

Abstract
Purpose.
Uveitis is a common cause of vision loss. The renin angiotensin system (RAS), which plays a vital role in cardiovascular system, is a potent mediator of inflammation and has been implicated in the pathogenesis of uveitis. A newly identified axis of RAS, ACE2/Ang-(1-7)/Mas, has emerged as a novel target because it counteracts the deleterious effect of angiotensin II. The purpose of this study was to investigate the effect of endogenous ACE2 activation in preventing endotoxin-induced uveitis (EIU) in mice.
Methods.
ACE2 activator diminazene aceturate (DIZE) was administered both systemically and locally. For systemic administration, female BALB/c mice received intraperitoneal injection of DIZE (60 mg/kg body weight [BW]) for 2 days prior to lipopolysaccharide (LPS) intravitreal injection (125 ng) to induce uveitis. For local study, DIZE was given at 0.5, 0.1, and 0 mg/mL as eyedrops six times per day for 2 days before LPS injection. The anterior segment of the mice was examined at 12, 24, 48, and 72 hours after LPS injection, and clinical scores were determined at the same time. Morphology and infiltrating inflammatory cells were evaluated after 24 hours. The mRNA levels of inflammatory cytokines were analyzed by real-time RT-PCR. ACE2 activity was determined using a self-quenching fluorescent substrate.
Results.
At 24 hours, the clinical score of mice treated with DIZE systemically was significantly lower (mean, ∼1.75) than the saline vehicle group (mean, ∼4) (P < 0.001). Histological examination showed 63.4% reduction of infiltrating inflammatory cells in the anterior segment and 57.4% reduction in the posterior segment of DIZE-treated eyes. The number of CD45+ inflammatory cells in the vitreous of the DIZE-treated group was decreased (43.3%) compared to the vehicle group (P < 0.01). The mRNA levels of inflammatory cytokines were significantly reduced in the DIZE-treated group (P < 0.01, P < 0.001). The number of infiltrating inflammatory cells was also significantly reduced in eyes that received topical administration of DIZE: 73.8% reduction in the 0.5 mg/mL group and 51.7% reduction in the 0.1mg/mL group compared to the control group. DIZE treatment resulted in significantly increased ACE2 activity in the retina (P < 0.001).
Conclusions.
Endogenous ACE2 activation by DIZE has a preventive effect on LPS-induced ocular inflammation in the EIU mouse model. These results support the notions that RAS plays a role in modulating ocular immune response and that enhancing ACE2 provides a novel therapeutic strategy for uveitis.
Keywords: uveitis, EIU, angiotensin-converting enzyme 2, renin angiotensin system, diminazene aceturate
This study supports the notion that RAS plays a role in modulating ocular immune response and that enhancing ACE2 provides a novel therapeutic strategy for uveitis.
Introduction
Uveitis is a common inflammatory ocular disease that compromises the vision of patients. It is one of the major causes of visual impairment, even blindness, within the working population worldwide.1 There are diverse causes that could lead to uveitis, which may include trauma, infection, and autoimmune disease.2 Uveitis can act either as an isolated intraocular inflammation or as a part of a systemic autoimmune disease, such as ankylosing spondylitis, Behcet's disease, or Reiter's diesease.3,4 The inflammation may involve not only the anterior segment, but also the posterior segment of the eyes. The conventional treatment of uveitis includes corticosteroids and immunosuppressive agents, which are highly efficacious but can be associated with serious systemic side effects.5 Furthermore, long-term application of corticosteroids is associated with numerous adverse effects including cataract, glaucoma, and metabolic disorders.6
Endotoxin-induced uveitis (EIU) is an animal model of human uveitis, induced by either a single systemic or local injection of lipopolysaccharide (LPS),7 a component of the outer membranes of gram-negative bacteria. Both the anterior and posterior segments can be involved. The inflammation in the anterior segment is characterized by the breakdown of the blood–aqueous barrier and infiltration of leukocytes into the anterior chamber. The inflammatory cytokines such as monocyte chemotactic protein (MCP)-1, tumor necrosis factor (TNF)-α, and interleukin (IL)-6 contribute to the development of EIU.8–10 The inflammation in the posterior segment of the eye in EIU is characterized by the blood–retinal barrier breakdown; increased expression of adhesion molecules, mainly intercellular adhesion molecule (ICAM)-111; increased adhesion of leukocytes in the retinal vascular endothelium; and infiltration into the vitreous cavity.12,13
The renin–angiotensin system (RAS) is a hormone system that plays an essential role in modulating various physiological processes such as fluid homeostasis, renal and cardiovascular function. Angiotensin II (Ang II), the main effector molecule of RAS, is produced from the precursor angiotensinogen through sequential enzymatic cleavages by renin and angiotensin-converting enzyme (ACE).14 Ang II acts on two receptors, Ang II type 1 receptor (AT1R) and AT2R,15 that belong to the superfamily of seven trans-membrane G-protein–coupled receptors, and it mediates diverse physiologic responses beyond renal and cardiovascular actions.16 Moreover, Ang II is also a potent peptide hormone that stimulates proinflammatory response,17 activates immune cells, and promotes cell infiltration into target organs.18 AT1R blockers have been shown to attenuate inflammation by suppressing the gene expression of inflammatory cytokines and subsequent macrophage infiltration in the EIU model.19 ACE inhibitor was also shown to suppress inflammation by decreasing the Ang II-induced overexpression of endothelial-derived adhesion molecules and chemokines as well as macrophage infiltration.20
Since the discovery of ACE2, which cleaves Ang II to produce Angiotensin-(1-7) [Ang-(1-7)],21 overwhelming evidence has established the existence of a protective axis of RAS, the ACE2/Ang-(1-7)/Mas axis. This axis, mediated via activation of the Mas receptor by Ang-(1-7), not only directly counter-regulates the deleterious effects of Ang II, but also has a broad range of beneficial effects including improving pathological conditions such as fibrosis and inflammation.22,23 Existing evidence from our group and others suggests that hyperactivity of the deleterious axis (ACE/Ang II/AT1R) of RAS plays an important role in retinal pathophysiology and increased expression of ACE2 or Ang-(1-7), two key members of the vasoprotective axis, via adeno-associated virus–mediated gene delivery to the retina diminishes diabetes-induced retinal inflammation and vascular pathology.24 This evidence provides a proof-of-principle that enhancing the ACE2/Ang-(1-7)/Mas axis may be a promising approach for ocular diseases in which RAS hyperactivity is implicated.
