

Abstract
Although magnesium is involved in a wide spectrum of vital functions in normal human physiology, the significance of hypomagnesemia and necessity for its treatment are under-recognized and underappreciated in clinical practice. In the current review, we first present an overview of the clinical significance of hypomagnesemia and normal magnesium metabolism, with a focus on renal magnesium handling. Subsequently, we review the literature for both congenital and acquired hypomagnesemic conditions that affect the various steps in normal magnesium metabolism. Finally, we present an approach to the routine evaluation and suggested management of hypomagnesemia.
Keywords: hypomagnesemia, magnesium, diabetes mellitus, alcohol, TRPM6, cisplatin
Introduction
Despite being the second most abundant intracellular and fourth most abundant extracellular cation in the body, hypomagnesemia has received relatively poor attention in the medical literature compared with hyponatremia, hypokalemia, and hypocalcemia. As of November 14, 2013, there were 2,100 versus 10,298, 1,107, and 12,345 citations related to the respective electrolyte disorders, recorded on PubMed (Figure 1). The low interest in hypomagnesemia may have stemmed from its relative lack of symptoms until plasma concentrations reach severely low levels, our poor understanding of magnesium metabolism until recent years, or both. Nonetheless, magnesium is involved in a wide spectrum of vital functions in human physiology. Magnesium is required for all enzymatic reactions requiring adenosine triphosphate (ATP), various reactions requiring kinases, neuromuscular excitability and cell permeability, regulation of ion channels and mitochondrial function, cellular proliferation and apoptosis, as well as immunity, among others.1–3
Figure 1.
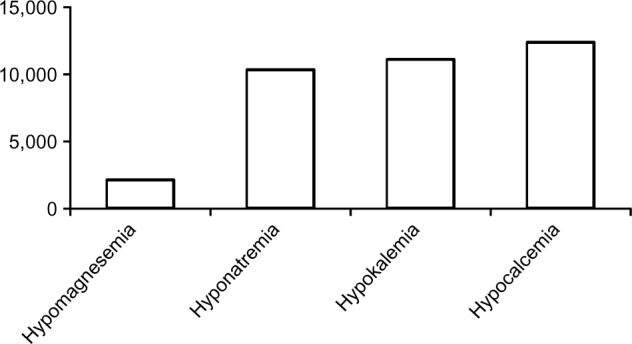
Number of PubMed citations (November 2013).
Clinical manifestations
Hypomagnesemia, while typically defined as having serum magnesium concentration below 0.66 mmol/L (1.6 mg/dL), with or without accompanying total body depletion, does not lead to clinically significant signs and symptoms until serum levels fall below 0.5 mmol/L (1.2 mg/dL).3 Nonetheless, as magnesium is involved in an array of structural and physiological functions, adverse effects associated with hypomagnesemia may occur in almost every organ system, whether they are clinically acute and overt, or chronic and subtle.
Clinical manifestations of hypomagnesemia that promptly lead to medical attention involve neuromuscular hyperexcitability that may range from tremors, fasciculation, tetany, to convulsions, and neuropsychiatric disturbances including apathy, delirium, and even coma. Other potentially life-threatening complications may arise not solely from hypomagnesemia, but also from the associated hypocalcemia and/or hypokalemia, and include atrial and ventricular arrhythmias, torsades de pointe, enhanced sensitivity to digoxin toxicity, and sudden death. In contrast, long-term adverse complications where the association with hypomagnesemia is not often recognized include altered glucose homeostasis, hypertension, atherosclerosis, osteoporosis, asthma, migraines, and other end-organ damage. Presumed mechanisms involved in various hypomagnesemia-associated signs/symptoms are listed in Table 1.2–5 The mechanisms of many, if not most, clinical signs and symptoms of hypomagnesemia are likely multifactorial and beyond the scope of discussion in the current review.
Table 1.
