underlying group 1B phospholipase A2 inhibition of fatty acid oxidation and promotion of metabolic diseases.
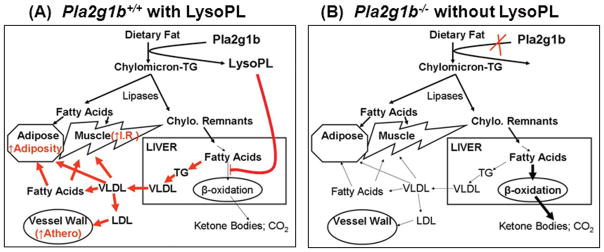
Panel A shows pathway in wild type Pla2g1b+/+ mice, where Pla2g1b-catalyzed hydrolysis of dietary fat and phospholipids generates lysophospholipids (LysoPL) that inhibits fatty acid oxidation in the liver. The increased fatty acid availability in the liver promotes triglyceride (TG) synthesis, leading to elevated VLDL secretion into the plasma circulation. Fatty acids generated directly from lipase-catalyzed hydrolysis of chylomicron-TG as well as from VLDL hydrolysis are deposited in adipose tissues to promote adiposity as well as transported to skeletal muscle to promote insulin resistance (I.R.). The LDL generated from increased lipoprotein secretion can also be accumulating in the vessel wall to promote atherosclerosis. Panel B shows pathway in Pla2g1b−/− mice, where reduced LysoPL absorption due to Pla2g1b inactivation results in increased hepatic fatty acid oxidation (bold arrow) and reduced TG synthesis and VLDL secretion, ultimately leading to reduced fatty acid deposition in adipose tissue and skeletal muscle and decreased LDL accumulation in the vessel wall.
The resistance of Pla2g1b gene inactivated mice to diet-induced obesity, diabetes, and hyperlipidemia suggests that the PLA2G1B protein may be a novel therapeutic target for treatment of these metabolic disorders. This possibility has been explored in a pre-clinical model testing the efficacy of the PLA2G1B inhibitor methyl indoxam to suppress body weight gain and hyperglycemia in response to a high fat/high carbohydrate diet in mice. When mixed with food and administered orally, methyl indoxam did not alter food intake or lipid nutrient absorption but significantly reduced postprandial lysophospholipid levels in a concentration-dependent manner, indicating the suppression of PLA2G1B activity in vivo [13]. Importantly, oral methyl indoxam administration reduced high fat/high carbohydrate diet-induced body weight gain in a dose-dependent manner [13]. Fasting blood glucose levels and glucose tolerance in high fat/high carbohydrate-fed mice were also dramatically improved with methyl indoxam treatment, to levels similar to those observed in mice fed a low-fat control diet [13]. Oral methyl indoxam administration had no effect on body weight and blood glucose levels in chow-fed animals, thus indicating that the reduced body weight gain and improved glucose tolerance with methyl indoxam treatment were not due to adverse effects but a direct consequence of suppressing PLA2G1B activity in vivo. Finally, whereas 10 weeks of feeding the high fat/high carbohydrate diet increased fasting plasma cholesterol levels in wild type C57BL/6 mice, the inclusion of methyl indoxam in the high fat/high carbohydrate meal suppressed diet-induced hypercholesterolemia, reducing plasma cholesterol levels to levels observed in chow-fed wild type mice or in Pla2g1b−/− mice fed either chow or high fat/high carbohydrate diet (Figure 2).
Figure 2. PLA2G1B inactivation protects against diet-induced hypercholesterolemia.

Fasting plasma cholesterol levels were measured from wild type (WT) Pla2g1b+/+ mice and Pla2g1b−/− (PLA2G1B KO) mice before (solid bars) and 10 weeks after feeding a high fat/high carbohydrate diet (open bars). Results showed elevation of plasma cholesterol levels in wild type mice after high fat/high carbohydrate feeding, and the diet-induced hypercholesterolemia was not observed in WT mice when PLA2G1B activity was inhibited with oral methyl indoxam administration or in mice with Pla2g1b gene inactivation.