Recently, diminazene aceturate (DIZE) was identified as an activator of ACE2,25 thus a potential enhancer of ACE2/Ang-(1-7)/Mas, the protective axis of RAS. It has been demonstrated that DIZE has protective effects under various pathologic conditions including pulmonary hypertension,26 ischemic stroke,27 and glaucoma.28 We hypothesized that DIZE, as an activator of ACE2, can attenuate the inflammatory response in the EIU mouse model by activating the ACE2/Ang-(1-7)/Mas pathway. In this study, we tested both systemic and topical treatment of DIZE, and we showed that pretreatment with DIZE administered either systemically or topically reduced ocular inflammation in the EIU mouse model.
Materials and Methods
Animals and Experimental Procedures
Female BALB/c mice at the age of 6 to 8 weeks were purchased from Jackson Laboratories (Bar Harbor, ME, USA). All animal procedures were carried out according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The protocol was approved by the Animal Care and Use Committee of the University of Florida. All animals were housed at the Animal Care Service at the University of Florida with a 12/12-hour light/dark cycle. For systemic administration of DIZE, BALB/c mice were divided into four groups: (1) DIZE + LPS group, in which mice were treated with DIZE, then injected with LPS; (2) saline + LPS group, in which mice were treated with vehicle then injected with LPS; (3) untreated group, in which normal mice received no treatment; and (4) DIZE group, in which mice were treated with DIZE alone. Mice received daily intraperitoneal (IP) injection of DIZE (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) at the daily dosage of 60 mg/kg body weight (BW) (dissolved in sterile saline) for 2 days until the injection of LPS. Age-matched normal mice treated with saline and untreated mice were used as controls. The choice for using BALB/c mice in our study was based on previous evidence that EIU has been successfully induced by intravitreal injection of LPS in this strain.29 To induce EIU, each mouse eye received a single intravitreal injection of LPS according to previous study, with some modification based on our preliminary experiment.29 For induction of EIU, we tested different doses of LPS from 15 to 250 ng per eye. The LPS concentration that induced robust infiltrating inflammatory cells without too much damage to the retina was chosen in our study. For systemic study, each mouse received a single intravitreal injection of LPS (125 ng in 1 μL PBS; Sigma-Aldrich, St. Louis, MO, USA) 30 minutes after the IP injection of DIZE or saline on the second day.
For local administration, DIZE was given as eyedrops to BALB/c mice in two concentrations, 0.5 and 0.1 mg/mL in diluted artificial tears. Each concentration was given six times per day for 2 days. The control group was given diluted artificial tears without DIZE. To induce EIU, each mouse eye received a single intravitreal injection of 15 ng LPS bilaterally 30 minutes after the sixth treatment of eyedrops on the second day.
Inflammation Evaluation in the Anterior Chamber
Inflammation in the anterior chamber was evaluated by slit lamp observation at 12, 24, 48, and 72 hours after LPS administration (n = 7 per group). The clinical severity of ocular inflammation was graded by two independent observers according to the criteria described previously,8 with some modification (Table 1).
Table 1.
Criteria of EIU Clinical Scoring in BALB/c Mice
| Clinical signs |
Grade of Signs |
Score |
| Iris hyperemia | Absent | 0 |
| Mild | 1 | |
| Moderate | 2 | |
| Severe | 3 | |
| Exudate in anterior chamber | Absent | 0 |
| Small | 1 | |
| Large | 2 | |
| Hypopyon | Absent | 0 |
| Present | 1 | |
| Pupil | Normal | 0 |
| Miosed | 1 | |
| Maximum possible score | 7 |
Histopathology
Mice were euthanized 24 hours after LPS-induced uveitis. Eyes were enucleated and fixed in 4% paraformaldehyde at 4°C overnight. Eyeballs were embedded in paraffin. Serial 4-μm sections were cut through the cornea–optic nerve axis and stained by hematoxylin and eosin. For histopathologic evaluation, the anterior chamber, vitreous, and retina were examined under light microscopy (n = 6 per group).
Immunohistochemistry
Mice were euthanized 24 hours after LPS administration. The eyes were enucleated immediately and fixed with 4% paraformaldehyde at 4°C overnight. Eyeballs were cryoprotected in 30% sucrose and embedded in OCT compound (Sakura Finetek, Inc., Torrance, CA, USA). Frozen sections were cut at 14 μm and mounted on slides. Sections were permeated with 1% Triton X100 made in PBS for 10 minutes, and blocked with 5% BSA for 30 minutes at room temperature. Sections were incubated with FITC-conjugated anti-mouse CD45 antibody (BD Biosciences, San Jose, CA, USA) diluted 1:200 in 1% BSA at 4°C overnight, followed by incubation with anti-FITC-alkaline phosphatase–conjugated secondary antibody (1:200; Sigma-Aldrich) at room temperature for 2 to 3 hours, and detected by NBT/BCIP substrate (Roche Applied Science, Indianapolis, IN, USA) in the presence of levamisole solution (Vector Laboratories, Inc., Burlingame, CA, USA) to block endogenous alkaline phosphatase activity (n = 6 per group).