Magnesium functions and hypomagnesemia related clinical manifestations
| General functions | Specific involvement | Signs/symptoms and altered metabolisms associated with hypomagnesemia* |
|---|---|---|
| Enzymatic activities: functioning as substrate or direct enzyme activation | Kinases, ATPases or GTPases, cyclases, phosphofructokinase, creatine kinase, 5-phosphoribosyl-pyrophosphate synthetase, adenylate cyclase, Na+-K+-ATPase | Altered glucose metabolism, electrolyte imbalances (hypokalemia, hypocalcemia), osteoporosis, enhanced digoxin sensitivity, enhanced apoptosis, arrhythmias, atherosclerosis |
| Membrane function | Cell adhesion, transmembrane electrolyte flux | Arrhythmias: atrial tachycardia and fibrillation, supraventricular and ventricular arrhythmias, Torsade de pointes, rhabdomyolysis, hemolysis, myocardial infarction |
| Calcium antagonist | Neuromuscular function | Neuromuscular hyperexcitability: tremors, fasciculation, tetany, convulsions, neuropsychiatric changes, eg, apathy, depression, psychosis, vertigo, nystagmus, athetoid movements and choreiform movements, migraine, asthma (reactive airways), impaired exercise performance, electrolyte imbalances (hypokalemia, hypocalcemia), hypertension, atherosclerosis |
| Structural function, ion complex formation | Proteins, polyribosomes, nucleic acids, multiple enzyme complexes, mitochondria, ion complexes | Enhanced apoptosis, osteoporosis, kidney stones |
Notes: Copyright © 2003. Australasian Association of Clinical Biochemists. Adapted from Swaminathan R. Magnesium metabolism and its disorders. Clin Biochem Rev. 2003;24(2):47–66.3
Signs and symptoms are listed based on possible and/or presumed mechanisms of hypomagnesemia-induced defective structural or physiological functions. Many signs and symptoms are multifactorial, not fully understood, and beyond the scope of the current paper.
Abbreviations: ATPase, adenosine triphosphatase; GTPase, guanosine triphosphatase; Na+-K+-ATPase, sodium-potassium adenosine triphosphatase.
Magnesium metabolism
Gastrointestinal absorption
While the Institute of Medicine recommends a daily magnesium intake of 310–420 mg/day in adults, with the end of normal range for women and higher range for men, it has been estimated that 300–350 mg of magnesium is consumed daily in a typical American diet.6 Twenty percent to 80% of dietary magnesium is absorbed in the intestines, where absorption depends on both intake and body magnesium status, and occurs via both passive and active pathways.6–8 Passive Mg2+ absorption occurs paracellularly and predominantly in the small intestines, a process driven by a favorable electrochemical gradient and solvent drag with dietary intake. At low dietary magnesium intake, Mg2+ absorption relies heavily on active transcellular uptake via Mg2+-specific transporters in the large intestines.7,9 Two Mg2+-specific channels identified over recent years include the transient receptor potential melastatin (TRPM) 6 and TRPM7. TRPM7 is ubiquitously expressed among tissues, whereas TRPM6 is predominantly expressed along the full length of the intestine, the distal convoluted tubules (DCTs), the lungs, and the testis tissue. TRPM6 and TRPM7 can form heterodimers and may influence trafficking and activity of the TRPM6 Mg2+ channel. However, the extent and significance of the interactions between TRPM6 and 7 remain to be fully elucidated.8 Nonetheless, it has been suggested that while TRPM6 plays an important role in epithelial Mg2+ transport, TRPM7 is involved in cellular Mg2+ homeostasis.10 Loss-of-function mutations of TRPM6 have been reported in patients with familial hypomagnesemia with secondary hypocalcemia.11
Magnesium exists as protein bound (20% to 30%), complexed with organic anions such as sulfates, phosphates, or bicarbonates and citrates (5% to 15%), or free ionized cations (55% to 70%), where the proportion of each form is dependent on plasma pH, ionic strength, and protein/organic anion contents. Intracellular magnesium concentration has been estimated to range from 5 t o 20 mmol/L (12–49 mg/dL), while extracellular magnesium concentration typically ranges from 0.70 to 1.05 mmol/L (1.7–2.6 mg/dL).5
Cellular shift
Unlike potassium, regulation of Mg2+ cellular uptake or release occurs slowly and likely does not occur in all cell types. Bone is the largest reservoir of magnesium but its role and regulation in maintaining plasma levels remain poorly understood.12
Kidney handling of magnesium
The kidneys are thought to play a key role in regulating and maintaining magnesium balance.