The effectiveness of oral methyl indoxam administration to reduce diet-induced obesity, hyperglycemia, and hypercholesterolemia in mice is consistent with the possibility that PLA2G1B inhibition may be a potentially effective oral therapeutic option for treatment of metabolic diseases. However, it is important to note that in vitro experiments have documented that methyl indoxam is a general inhibitor capable of inhibiting the activities of other secretory phospholipase A2 enzymes in addition to PLA2G1B [14]. Although cell culture studies failed to detect methyl indoxam transport from the apical to the basolateral side of intestinal cells [13, 15], thus suggesting that methyl indoxam protection against diet-induced metabolic diseases through direct inhibition of PLA2G1B in the intestinal lumen, in vivo measurements revealed limited but significant bioavailability of orally fed methyl indoxam [13]. Whether a non-absorbable PLA2G1B inhibitor, which presumably would be more advantageous with potentially less systemic side effects, can be an effective option, or if inhibition of other phospholipase A2 enzymes in addition to PLA2G1B inhibition is necessary to reduce diet-induced obesity, diabetes, and hypercholesterolemia remains to be determined.
Lipoprotein-Associated Phospholipase A2
The lipoprotein-associated PLA2 (Lp-PLA2) is also a secreted protein and is made by lymphocytes, macrophages and mast cells. In addition to the hydrolysis of phospholipids, Lp-PLA2 is also capable of degrading biologically active oxidized phospholipids (oxPLs) and platelet activating factor. Studies exploring the role of Lp-PLA2 in metabolic diseases have focused primarily on atherosclerosis. Nevertheless, the role of Lp-PLA2 in atherosclerosis has not been resolved completely. The hydrolysis of oxPLs by Lp-PLA2 generates lysophospholipids and oxidized fatty acids, which are pro-inflammation and promote atherosclerosis. The oxPLs have also been reported to have anti-inflammatory properties and function in a negative feedback manner to blunt macrophage inflammatory response [16]. These observations suggest that Lp-PLA2 is pro-atherogenic by increasing inflammatory response as well as reducing the ability to tempered inflammation in response to vascular insults. Surprisingly, adenovirus-mediated transfer of the Lp-PLA2 gene suppressed injury-induced neointimal hyperplasia and reduced spontaneous atherosclerosis in apoE−/− mice [17], suggesting that Lp-PLA2 is atheroprotective in mice. However, a recent study reported that pharmacological inhibition of Lp-PLA2 activity also ameliorated inflammation and atherosclerotic plaque formation in apoE−/− mice [18]. The differences between these two studies have not been resolved. Thus, the role of Lp-PLA2 in atherosclerosis development in mice remains unclear. Mallat and colleagues suggested that the role of Lp-PLA2 in atherosclerosis may be different between humans and mice because the enzyme is associated with HDL in mice and with LDL in humans [1]. In humans, high Lp-PLA2 levels and activities are associated with coronary heart disease and inflammation [19]. Reducing circulating Lp-PLA2 levels in patients with acute coronary syndrome was associated with plaque regression [20]. Plasma Lp-PLA2 levels are also associated with high waist-hip ratio, plasma triglyceride- and LDL-cholesterol levels, and Lp-PLA2 levels are higher in subjects with metabolic syndrome than in normal subjects [21]. Taken together, these results indicate that Lp-PLA2 promotes metabolic diseases in humans, suggesting that Lp-PLA2 may also be a target for intervention. Pre-clinical and clinical testing of efficacy of the Lp-PLA2-specific inhibitor darapladib in reducing cardiovascular and metabolic diseases are currently underway [22–24].
Other Secreted Phospholipase A2 Enzymes
Other secreted PLA2s are similar to PLA2G1B and Lp-PLA2 in the production of pro-inflammatory mediators such as non-esterified fatty acids and lysophospholipids. The group IIa PLAs (PLA2G2A) is the most abundant phospholipase A2 enzyme in human serum. The most commonly used mouse strain for study cardiovascular and metabolic diseases, C57BL/6 mouse is a natural mutant of PLA2G2A. Over-expression of the human PLA2G2A gene increases atherosclerosis in wild type and LDL receptor-deficient C57BL/6 mice [25, 26], indicating that PLA2G2A is pro-atherogenic. This enzyme accelerates atherosclerosis through multiple mechanisms including: (i) the hydrolysis of LDL-associated phospholipids to promote remodeling of LDL to smaller and denser LDL particles with increased retention to matrix proteoglycans at the vessel wall [27], (ii) induction of LDL oxidation and cellular oxidative stress [28], and (iii) direct binding to sPLA2 receptors on cell surfaces to promote inflammatory cytokine secretion [29]. In the vessel wall, PLA2G2A can be localized to atherosclerotic lesion areas but the enzyme is not present in normal arteries [30]. The expression of PLA2G2A by vascular smooth muscle cells is induced during inflammation [31, 32]. Tissue inflammation also elevates PLA2G2A expression in adipose tissues [32], suggesting that PLA2G2A not only contributes to atherosclerosis but may also be a contributing factor in other inflammatory metabolic diseases such as diabetes. The potential of sPLA2 inhibitors for treatment of both of these metabolic disorders has been suggested in a recent review article [23].