Real-Time RT-PCR Analysis of Inflammatory Cytokines
Total RNA was extracted from the freshly enucleated eyeball by using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Complementary DNA (cDNA) was generated using PrimeScript RT reagent Kit (Takara Biotechnology, Dalian, China). Real-time PCR was performed according to manufacturer's instruction with Bio-Rad C-1000 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA). Each reaction was run in duplicate, and relative quantification was achieved by the comparative 2−ΔΔCt method. The relative fold changes of mRNA of target gene X in the experimental group (EG) was calculated using the control group as the calibrator, ΔΔ Ct = (Ct.X [EG] − Ct. GAPDH [EG]) − (Ct.X[control] − Ct. GAPDH [control]), and primers specific for mouse ICAM-1 (primer ID Mm-QRP-20301), MCP-1 (primer ID Mm-QRP-20287), TNF-α (primer ID Mm-QRP-20147), and IL-6 (primer ID Mm-QRP-20026), standardized to the housekeeping gene GAPDH (primer ID Mm-QRP-20043) (GeneCopoeia Inc., Rockville, MD, USA). The sequences and accession numbers of the primers are shown in Table 2. The specificity of all primers was verified by GeneCopoeia Inc. Real-time PCR was performed in a volume of 20 μL by using all-in-one qPCR Mix (GeneCopoeia Inc.). The conditions were 95°C for 10 minutes, followed by 40 cycles of 10 seconds at 95°C, 20 seconds at 60°C, and 15 seconds at 72°C. Fluorescence data were acquired at 72°C to 95°C to decrease the amount of nonspecific signal, and amplification of specific transcripts was confirmed by melting curve profiles at the end of each PCR (n = 4 per group).
Table 2.
Sequences and Accession Numbers of Primers for Real-Time RT-PCR
| Gene |
Forward |
Reverse |
Accession No. |
| ICAM-1 | 5′-AGATGACCTGCAGACGGAAG-3′ | 5′-GGCTGAGGGTAAATGCTGTC-3′ | NM_010493.2 |
| MCP-1 | 5′-CCCCACTCACCTGCTGCTACT-3′ | 5′-GGCATCACAGTCCGAGTCACA-3 | NM_011333 |
| TNF-α | 5′-AGGCGCCACATCTCCCTCCA-3′ | 5′-CGGTGTGGGTGAGGAGCACG-3′ | NM_013693.2 |
| IL-6 | 5′-TCGGCAAACCTAGTGCGTTA-3′ | 5′-CCAAGAAACCATCTGGCTAGG-3′ | NM_031168 |
| GAPDH | 5′-ATGGTGAAGGTCGGTGTGAAC-3′ | 5′-TTACTCCTTGGAAG-3′ | NM_008084 |
ACE2 Activity Assay
Retinas in each group were dissected 24 hours after LPS administration. All the retinas were homogenized with sonication in ACE2 assay buffer (75 mM Tris, 1 M NaCl, 0.5 μM ZnCl2, pH 7.5), and the homogenates were centrifuged at 13,000 rpm for 10 minutes at 4°C. The protein concentration in the supernatant was determined using BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific, Inc., Rockford, IL, USA). ACE2 enzymatic activity assay was performed in black 96-well plates with 50 μM ACE2-specific fluorogenic peptide substrate (R&D Systems, Inc., Minneapolis, MN, USA) in a final volume of 100 μL per well. For each well, 50 μg protein was loaded. Reaction mixture and fluorescent intensity was measured using SpectraMax M3 fluorescence microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA) for every 30 seconds with excitation at 340 nm and emission at 400 nm at 37°C for 4 hours (n = 4 per group).
Statistical Analysis
All results were expressed as mean ± SD. Experimental data of clinical scores were analyzed by two-way ANOVA followed by Bonferroni correction. The statistical significance between multiple groups was analyzed by one-way ANOVA followed by Bonferroni correction. P < 0.05 was considered to be significantly different.
Results
Systemic DIZE Treatment Ameliorated Inflammation in the Anterior Chamber
Inflammation in the anterior chamber was assessed by slit lamp at different time points after LPS injection. The clinical scores were evaluated based on the criteria of Table 1 at the same time by the slit lamp. Obvious inflammatory reaction, as evidenced by iris hyperemia, exudation into the anterior chamber, and hypopyon, was observed in both the DIZE + LPS group and the saline + LPS group. There was no inflammation seen in the untreated group or the DIZE group. There was significant iris hyperemia and exudation into the anterior chamber in both the DIZE + LPS group and the saline + LPS group 12 hours after LPS injection compared to the untreated group; however, the saline + LPS group had more severe inflammation than the DIZE + LPS group. At 24 hours, the clinical signs of saline + LPS group became worse. The iris hyperemia was much more severe, and there was more exudation into the anterior chamber than in the DIZE-treated group; some mice even had hypopyon (Fig. 1).
Figure 1.

Inflammatory reactions in the anterior chamber after LPS administration. The ocular inflammation was assessed by slit lamp microscopy at 12 (A) and 24 hours (B) after LPS injection. (A) Representative images of the anterior chamber from different treatment groups at 12 hours after LPS injection. (B) Representative images of the anterior chamber from different treatment groups at 24 hours after LPS injection (n = 7): (a, b) DIZE + LPS group; (c, d) saline + LPS group; (e, f) untreated group; (g, h) DIZE group.
Evaluation of the clinical score showed that inflammation of the anterior segment in the EIU mice was obvious 12 hours after LPS administration and reached its peak at 24 hours, then gradually subsided thereafter. At 48 hours, the inflammation decreased sharply and almost completely subsided by 72 hours. At 24 hours, the clinical score of mice in the DIZE + LPS group was significantly lower (mean, ∼1.75) than in the saline + LPS group (mean, ∼4) (P < 0.001). At 48 and 72 hours, there were no statistical differences between the DIZE + LPS group and the saline + LPS group (Fig. 2).
Figure 2.