Approximately 70% to 80% of plasma magnesium in ionized or complexed forms is ultrafilterable in the kidneys. Once filtered, 15% to 25% is reabsorbed passively with sodium and water in the proximal tubules.13 Sixty-five percent to 75% of the filtered magnesium load is reabsorbed paracellularly in the thick ascending limb of the loop of Henle (TAL), a process facilitated by the tight junction protein claudin-16, also known as paracellin-1.14,15 Mutation of claudin-16 is associated with severe hypomagnesemia with hypercalciuria and nephrolithiasis.16 More recently, mutations encoding the tight junction protein claudin-19 have also been reported to cause the inherited human renal disorder, familial hypomagnesemia with hypercalciuria and nephrocalcinosis.17 Although further studies are required, current evidence from both in vitro and in vivo studies suggests that claudin-16 and claudin-19 cogenerate a cation channel at the TAL tight junction necessary for normal Mg2+ reabsorption at this nephron segment.18
Five percent to 10% of the filtered magnesium, or 70% to 80% of magnesium delivered from the TAL, is reabsorbed at the DCT subsegment 1, the final nephron segment where Mg2+ can be reabsorbed, via an active and regulated transcellular pathway through the apical TRPM6 (Figure 2).19,20 Although the percentage of filtered magnesium reabsorbed in the DCT is lower than that at more proximal segments, regulated reabsorption at this segment is essential to magnesium balance because it determines the final urinary Mg2+ loss.21
Figure 2.
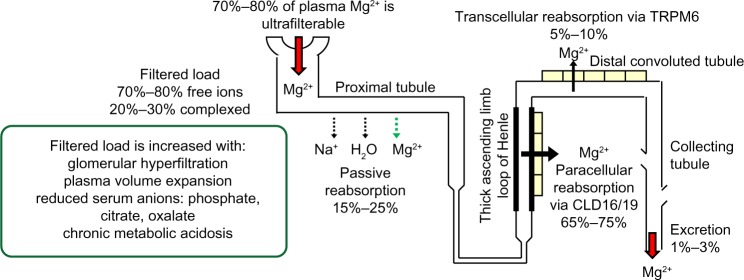
Kidney handling of magnesium.
Abbreviations: CLD, claudin; TRPM6, transient receptor potential channel 6.
DCT magnesium reabsorption
Apical factors
In Gitelman syndrome, where there is a homozygous or compound heterozygous mutation or deletion in the SLC12A3 gene encoding the apical thiazide-sensitive NaCl cotransporter (NCC), significant hypomagnesemia occurs in association with hypokalemic metabolic alkalosis and hypocalciuria.22 Although mechanistically unclear, Nijenhuis et al demonstrated reduced TRPM6 expression in mice chronically treated with thiazide, a Gitelman-equivalent condition.23 Additionally, it has been suggested that the compensatory hyperaldosteronism associated with reduced NCC sodium reabsorption in Gitelman may lead to reduced TRPM6 activity, particularly in concert with low dietary magnesium intake.24 It may also be speculated that reduced NCC sodium reabsorption could lead to suboptimal basolateral (3)Na+-(2)K+-ATPase activity, hence reduced generation of the favorable potential difference (ie, more negative intracellular voltage) to facilitate apical Mg2+ reabsorption (Figure 3). Whatever the mechanism(s) is/are, normal NCC function is likely necessary for DCT Mg2+ reabsorption.
Figure 3.
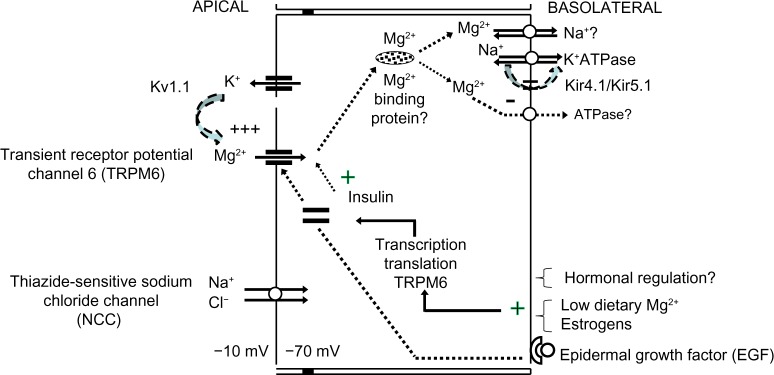
Magnesium reabsorption at the distal convoluted tubule.