Intracellular Phospholipase A2 Enzymes
The two most prominent intracellular PLA2 enzymes are the calcium-dependent cPLA2α (PLA2G4A) and the calcium-independent iPLA2 (PLA2G6). Both enzymes are important inflammatory mediators, responding to extracellular signaling to provide distinct lipid mediators at different intracellular sites that are required for leukocyte chemotaxis [4]. The cPLA2α enzyme is selective for liberating sn-2 arachidonic acids from phospholipids, thereby is essential for signal-activated generation of arachidonic acid-derived lipid metabolites during cell activation. In contrast, iPLA2 lacks acyl chain specificity but prefers phosphatidic acid instead of phospholipid substrates. Both cPLA2α-generated arachidonic acid and iPLA2-mediated lysophosphatic acid production are required for monocyte chemotaxis to monocyte chemoattractant protein-1. Thus, activation of these intracellular PLA2s by extracellular stimulation is also pro-inflammatory, and potentially contributory to both atherosclerosis and diabetes. Although these results suggest intracellular PLA2 inhibition may be another approach for treatment of these inflammatory metabolic diseases, a recent report showing cPLA2 inhibition increases CD36 expression and foam cell formation in a macrophage cell line raises caution to this strategy [33].
Conclusion
Depending on the anatomic site of expression, each member of the phospholipase A2 family of enzymes serves a distinct role in normal physiology in response to meal consumption and infection, but also generates active lipid metabolites that promote inflammatory metabolic diseases including atherosclerosis, hyperlipidemia, obesity, and diabetes. The PLA2G1B is expressed primarily in the pancreatic acinar cells and participates in remodeling of dietary lipids in the intestinal lumen prior to lipid nutrient absorption. However, PLA2G1B elevates lysopholipid levels postprandially leading to increase susceptibility of obesity, diabetes, and hyperlipidemia. The other secretory PLA2s generates active lipid metabolites that promote oxidative stress and inflammatory cytokine production, whereas the major intracellular PLA2 enzymes cPLA2α and iPLA2 are activated in response to extracellular stimuli in relaying signals for leukocyte chemotactic responses in accentuating inflammatory metabolic diseases. The development of specific drugs that target one or more of these PLA2 enzymes should be considered as novel strategies to treat chronic inflammatory metabolic diseases such as diabetes and atherosclerosis.
Key Points.
The group 1B PLA2 (PLA2G1B) promotes lysophospholipid absorption, which suppresses hepatic fatty acid oxidation and leading to promotion of obesity, diabetes, and hyperlipidemia.
Unique role of each phospholipase A2 in generating bioactive lipid metabolites that promote inflammation and inflammatory metabolic diseases.
Pharmacological inhibition of PLA2 enzyme activities are effective strategies to reduce diet-induced obesity, diabetes, hyperlipidemia, and atherosclerosis.
Acknowledgments
This work was supported by NIH grant RO1-DK069967.
Footnotes
Conflicts of interest
There is no conflict of interest.
Funding Disclosure: This work was supported by NIH grant RO1-DK069967.