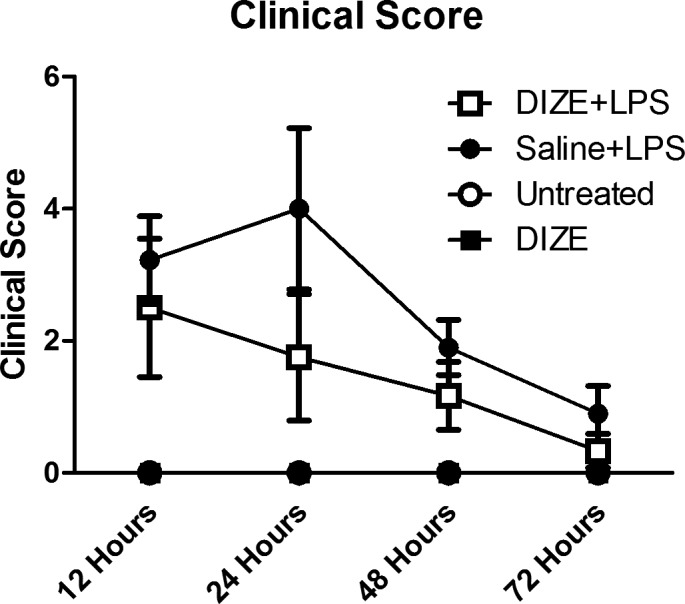
Clinical score of EIU at different time points after LPS injection. The clinical scores were evaluated at 12, 24, 48, and 72 hours after LPS administration using the criteria of EIU clinical score (Table 1). For each time point the clinical score of ocular inflammation was assessed by two independent observers according to the criteria (n = 7).
Systemic DIZE Treatment Reduced Infiltrating Inflammatory Cells in the Vitreous Cavity
Histological examination 24 hours after LPS injection showed severe intraocular inflammation in vehicle-treated group as evidenced by the large number of inflammatory cells that had infiltrated into the vitreous cavity and the ciliary body. DIZE-treated eyes had a 63.4% reduction of infiltrating inflammatory cells in the anterior segment and a 57.4% reduction in the posterior segment compared to the vehicle-treated group (Fig. 3). LPS-injected mice had increased CD45+ inflammatory cells infiltrating into the vitreous cavity and the retina. DIZE treatment resulted in a 43.3% reduction in vitreous (P < 0.01), but there was no significant decrease of the CD45+ inflammatory cells in the retina between the DIZE-treated group and the vehicle group (Fig. 4).
Figure 3.

Histological evaluation of EIU from hematoxylin and eosin–stained paraffin sections with and without systemic DIZE treatment. (A) Representative images showing infiltrating inflammatory cells in the posterior and anterior segments of the eyes in the DIZE + LPS group (a, e), the saline +LPS group (b, f), untreated group (c, g), and DIZE group (d, h). (B) Quantification of the percentage of inflammatory cells in the posterior and anterior segments of the eyes of all groups. ***P < 0.001 (n = 6, versus saline + LPS group; ×100 magnification for a–h).
Figure 4.

Systemic administration of DIZE reduced LPS induced inflammation. Quantification of the CD45+ macrophage cells in the vitreous (A) and in the retinas (B) 24 hours after LPS injection from the untreated group, DIZE group, saline + LPS group, and DIZE + LPS group. **P < 0.01, ***P < 0.001 (n = 6, versus saline + LPS group).
Local Instillation of DIZE Reduced the Number of Infiltrating Inflammatory Cells in the Vitreous
Eyedrops containing different concentrations of DIZE were given bilaterally to each mouse. Each eye was given 10-μL eyedrops. After instillation, mice were held for several seconds to avoid the eyedrops being wiped off. The number of infiltrating inflammatory cells were significantly reduced in eyes received topical administration of DIZE: 73.8% reduction in the 0.5 mg/mL group (P < 0.001) and 51.7% reduction in the 0.1 mg/mL group (P <0.01) compared to the control group that received artificial tears. However, there was no significant difference between the 0.5 mg/mL group and the 0.1 mg/mL group (Fig. 5). These experiments were also repeated in C57BL/6J mice with similar protective results (data not shown).
Figure 5.
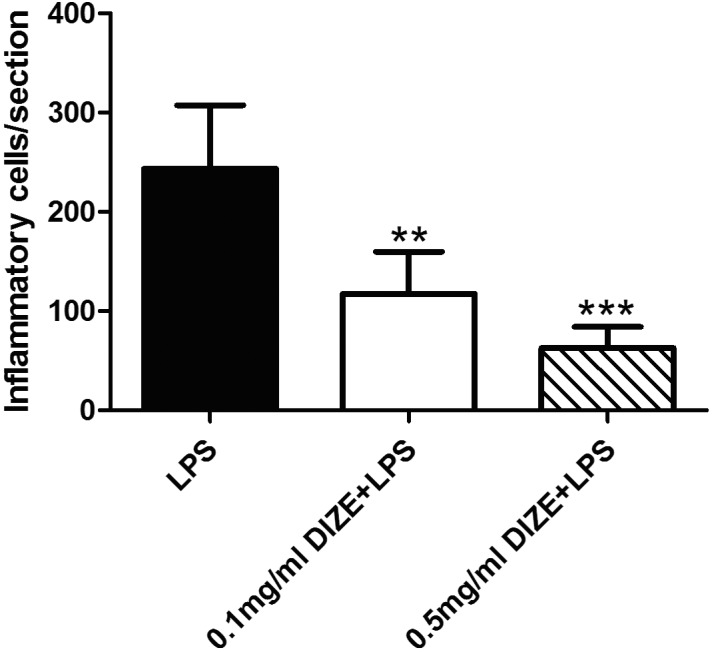
Quantification of infiltrating inflammatory cells in eyes with and without DIZE instillation. The numbers of inflammatory cells in the vitreous were counted as five sections per eye and the number of mice in each group was 0.5 mg/mL DIZE + LPS group (n = 4), 0.1 mg/mL DIZE + LPS group (n = 4), and the LPS only group (n = 6). The number of inflammatory cells decreased in the 0.5 mg/mL DIZE-treated group and the 0.1 mg/mL group compared to the LPS group. **P < 0.01, ***P < 0.001 (versus LPS group).
Systemic DIZE Treatment Reduced Expression of Inflammatory Cytokines
The inflammation in the EIU model involves both anterior and posterior segments of eyes. The inflammatory cytokines MCP-1, TNF-α, and IL-6 contribute to the development of anterior inflammation of EIU, whereas ICAM-1 plays a vital role in the posterior inflammation. Total RNA was isolated from the eyeballs, and real-time RT-PCR was performed to quantitatively measure the levels of these inflammatory cytokines. Compared with the saline + LPS group, the mRNA levels of the inflammatory cytokines were significantly reduced in the DIZE + LPS group. There was approximately a 4-fold decrease in the mRNA level of ICAM-1 and approximately a 5-fold decrease of TNF-α. Furthermore, the mRNA levels of IL-6 and MCP-1 were also significantly reduced in the DIZE + LPS group (P < 0.001). There was approximately 4.5-fold decrease of IL-6 and 2-fold decrease of MCP-1 (Fig. 6).