The apical voltage-gated potassium channel Kv1.1 has also been implicated to play a role in normal DCT Mg2+ reabsorption. A mutation involving substitution of asparagine for aspartic acid leading to nonfunctional Kv1.1 channels in the DCT has been reported in a family with isolated autosomal-dominant hypomagnesemia paired with signs and symptoms of neuromuscular dysfunction including recurrent muscle cramps and weakness, tremors, tetany, cerebellar atrophy, and myokymia.25 It was suggested that the loss of Kv1.1 function reduces apical K+ intraluminal secretion, hence the associated favorable intraluminal positive voltage to facilitate Mg2+ reabsorption via TRPM6.
Basolateral factors
Renal magnesium handling at the DCT occurs via the apical TRPM6, whose shuttling from intracellular vesicles toward apical membranes requires binding of epidermal growth factor (EGF) to its basolateral receptor. Mutations of the EGF gene have been shown to be the causative defect in recessive isolated renal hypomagnesemia, where apical TRPM6 expression is reduced.26,27
It is speculated that following apical reabsorption, Mg2+ binds to Mg2+-binding proteins for transport to the basolateral sides, where eventual reabsorption into the interstitium occurs via transporters such as Na+/Mg2+ exchangers and/or Mg2+-ATPase. While basolateral transport of Mg2+ has not been elucidated, mutations involving the γ-subunit of the basolateral Na+-K+-ATPase or the regulatory protein of Na+-K+-ATPase, the hepatocyte nuclear factor 1 homeobox B (HNF1B), are associated with renal Mg2+ wasting.28–30 Mutation in the FXYD2 gene encoding the γ-subunit of Na+-K+-ATPase is thought to affect normal routing, hence activity of the transporter. Suboptimal Na+-K+-ATPase activity theoretically leads to depolarization of the DCT due to the 3Na+-to-2K+ exchange ratio (ie, reduced intracellular negative voltage that normally favors reabsorption of the divalent cation Mg2+) thus reduced Mg2+ reabsorption via TRPM6. If basolateral Mg2+ reabsorption significantly relies on Na+/Mg2+ exchangers, it may be also be deduced that reduced Na+-K+-ATPase activity, hence reduced basolateral Na+ recycling, would also lead to reduced basolateral Mg2+ reabsorption. Heterozygous mutations of the HNF1B gene, either whole-gene deletion or point mutations, are linked to a dominant renal cysts and diabetes syndrome, where up to 50% of patients also present with renal magnesium wasting with hypocalciuria.31 HNF1B is thought to play a regulatory role in the transcription of the FXYD2 gene, specifically at the promoter responsible for the transcription the γa-subunit of Na+-K+-ATPase.30
The basolateral, heteromeric, inwardly rectifying Kir4.1/Kir5.1 K+ channel has also been found to affect Mg2+ reabsorption, presumably via its K+ recycling function necessary to maintain optimal Na+-K+-ATPase activity. Mutations involving the KCNJ10 gene encoding Kir4.1 have been reported to cause a clinical constellation involving hypomagnesemia with associated SEizures, Sensorineural deafness, Ataxia, Mental retardation, and Electrolyte imbalance Epilepsy, Ataxia, Sensorineural deafness, and renal Tubulopathy, known as (SeSAME/EAST) syndrome.32,33 It is possible that the associated suboptimal Na+-K+-ATPase function leads to reduced sodium reabsorption at the DCT thiazide-sensitive sodium chloride channel, a defect observed with Gitelman syndrome. Accordingly, with the exception of hypocalcemia, a Gitelman-like clinical syndrome including sodium wasting, hypokalemia, hypomagnesemia, and metabolic alkalosis may be observed with Kir4.1 loss-of-function mutation. Additionally, it has been reported that the Kir4.1/Kir5.1 K+ channel is extremely sensitive to inhibition by intracellular pH, a characteristic thought to be conferred by the intact Kir5.1 subunit.34 Interestingly, activation or gain-of-function mutation of the calcium-sensing receptor (CaSR) has been shown to reduce basolateral Kir4.1 expression, presumably via altered caveolin-mediated trafficking of the channel, with resultant reduction in Mg2+ reabsorption and salt-wasting at the DCT.35
Finally, although mechanistically unclear, both deletion and missense mutations involving cyclin M2, a protein localized to the basolateral side in both the TAL and the DCT, have been identified in two unrelated families with unexplained dominant hypomagnesemia.36