References and recommended reading
- 1.Mallat Z, Lambeau G, Tedgui A. Lipoprotein-associated and secreted phospholipase A2 in cardiovascular disease. Roles as biological effectors and biomarkers. Circulation. 2010;122:2183–2200. doi: 10.1161/CIRCULATIONAHA.110.936393. [DOI] [PubMed] [Google Scholar]
- 2.Karabina S-A, Gora S, Atout R, Ninio E. Extracellular phospholipases in atherosclerosis. Biochimie. 2010;92:594–600. doi: 10.1016/j.biochi.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Murakami M, Taketomi Y, Sato H, Yamamoto K. Secreted phospholipase A2 revisited. J Biochem. 2011;150:233–255. doi: 10.1093/jb/mvr088. [DOI] [PubMed] [Google Scholar]
- 4.Cathcart MK. Signal-activated phospholipase regulation of leukocyte chemotaxis. J Lipid Res. 2009;50:S231–S236. doi: 10.1194/jlr.R800096-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson SG, Adam G, Langdown M, et al. Linkage and potential association of obesity-related phenotypes with two genes on chromosome 12q24 in a female dizygous twin cohort. Eur J Hum Genet. 2006;14:340–348. doi: 10.1038/sj.ejhg.5201551. [DOI] [PubMed] [Google Scholar]
- 6.Huggins KW, Boileau AC, Hui DY. Protection against diet-induced obesity and obesity-related insulin resistance in Group 1B PLA2-deficient mice. Am. J. Physiol. 2002;283:E994–E1001. doi: 10.1152/ajpendo.00110.2002. [DOI] [PubMed] [Google Scholar]
- 7.Cash JG, Kuhel DG, Goodin C, Hui DY. Pancreatic acinar cell-specific overexpression of group 1B phospholipase A2 exacerbates diet-induced obesity and insulin resistance in mice. Int J Obes. 2011;35:877–881. doi: 10.1038/ijo.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Labonte ED, Kirby RJ, Schildmeyer NM, et al. Group 1B phospholipase A2-mediated lysophospholipid absorption directly contributes to postprandial hyperglycemia. Diabetes. 2006;55:935–941. doi: 10.2337/diabetes.55.04.06.db05-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Labonte ED, Pfluger PT, Cash JG, et al. Postprandial lysophospholipid suppresses hepatic fatty acid oxidation: the molecular link between group 1B phospholipase A2 and diet-induced obesity. FASEB J. 2010;24:2516–2524. doi: 10.1096/fj.09-144436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollie NI, Hui DY. Group 1B phospholipase A2 deficiency protects against diet-induced hyperlipidemia in mice. J Lipid Res. 2011;52:2005–2011. doi: 10.1194/jlr.M019463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson BS, Yao Z, Baisted DJ, Vance DE. Lysophosphatidylcholine metabolism and lipoprotein secretion by cultured rat hepatocytes deficient in choline. Biochem J. 1989;260:207–214. doi: 10.1042/bj2600207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wanninger J, Neumeier M, Weigert J, et al. Metformin reduces cellular lysophosphatidylcholine and thereby may lower apolipoprotein b secretion in primary human hepatocytes. Biochim Biophys Acta. 2008;1781:321–325. doi: 10.1016/j.bbalip.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Hui DY, Cope MJ, Labonte ED, et al. The Phospholipase A2 inhibitor methyl indoxam suppresses diet-induced obesity and glucose intolerance in mice. Br J Pharmacol. 2009;157:1263–1269. doi: 10.1111/j.1476-5381.2009.00308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singer AG, Ghomashchi F, Le Calvez C, et al. Interfacial Kinetic and Binding Properties of the Complete Set of Human and Mouse Groups I, II, V, X, and XII Secreted Phospholipases A2. J Biol Chem. 2002;277:48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 15.Mounier CM, Ghomashchi F, Lindsay MR, et al. Arachidonic acid release from mammalian cells transfected with human Group IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2-alpha. J Biol Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 16.BockKov VN, Kadl A, Huber J, et al. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419:77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- 17.Quarck R, De Geest B, Stengel D, et al. Adenovirus-mediated gene transfer of human platelet-activating factor-acetylhydrolase prevents injury-induced neointima formation and reduces spontaneous atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;103:2495–2500. doi: 10.1161/01.cir.103.20.2495. [DOI] [PubMed] [Google Scholar]