Figure 6.

Real-time RT-PCR analysis of mRNA expression for inflammatory cytokines. The whole eyecups were used for analyzing the mRNA levels of the inflammatory cytokines: ICAM-1, MCP-1, TNF-α, and IL-6. Values on the y-axis represent the fold changes at 24 hours after LPS injection. All values were normalized to the untreated control. **P < 0.01, ***P < 0.001 (n = 4, versus saline + LPS group).
ACE2 Activity Increased With the Systemic Treatment of DIZE
DIZE treatment resulted in significantly increased ACE2 activity in the retina. The increase was even more in normal eyes without LPS injection than in the saline + LPS group (P < 0.001) and the untreated group (P < 0.001). The DIZE + LPS group showed approximately 16% and 20% increases in ACE2 activity compared to the saline + LPS group and the untreated group, respectively. The DIZE group showed 26% higher ACE2 activity than the saline + LPS group (Fig. 7).
Figure 7.
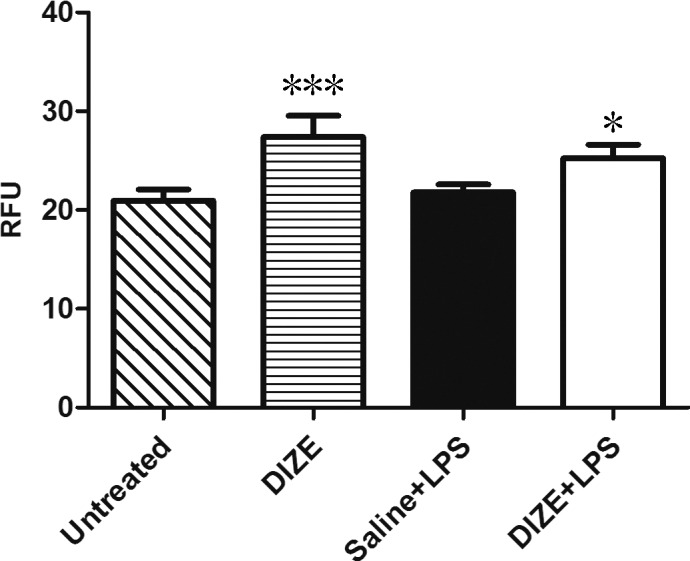
ACE2 activities in the retina. Quantification of ACE2 activity changes for the dissected retinas of the untreated group, DIZE group, and DIZE + LPS group compared to the saline + LPS group with the ACE2-specific fluorogenic peptide substrate. Values on the y-axis represent the relative fluorescent intensity unit (RFU) for every 30 seconds with excitation at 340 nm and emission at 400 nm at 37°C. *P < 0.05, ***P < 0.001 (n = 4, versus saline + LPS group).
Discussion
In the present study, we found that DIZE pretreatment resulted in increased retinal ACE2 activity. Administration of DIZE, either systemically or topically, ameliorated the inflammation in the EIU model. DIZE pretreatment reduced anterior segment inflammation, decreased the infiltration of inflammatory cells in both anterior and posterior segments, and decreased the expression of inflammatory cytokines.
It is well known that the classical RAS component, Ang II, contributes to the inflammatory process by increasing the expression of proinflammatory cytokines, chemokines, and cell adhesion molecules via AT1R.30 It is also a regulator of proinflammatory transcription factors that are responsible for triggering the activation of the inflammatory network. Within the transcription factors that are induced by Ang II, nuclear factor κB (NF-κB) plays a vital role in regulating several proinflammatory genes at the transcriptional level.31 On the one hand, Ang II influences NF-κB activation, stimulating nuclear translocation of the p65 subunit, DNA binding, transcription of a NF-κB reporter gene, and degradation of transcription inhibition factor IκB. On the other hand, NF-κB stimulates the expression of the gene encoding angiotensinogen, amplifying AngII-mediated inflammatory responses.32 Moreover, Ang II directly induces the proliferation and differentiation of inflammatory cell per se.30,33 As a result, AT1R blockers and ACE inhibitors have been shown to reduce the expression of inflammatory cytokines and thus to suppress ocular inflammation19 as well as vascular inflammation.20
More recently, several studies have shown that the ACE2/Ang-(1-7)/Mas axis may also influence inflammatory responses. Indeed, there is much evidence to indicate that this beneficial axis negatively regulates leukocyte migration and cytokine expression and release.34 Furthermore, there is direct evidence that ACE2/Ang-(1-7)/Mas axis evokes an anti-inflammatory response at the cellular level. An inflammatory response is orchestrated by the interaction between several inflammatory mediators and immune cells.35 After the exposure to LPS, many inflammatory cells can be infiltrating, such as macrophages, dendritic cells, and neutrophils. These infiltrating inflammatory cells can secrete a broad spectrum of proinflammatory cytokines and chemokines, thus triggering the inflammation process in the EIU model.36 Therefore, these infiltrating inflammatory cells can serve as an indicator of inflammation in the EIU model. Macrophages are a type of key innate immune cells that play a critical role in inflammation. They participate in the immune response via the production of inflammatory and immunomodulatory cytokines.37 Under stimulation with LPS, induced macrophages start to produce potent proinflammatory mediators, including TNF-α and IL-6 as well as other inflammatory mediators.38 A very recent study showed hyperactivated ACE2/Ang-(1-7)/Mas axis reduced the expression of proinflammatory cytokines including TNF-α and IL-6 after LPS stimulation in mouse peritoneal macrophages, with the Mas antagonist A779 abrogating the anti-inflammatory effect.39 Moreover, in response to inflammatory stimuli, a series of intracellular signaling cascades are initiated that ultimately lead to activation of macrophages and their release of proinflammatory cytokines. The major cascades among these intracellular signaling pathways include the phosphorylation of mitogen-activated protein kinase (MAPK) as well as signal transducer and activator of transcription (STAT) pathways, which lead to the activation of NF-κB,40 a crucial transcription factor for proinflammatory cytokine production. Indeed, LPS-induced production of inflammatory cytokines by macrophages involves activation of MAPK.41
In addition, treatment with Ang-(1-7) ameliorates the inflammatory conditions of the respiratory tract in allergic asthma mouse model, revealing one possible mechanism by which the anti-inflammatory property of Ang-(1-7) is through one of the MAPK family members, the extracellular signal-regulated kinase (ERK) or NF-κB signaling pathway, because Ang-(1-7) diminished the phosphorylation of ERK1 and IκBα (inhibitor of NF-κB).42 Most recently, there is evidence showing that DIZE treatment remarkably suppresses the production of proinflammatory cytokines by macrophages, leading to a reduction in serum levels of these cytokines after LPS treatment. The study also showed that DIZE down-regulated the phosphorylation of key signaling molecules and transcription factors, such as MAPKs, STATs, and NF-κB p65 subunit, which are involved in the production of proinflammatory cytokines by immune cells, and also directly affected transcription of proinflammatory genes.43 Consistent with these observations, our results demonstrate that DIZE alleviated the ocular inflammation not only by reducing the amount of infiltrating CD45+ inflammatory cells, but also by decreasing the retinal expression of the inflammatory cytokines.