Hormonal regulation
Regulatory hormones reported to influence magnesium homeostasis via TRPM6 and TRPM7 protein expression and activity include angiotensin II, aldosterone, bradykinin, thrombin, estrogen, and insulin. Parathyroid hormone and 1,25 vitamin D, however, do not appear to have any effect on TRPM6 expression.8,37–41 Of interest, the inability of insulin activation of TRPM6 in two single nucleotide TRPM6 polymorphisms, Ile1393Val and Lys1584Glu, has been linked to hypomagnesemia and presumed hypomagnesemia-induced glucose intolerance in women on low dietary magnesium intake.37 Low dietary magnesium intake and estrogens have been shown to upregulate renal TRPM6 expression and reduce urinary magnesium excretion.38
Aldosterone has been reported to induce renal Mg2+ wasting, an effect that may be ameliorated by aldosterone antagonists.39,40 Additionally, the use of aldosterone antagonists has also been shown to maintain plasma magnesium levels in patients receiving routine therapy for congestive heart failure, presumably via enhancing cellular Mg2+ efflux, thus implicating a role for aldosterone in cellular Mg2+ shift.40 Sontia et al have suggested that aldosterone may influence urinary Mg2+ excretion through redistribution of Mg2+ (Mg2+ efflux) in muscle, bone, and the gastrointestinal tract via stimulation of the Na+/Mg2+ exchanger and downregulation of renal TRPM7.41
Congenital conditions associated with hypomagnesemia are summarized in Table 2.
Table 2.
Congenital causes of hypomagnesemia
| Sources of magnesium loss | Sites of defect in magnesium metabolism | Congenital | |
|---|---|---|---|
| Gastro-intestinal absorption | Passive reabsorption | ||
| Active reabsorption | TRPM6 | Hypomagnesemia with secondary hypocalcemia | |
| Cellular shift; tissue sequestration | – | – | |
| Kidney handling | Glomerular filtration | – | |
| Proximal tubular reabsorption | Proximal renal tubular acidosis with or without associated Fanconi’s syndrome | ||
| Thick ascending limb of loop of Henle | Claudin-16 | Familial hypomagnesemia with hypercalciuria, and nephrocalcinosis | |
| Claudin-19 | Familial hypomagnesemia, with hypercalciuria, nephrocalcinosis, and ocular manifestation | ||
| NKCC2 | Antenatal Bartter syndrome type 1 with low-normal serum magnesium | ||
| ROMK | Antenatal Bartter syndrome type II with low-normal serum magnesium | ||
| ClC-Kb | Classic Bartter syndrome type III, hypomagnesemia in 20% | ||
| CaSR | Bartter syndrome type V | ||
| Distal convoluted tubule: | TRPM6 | Hypomagnesemia, secondary hypocalcemia | |
| Apical regulators/effectors of TRPM6 | |||
| Kv1.1 | Isolated autosomal dominant hypomagnesemia | ||
| Kir4.1/Kir5.1 | Kir4.1 mutation: SeSAME/EAST syndrome; Kir5.1 mutation: hypomagnesemia, hypokalemia, hyperchloremic metabolic acidosis and hypercalciuria | ||
| NCCT | Gitelman syndrome | ||
| Basolateral regulators/effectors of TRPM6 | |||
| Na+-K+-ATPase | FXYD2: autosomal dominant hypomagnesemia with hypocalciuria; HNF1B: renal cysts and diabetes mellitus with renal magnesium wasting and hypocalciuria | ||
| EGF | Isolated recessive hypomagnesemia with normocalciuria | ||
| CaSR | Activating mutations | ||
| CNNM2 | Autosomal dominant hypomagnesemia | ||
| Hormonal regulators of TRPM6 | |||
| Insulin | TRPM6 polymorphisms Ile1393Val and Lys1584Glu: reduced insulin activation of TRPM6, particularly if low dietary magnesium intake; tendency for diabetes mellitus | ||
| Other sources of loss | Nonspecific tubular injury/cellular leak | – | |
| Other | – | ||
Abbreviations: NKCC2, sodium potassium 2 chloride cotransporter; ROMK, renal outer medullary potassium channel; ClC-Kb, chloride channel Kb; CaSR, calcium sensing receptor; Kv1.1, potassium voltage-gated channel subfamily A member 1; Kir4.1/Kir5.1, inward-rectifier type potassium channel 4.1/5.1 dimer; NCCT, sodium chloride cotransporter; Na+-K+-ATPase, sodium-potassium adenosine triphosphatase; HNF1B, hepatocyte nuclear factor 1 homeobox B; EGF, epidermal growth factor; CNNM2, cyclin M2; TPRM 6, transient receptor potential melastatin 6; SeSAME/EAST syndrome, SEizures, Sensorineural deafness, Ataxia, Mental retardation, and Electrolyte imbalance Epilepsy, Ataxia, Sensorineural deafness, and renal Tubulopathy.