- 18.Wang W-y, Zhang J, Wu W-y, et al. Inhibition of lipoprotein-associated phospholipase A2 ameliorates inflammation and decreases atherosclerotic plaque formation in apoE-deficient mice. PLoS One. 2011;6:e23425. doi: 10.1371/journal.pone.0023425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Packard CJ. Lipoprotein-associated phospholipase A2 as a biomarker of coronary heart disease and a therapeutic target. Curr Opin Cardiol. 2009;24:358–363. doi: 10.1097/HCO.0b013e32832bcb22. [DOI] [PubMed] [Google Scholar]
- 20.Dohi T, Miyauchi K, Okazaki S, et al. Decreased circulating lipoprotein-associated phospholipase A2 levels are associated with coronary plaque regression in patients with acute coronary syndrome. Atherosclerosis. 2011;219:907–912. doi: 10.1016/j.atherosclerosis.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 21.Noto H, Chitkara P, Raskin P. The role of lipoprotein-associated phospholipase A2 in the metabolic syndrome and diabetes. J Diabetes Complications. 2006;20:343–348. doi: 10.1016/j.jdiacomp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Wilensky RL, Shi Y, Mohler ER, et al. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat Med. 2008;14:1059–1066. doi: 10.1038/nm.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magrioti V, Kokotos G. Phospholipase A2 inhibitors as potential therapeutic agents for the treatment of inflammatory diseases. Expert Opin Ther Patents. 2009;20:1–18. doi: 10.1517/13543770903463905. [DOI] [PubMed] [Google Scholar]
- 24.Colley KJ, Wolfert RL, Cobble ME. Lipoprotein associated phospholipase A2: role in atherosclerosis and utility as a biomarker for cardiovascular risk. EPMA J. 2011;2:27–38. doi: 10.1007/s13167-011-0063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivandic B, Castellani LW, Wang X-P, et al. Role of Group II Secretory Phospholipase A2 in Atherosclerosis : 1. Increased Atherogenesis and Altered Lipoproteins in Transgenic Mice Expressing Group IIa Phospholipase A2. Arterioscler Thromb Vasc Biol. 1999;19:1284–1290. doi: 10.1161/01.atv.19.5.1284. [DOI] [PubMed] [Google Scholar]
- 26.Webb NR, Bostrom MA, Szilvassy SJ, et al. Macrophage-Expressed Group IIA Secretory Phospholipase A2 Increases Atherosclerotic Lesion Formation in LDL Receptor–Deficient Mice. Arterioscler Thromb Vasc Biol. 2003;23:263–268. doi: 10.1161/01.atv.0000051701.90972.e5. [DOI] [PubMed] [Google Scholar]
- 27.Hurt-Camejo E, Camejo G, Sartipy P. Phospholipase A2 and small dense low density lipoproteins. Curr Opin Lipidol. 2000;11:465–471. doi: 10.1097/00041433-200010000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Tietge UJF, Pratico D, Ding T, et al. Macrophage-specific expression of group IIA sPLA2 results in accelerated atherogenesis by increasing oxidative stress. J Lipid Res. 2005;46:1604–1614. doi: 10.1194/jlr.M400469-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Rouault M, Le Calvez C, Boilard E, et al. Recombinant Production and Properties of Binding of the Full Set of Mouse Secreted Phospholipases A2 to the Mouse M-Type Receptor. Biochemistry. 2007;46:1647–1662. doi: 10.1021/bi062119b. [DOI] [PubMed] [Google Scholar]
- 30.Bobryshev YV, Crozier JA, Lord RS, et al. Expression of Secretory Group II Phospholipase A2 by CD1a positive cells in human atherosclerotic plaques. Atherosclerosis. 1996;127:283–285. doi: 10.1016/s0021-9150(96)05946-1. [DOI] [PubMed] [Google Scholar]
- 31.Jaulmes A, Thierry S, Janvier B, et al. Activation of sPLA2-IIA and PGE2 production by high mobility group protein B1 in vascular smooth muscle cells sensitized by IL-1β. FASEB J. 2006;20:1727–1729. doi: 10.1096/fj.05-5514fje. [DOI] [PubMed] [Google Scholar]
- 32.Dutour A, Achard V, Sell H, et al. Secretory Type II Phospholipase A2 Is Produced and Secreted by Epicardial Adipose Tissue and Overexpressed in Patients with Coronary Artery Disease. Journal of Clinical Endocrinology & Metabolism. 2010;95:963–967. doi: 10.1210/jc.2009-1222. [DOI] [PubMed] [Google Scholar]
- 33.Anwar K, Voloshyna I, Littlefield MJ, et al. COX2 inhibition and inhibition of cytosolic phospholipase A2 increase CD36 expression and foam cell formation in THP-1 cells. Lipids. 2011;46:131–142. doi: 10.1007/s11745-010-3502-4. [DOI] [PubMed] [Google Scholar]