The role of ACE2 in metabolizing Ang II and generating Ang-(1-7) has been well established since the discovery of the ACE2 gene.44 A critical question addressed in our study was whether DIZE can activate ACE2 and whether such activation can exert the anti-inflammatory effects. Although DIZE has been tested in several studies in animal models for different diseases, it has not been studied in ocular inflammation. Our study provides the first experimental evidence that DIZE can activate ACE2 and such activation plays an anti-inflammatory role in ocular inflammation model. Moreover, previous studies have demonstrated that DIZE activates ACE2 and the beneficial effects by DIZE are abolished by ACE2/Ang-(1-7)/Mas antagonists,26,28,45 suggesting that these beneficial effects are mediated by activating the ACE2/Ang-(1-7)/Mas axis. In fact the anti-inflammatory effects of DIZE in most recent studies43,46 may also involve the ACE2/Ang-(1-7)/Mas axis, since Ang II-mediated signaling is known to activate MAPK and the NF-κB signaling pathway.47,48 Nevertheless, since DIZE is a small molecular compound and has been used as a drug for the treatment of trypanosomiasis and babesiosis.49 Possible off-targets effects unrelated to the renin–angiotensin system cannot be ruled out and future studies should be conducted for further understanding of the underlying mechanisms. This evidence supports the notion that DIZE may have anti-inflammatory activity by regulating key pathways for proinflammatory cytokine production via activation of the protective ACE2/Ang-(1-7)/Mas axis.
In our present study, we also showed that systemic pretreatment with DIZE significantly increased the ACE2 activity in the retina, suggesting that this low molecular weight compound may pass through multiple barriers of the eye and play a protective role under the modulation of the local ACE2/Ang-(1-7) /Mas axis. Indeed, in addition to the circulating RAS, various members of RAS have already been identified in the eyes.50 More importantly, ACE2, Ang-(1-7), and Mas were also observed in the eyes,50–52 indicating that the ACE2/Ang-(1-7)/Mas axis is present in ocular tissues. In this current study, since systemic pretreatment with DIZE could activate local ACE2 effectively, we hypothesized that topical pretreatment with DIZE might activate the local ACE2/Ang-(1-7)/Mas axis directly, thus the concentration of DIZE in the eye may be higher than with the systemic treatment and may protect the eye more efficiently without undesirable systemic influence. To test this hypothesis, we pretreated EIU mice with DIZE in eyedrops. The results showed that topical treatment with DIZE also has an anti-inflammatory effect because the number of inflammatory cells was significantly reduced. Moreover, the dose of DIZE eyedrops was much less than the dose for systemic treatment. Ocular inflammation is a local eye disease, and topical administration of DIZE at a much lower dose offers a greater advantage for therapeutic intervention in patients with uveitis because systemic drug administration potentially has off-target side effects on other organs. Nonetheless, the detailed pharmacokinetic characteristics of DIZE in the eyes still need to be elucidated.
Our study provides a proof-of-concept that activating ACE2 or enhancing the activity of ACE2 is protective in ocular inflammation, consistent with the observations from many other studies.39,53,54 Moreover, it is significant for providing a potential strategy to enhance the protective axis for treatment of ocular inflammatory diseases. In particular, the prolonged inflammation often results in a chronic inflammatory condition, which is the major pathogenic mechanism in a number of autoimmune diseases, such as rheumatoid arthritis as well as uveitis. In addition, given that uveitis tends to recur, patients often develop chronic inflammation. Thus, long-term application is needed, regardless of whether it is systemic or topical treatment. Undesirable side effects or complication must be avoided though, so a therapeutic strategy with a long-lasting effect that does not require frequent repetition is desirable. Topical treatment with slow-release technique or other long-lasting therapeutic methods, such as delivering the gene of the members from ACE2/Ang-(1-7)/Mas axis and/or silencing the genes of the members from ACE/Ang II/AT1R, maybe a preferable choice for preventing the inflammation process of uveitis.
In summary, our research demonstrated that DIZE has a preventive effect on LPS-induced ocular inflammation in the EIU mouse model by activating ACE2. These results support the notion that RAS plays a role in modulating ocular immune response and that enhancing ACE2 provides a novel therapeutic strategy for uveitis.
Acknowledgments
Supported by American Diabetes Association, American Heart Association, Research to Prevent Blindness, National Institutes of Health Grants EY021752 and EY021721, and National Natural Science Foundation of China (81271033). The authors alone are responsible for the content and writing of the paper.