Etiologies of acquired hypomagnesemia
Similar to congenital conditions, acquired etiologies of hypomagnesemia may be categorized based on the presumed defects in the various steps of magnesium metabolism and are summarized in Table 3.
Table 3.
Acquired causes of hypomagnesemia
| Sources of magnesium loss | Sites of defect in magnesium metabolism | Acquired | |
|---|---|---|---|
| Gastro-intestinal absorption | Passive reabsorption | Low dietary Mg2+ intake; prolonged nasogastric suction; diarrhea; enteric fistulas; steatorrhea; short-gut syndrome; alcoholism | |
| Active reabsorption | TRPM6 | See DCT TRPM6 below | |
| Cellular shift; tissue sequestration | – | Acute pancreatitis (magnesium and calcium saponification in necrotic fat); hungry bone syndrome; refeeding syndrome; foscarnet (magnesium incorporation into bone matrix following complex formation with foscarnet); massive blood transfusions; third trimester pregnancy; lactation; excessive parenteral alimentation; cardiopulmonary bypass | |
| Kidney handling | Glomerular filtration | Hyperfiltration (diabetes mellitus, post-obstructive or acute tubular necrosis diuresis, osmotic diuresis, post-kidney transplantation); extracellular volume expansion; increased filterable Mg (metabolic acidosis, low organic anions) | |
| Proximal tubular reabsorption | Acquired Fanconi’s syndrome, drug toxicity (cisplatin, gentamicin, pentamidine); high dietary salt intake | ||
| Thick ascending limb of loop of Henle | Claudin-16 | – | |
| Claudin-19 | – | ||
| NKCC2 | Loop diuretics; hypokalemia | ||
| ROMK | Hypokalemia | ||
| ClC-Kb | – | ||
| CaSR | Increase in serum calcium and magnesium; aminoglycosides | ||
| Distal convoluted tubule: | TRPM6 | Calcineurin inhibitors (cyclosporine and tacrolimus shown to reduce renal TRPM6 expression); rapamycin (reduced TRPM6 messenger (m)RNA stability); chronic metabolic acidosis | |
| Apical regulators/effectors of TRPM6 | |||
| Kv1.1 | Hypokalemia | ||
| Kir4.1/Kir5.1 | – | ||
| NCCT | Thiazide diuretics; cyclosporine | ||
| Basolateral regulators/effectors of TRPM6 | |||
| Na+-K+-ATPase | Hypophosphatemia; calcineurin inhibitors (cyclosporin shown to inhibit Na+-K+-ATPase); ethanol (inhibits Na+-K+-ATPase activity) | ||
| EGF | Anti-EGF receptor antibodies (cetuximab, panitumumab); cisplatin (reduced EGF and TRPM6 mRNA); cyclosporine (reduce TRPM6, NCC, EGF mRNA) | ||
| CaSR | Increase in serum calcium and magnesium; aminoglycoside (gentamicin, amikacin); ?cinacalcet | ||
| CNNM2 | – | ||
| Hormonal regulators of TRPM6 | |||
| Insulin | Diabetes mellitus? | ||
| Other sources of loss | Nonspecific tubular injury/cellular leak | Amphotericin; pentamidine | |
| Other | Severe burns (skin loss) | ||
Abbreviations: NKCC2, sodium potassium 2 chloride cotransporter; ROMK, renal outer medullary potassium channel; ClC-Kb, chloride channel Kb; CaSR, calcium sensing receptor; Kv1.1, potassium voltage-gated channel subfamily A member 1; Kir4.1/Kir5.1, inward-rectifier type potassium channel 4.1/5.1 dimer; NCCT, sodium chloride cotransporter; Na+-K+-ATPase, sodium-potassium adenosine triphosphatase; EGF, epidermal growth factor; CNNM2, cyclin M2; NCC, Thiazide-sensitive sodium chloride channel; TPRM6, transient receptor potential melastatin 6; DCT, distal convoluted tubule.
Gastrointestinal causes
Common conditions include severely low dietary Mg2+ intake; prolonged nasogastric suction; and overall malabsorptive states including diarrhea, steatorrhea, celiac disease, regional enteritis, and short-gut syndrome. Gastrointestinal magnesium loss due to malabsorptive states may be due to rapid intestinal transit, Mg2+ binding to undigested free fatty acids, and/or reduction in both passive and active absorption.42–44 Of interest, diarrhea has been attributed as an important cause of hypomagnesemia in patients with chronic alcoholism. In a study involving 127 patients with chronic alcoholism, 20 out of 38 cases of hypomagnesemia were thought to be due to alcohol withdrawal syndrome and diarrhea. The remaining patients with hypomagnesemia had evidence of hypermagnesiuria, thought to be associated with hypophosphatemia and/or metabolic acidosis.45
Cellular shift/tissue sequestration
Acute pancreatitis is known to be associated with hypocalcemia and hypomagnesemia, both presumably via saponification in necrotic fat. The accompanying hypocalcemia-induced hyperparathyroidism and resultant hypophosphatemia may also lead to concurrent renal Mg2+ loss.43 Post-parathyroidectomized patients are thought to develop hypomagnesemia predominantly due to increased bone formation and mineralization, and in some cases, concurrent hypermagnesiuria.46 Conditions associated with increased anabolic states and cellular magnesium uptake leading to reduced plasma magnesium concentration include refeeding syndrome, excessive parenteral alimentation, advanced pregnancy, and lactation.47–49 Hypomagnesemia associated with massive blood transfusions is not uncommon and presumed to be due to citrate toxicity, hemodilution, and/or associated comorbidities.50 Cardiopulmonary bypass surgeries without magnesium supplementation may be associated with hypomagnesemia, thought to be due to increased cellular uptake with the high catecholamine state and chelation by free fatty acids and/or citrate.51 Hypomagnesemia associated with foscarnet has been proposed to occur via magnesium incorporation into bone matrix following complex formation with foscarnet.52
Renal wasting
Hyperfiltration
Hyperfiltration associated with various conditions (including diabetes mellitus; post-obstructive, osmotic, or acute tubular necrosis diuresis; post-kidney transplantation; or excessive volume expansion) may lead to enhanced filterable magnesium load, thus overwhelming the kidneys’ Mg2+ reabsorption capacity, and resultant hypermagnesiuria. Conditions favoring magnesium in its ionized form such as chronic metabolic acidosis or states with low organic anions may also contribute to a high filterable magnesium load and renal Mg2+ wasting.53
Reduced proximal tubular reabsorption
As Mg2+ is reabsorbed passively at the proximal tubules, severe injury to this nephron segment in the presence of normal glomerular filtration can lead to hypermagnesiuria. Acquired Fanconi’s syndrome and tubular toxicities induced by drugs such as cisplatin, aminoglycosides, and pentamidine, have all been reported to be associated with hypomagnesemia. Of note, pentamidine may also be associated with acute pancreatitis, which could be contributory to hypomagnesemia.54 Dietary salt loading has been shown in rats to increase distal tubular Mg2+ delivery, presumably via reduced proximal passive reabsorption, and upregulation of TRPM6 in the DCT, but overall increases urinary Mg2+ excretion.55
Reduced TAL reabsorption
Acquired causes of hypomagnesemia that target the TAL include loop diuretics, hypokalemia, hypercalcemia, and aminoglycosides. Effective reabsorption of Mg2+ at this nephron segment relies on luminal K+ recycling, which requires the functional Na+-K+-2Cl− cotransporter (NKCC2) for cellular K+ uptake, the renal outer medullary K+ channel (ROMK) for K+ recycling back into the lumen, and the CaSR for regulation of the ROMK. Potassium recycling into the lumen via the ROMK creates a more positively charged lumen, which facilitates divalent cation (Ca2+, Mg2+) reabsorption paracellularly. This process is facilitated by the tight junction proteins claudin-16 and claudin-19. Any transporter defect leading to suboptimal