Disclosure: Y. Qiu, None; P.K. Shil, None; P. Zhu, None; H. Yang, None; A. Verma, None; B. Lei, None; Q. Li, None
References
- 1. Miserocchi E, Fogliato G, Modorati G, Bandello F. Review on the worldwide epidemiology of uveitis. Eur J Ophthalmol. 2013; 23: 705–717 [DOI] [PubMed] [Google Scholar]
- 2. Read RW. Uveitis: advances in understanding of pathogenesis and treatment. Curr Rheumatol Rep. 2006; 8: 260–266 [DOI] [PubMed] [Google Scholar]
- 3. Munoz-Fernandez S, Martin-Mola E. Uveitis. Best Pract Res Clin Rheumatol. 2006; 20: 487–505 [DOI] [PubMed] [Google Scholar]
- 4. Tabbara KF. Infectious uveitis: a review. Arch Soc Esp Oftalmol. 2000; 75: 215–259 [PubMed] [Google Scholar]
- 5. Servat JJ, Mears KA, Black EH, Huang JJ. Biological agents for the treatment of uveitis. Expert Opin Biol Ther. 2012; 12: 311–328 [DOI] [PubMed] [Google Scholar]
- 6. Gomes Bittencourt M, Sepah YJ, Do DV, et al. New treatment options for noninfectious uveitis. Dev Ophthalmol. 2012; 51: 134–161 [DOI] [PubMed] [Google Scholar]
- 7. Rosenbaum JT, Rosenzweig HL, Smith JR, Martin TM, Planck SR. Uveitis secondary to bacterial products. Ophthalmic Res. 2008; 40: 165–168 [DOI] [PubMed] [Google Scholar]
- 8. Hoekzema R, Murray PI, van Haren MA, Helle M, Kijlstra A. Analysis of interleukin-6 in endotoxin-induced uveitis. Invest Ophthalmol Vis Sci. 1991; 32: 88–95 [PubMed] [Google Scholar]
- 9. Koizumi K, Poulaki V, Doehmen S, et al. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis in vivo. Invest Ophthalmol Vis Sci. 2003; 44: 2184–2191 [DOI] [PubMed] [Google Scholar]
- 10. Tuaillon N, Shen DF, Berger RB, et al. MCP-1 expression in endotoxin-induced uveitis. Invest Ophthalmol Vis Sci. 2002; 43: 1493–1498 [PubMed] [Google Scholar]
- 11. Becker MD, Garman K, Whitcup SM, Planck SR, Rosenbaum JT. Inhibition of leukocyte sticking and infiltration, but not rolling, by antibodies to ICAM-1 and LFA-1 in murine endotoxin-induced uveitis. Invest Ophthalmol Vis Sci. 2001; 42: 2563–2566 [PubMed] [Google Scholar]
- 12. Miyamoto K, Ogura Y, Hamada M, et al. In vivo quantification of leukocyte behavior in the retina during endotoxin-induced uveitis. Invest Ophthalmol Vis Sci. 1996; 37: 2708–2715 [PubMed] [Google Scholar]
- 13. Yamashiro K, Kiryu J, Tsujikawa A, et al. Suppressive effects of selectin inhibitor SKK-60060 on the leucocyte infiltration during endotoxin induced uveitis. Br J Ophthalmol. 2003; 87: 476–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herichova I, Szantoova K. Renin-angiotensin system: upgrade of recent knowledge and perspectives. Endocr Regul. 2013; 47: 39–52 [DOI] [PubMed] [Google Scholar]
- 15. de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000; 52: 415–472 [PubMed] [Google Scholar]
- 16. Campbell DJ. Evolving concepts of the renin angiotensin system. Clin Exp Pharmacol Physiol. 2013; 40: 525–526 [DOI] [PubMed] [Google Scholar]
- 17. Ruiz-Ortega M, Lorenzo O, Suzuki Y, Ruperez M, Egido J. Proinflammatory actions of angiotensins. Curr Opin Nephrol Hypertens. 2001; 10: 321–329 [DOI] [PubMed] [Google Scholar]
- 18. Luft FC, Dechend R, Muller DN. Immune mechanisms in angiotensin II-induced target-organ damage. Ann Med. 2012; 44 (suppl 1): S49–S54 [DOI] [PubMed] [Google Scholar]
- 19. Nagai N, Oike Y, Noda K, et al. Suppression of ocular inflammation in endotoxin-induced uveitis by blocking the angiotensin II type 1 receptor. Invest Ophthalmol Vis Sci. 2005; 46: 2925–2931 [DOI] [PubMed] [Google Scholar]
- 20. da Cunha V, Tham DM, Martin-McNulty B, et al. Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis. 2005; 178: 9–17 [DOI] [PubMed] [Google Scholar]
- 21. Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000; 87: E1–E9 [DOI] [PubMed] [Google Scholar]
- 22. Passos-Silva DG, Verano-Braga T, Santos RA. Angiotensin-(1-7): beyond the cardio-renal actions. Clin Sci (Lond). 2013; 124: 443–456 [DOI] [PubMed] [Google Scholar]
- 23. Simoes e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. 2013; 169: 477–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verma A, Shan Z, Lei B, et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol Ther. 2012; 20: 28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gjymishka A, Kulemina LV, Shenoy V, Katovich MJ, Ostrov DA, Raizada MK. Diminazene aceturate is an ACE2 activator and a novel antihypertensive drug. FASEB J. 2010; 24:1032.3 [Google Scholar]
- 26. Shenoy V, Gjymishka A, Jarajapu YP, et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013; 187: 648–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mecca AP, Regenhardt RW, O'Connor TE, et al. Cerebroprotection by angiotensin-(1-7) in endothelin-1-induced ischaemic stroke. Exp Physiol. 2011; 96: 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Foureaux G, Nogueira JC, Nogueira BS, et al. Antiglaucomatous effects of the activation of intrinsic Angiotensin-converting enzyme 2. Invest Ophthalmol Vis Sci. 2013; 54: 4296–4306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang P, Smith JR, Damodar KS, Planck SR, Rosenbaum JT. Visualization of cell death in vivo during murine endotoxin-induced uveitis. Invest Ophthalmol Vis Sci. 2003; 44: 1993–1997 [DOI] [PubMed] [Google Scholar]
- 30. Suzuki Y, Ruiz-Ortega M, Lorenzo O, et al. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003; 35: 881–900 [DOI] [PubMed] [Google Scholar]
- 31. Savoia C, Schiffrin EL. Inflammation in hypertension. Curr Opin Nephrol Hypertens. 2006; 15: 152–158 [DOI] [PubMed] [Google Scholar]
- 32. Costanzo A, Moretti F, Burgio VL, et al. Endothelial activation by angiotensin II through NFkappaB and p38 pathways: involvement of NFkappaB-inducible kinase (NIK), free oxygen radicals, and selective inhibition by aspirin. J Cell Physiol. 2003; 195: 402–410 [DOI] [PubMed] [Google Scholar]
- 33. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010; 2: 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. da Silveira KD, Coelho FM, Vieira AT, et al. Anti-inflammatory effects of the activation of the angiotensin-(1-7) receptor, MAS, in experimental models of arthritis. J Immunol. 2010; 185: 5569–5576 [DOI] [PubMed] [Google Scholar]
- 35. Sacca R, Cuff CA, Ruddle NH. Mediators of inflammation. Curr Opin Immunol. 1997; 9: 851–857 [DOI] [PubMed] [Google Scholar]
- 36. McMenamin PG, Crewe J. Endotoxin-induced uveitis. Kinetics and phenotype of the inflammatory cell infiltrate and the response of the resident tissue macrophages and dendritic cells in the iris and ciliary body. Invest Ophthalmol Vis Sci. 1995; 36: 1949–1959 [PubMed] [Google Scholar]
- 37. Fujiwara N, Kobayashi K. Macrophages in inflammation. Curr Drug Targets Inflamm Allergy. 2005; 4: 281–286 [DOI] [PubMed] [Google Scholar]
- 38. Fujihara M, Muroi M, Tanamoto K, et al. Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: roles of the receptor complex. Pharmacol Ther. 2003; 100: 171–194 [DOI] [PubMed] [Google Scholar]
- 39. Souza LL, Costa-Neto CM. Angiotensin-(1-7) decreases LPS-induced inflammatory response in macrophages. J Cell Physiol. 2012; 227: 2117–2122 [DOI] [PubMed] [Google Scholar]
- 40. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012; 92: 689–737 [DOI] [PubMed] [Google Scholar]
- 41. Rao KM, Meighan T, Bowman L. Role of mitogen-activated protein kinase activation in the production of inflammatory mediators: differences between primary rat alveolar macrophages and macrophage cell lines. J Toxicol Environ Health A. 2002; 65: 757–768 [DOI] [PubMed] [Google Scholar]
- 42. El-Hashim AZ, Renno WM, Raghupathy R, et al. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-kappaB-dependent pathways. Br J Pharmacol. 2012; 166: 1964–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuriakose S, Muleme H, Onyilagha C, Okeke E, Uzonna JE. Diminazene aceturate (Berenil) modulates LPS induced pro-inflammatory cytokine production by inhibiting phosphorylation of MAPKs and STAT proteins [ published online ahead of print October 31, 2013]. Innate Immun. doi:10.1177/1753425913507488 [DOI] [PubMed] [Google Scholar]
- 44. Marian AJ. The discovery of the ACE2 gene. Circ Res. 2013; 112: 1307–1309 [DOI] [PubMed] [Google Scholar]
- 45. Qi Y, Zhang J, Cole-Jeffrey CT, et al. Diminazene aceturate enhances angiotensin-converting enzyme 2 activity and attenuates ischemia-induced cardiac pathophysiology. Hypertension. 2013; 62: 746–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kuriakose S, Muleme HM, Onyilagha C, et al. Diminazene aceturate (Berenil) modulates the host cellular and inflammatory responses to Trypanosoma congolense infection. PLoS One. 2012; 7: e48696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yaghooti H, Firoozrai M, Fallah S, Khorramizadeh MR. Angiotensin II induces NF-kappaB, JNK and p38 MAPK activation in monocytic cells and increases matrix metalloproteinase-9 expression in a PKC- and Rho kinase-dependent manner. Braz J Med Biol Res. 2011; 44: 193–199 [DOI] [PubMed] [Google Scholar]
- 48. Rajapurohitam V, Kilic A, Javadov S, Karmazyn M. Role of NF-kappaB and p38 MAPK activation in mediating angiotensin II and endothelin-1-induced stimulation in leptin production and cardiomyocyte hypertrophy. Mol Cell Biochem. 2012; 366: 287–297 [DOI] [PubMed] [Google Scholar]
- 49. Peregrine AS, Mamman M. Pharmacology of diminazene: a review. Acta Trop. 1993; 54: 185–203 [DOI] [PubMed] [Google Scholar]
- 50. Vaajanen A, Lakkisto P, Virtanen I, et al. Angiotensin receptors in the eyes of arterial hypertensive rats. Acta Ophthalmol. 2009; 88: 431–438 [DOI] [PubMed] [Google Scholar]
- 51. Luhtala S, Vaajanen A, Oksala O, Valjakka J, Vapaatalo H. Activities of angiotensin-converting enzymes ACE1 and ACE2 and inhibition by bioactive peptides in porcine ocular tissues. J Ocul Pharmacol Ther. 2009; 25: 23–28 [DOI] [PubMed] [Google Scholar]
- 52. Senanayake P, Drazba J, Shadrach K, et al. Angiotensin II and its receptor subtypes in the human retina. Invest Ophthalmol Vis Sci. 2007; 48: 3301–3311 [DOI] [PubMed] [Google Scholar]
- 53. Sukumaran V, Veeraveedu PT, Gurusamy N, et al. Telmisartan acts through the modulation of ACE-2/ANG 1-7/mas receptor in rats with dilated cardiomyopathy induced by experimental autoimmune myocarditis. Life Sci. 2012; 90: 289–300 [DOI] [PubMed] [Google Scholar]
- 54. Sukumaran V, Veeraveedu PT, Gurusamy N, et al. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1-7/mas receptor axis. Int J Biol Sci. 2011; 7: 1077–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]