K+ recycling or claudin-16/claudin-19 function may induce Ca2+ and Mg2+ wasting. While loop diuretics specifically block NKCC2, hypokalemia may cause both suboptimal NKCC2 functioning as well as K+ recycling. Activation of the CaSR with Ca2+ can inhibit ROMK, and hence cause ineffective K+ tubular recycling. Reduction in K+ recycling, hence favorable intraluminal positive voltage necessary to facilitate Ca2+ and Mg2+ paracellular reabsorption, leads to wasting of both divalent cations. Aminoglycosides have been suggested to directly inhibit tubular Mg2+ reabsorption via binding to and activation of the CaSR. Of interest, this class of antibiotics has been suggested to mimic type 5 Bartter syndrome where there is a gain-of-function mutation of the CaSR.56–58
Reduced DCT reabsorption
Drugs known to be associated with hypomagnesemia and likely exert their magnesiuric effect at the DCTs include calcineurin inhibitors and rapamycin. Both cyclosporine and tacrolimus have been shown to reduce TRPM6 expression in the DCT in animal studies.59–61 Additionally, cyclosporine has been shown to induce reduced messenger (m)RNA expression of NCC.59 Whether the latter effect affects TRPM6 expression or plays a contributory role in magnesiuria is not known. Clinically, tacrolimus has been observed to induce more significant magnesiuria compared with cyclosporine.62
While rapamycin (sirolimus) has been reported to induce hypomagnesemia, the mechanism(s) whereby rapamycin causes magnesiuria is (are) not well defined. In an in vitro study using NRK-52E cells derived from normal rat renal tubules, Ikari et al revealed that sirolimus reduces mRNA expression TRPM6 at the DCT via inhibition of EGF-induced increase in TRPM6 expression, presumably by reducing the stability of TRPM6 mRNA.63 In contrast, in a study using male Wistar rats treated with rapamycin, da Silva et al reported increased TRPM6 expression.64 The magnesiuric effect of rapamycin in the latter study was thought to be due to the primary downregulation of NKCC2 protein expression leading to magnesiuria, followed by a secondary compensatory response with increased DCT TRPM6 protein expression. A direct rapamycin stimulatory effect on TRPM6 expression, however, could not be ruled out. Interestingly, rosiglitazone was shown to ameliorate the rapamycin-associated electrolyte disturbances including hypokalemia and downregulation of NKCC2.64
Although both calcineurin and mammalian target of rapamycin (mTOR) inhibitors may induce renal Mg2+ wasting, mTOR inhibitors may have lower magnesiuric effect compared with calcineurin inhibitors. In a retrospective review involving 138 renal transplant patients who were converted from calcineurin inhibitors to mTOR inhibitors over a 6-month period, magnesium levels significantly improved in association with reduced fractional excretion of Mg2+.65 In addition to drugs, acid-base status has also been shown to determine renal expression of TRPM6; whereas chronic metabolic acidosis reduces, chronic metabolic alkalosis enhances TRPM6 expression.66
To our knowledge, acquired causes of hypomagnesemia associated with inhibition of basolateral Kir4.1/5.1 have not been reported. Although there is evidence of direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine and antibody production against the potassium channel Kir4.1 in patients with multiple sclerosis, hypomagnesemia is not associated with these conditions.67,68 Similarly, there are no known reported cases of acquired hypomagnesemia due to direct inhibition of Kv1.1.
As previously discussed, thiazide-, and to some extent, cyclosporine-induced hypomagnesemia may occur via downregulation of TRPM6 expression. Moreover, Loffing et al have shown that thiazide administration over three days in rats can provoke apoptosis of distal tubule cells and associated focal peritubular inflammation.69 The direct effect of thiazides on the terminal nephron segment for magnesium reabsorption may explain the severe hypomagnesemia associated with Gitelman and not necessarily Bartter syndrome. As observed in animal studies where high-salt treatment was associated with increased distal delivery of Mg2+ and upregulation of TRPM6, the increased distal delivery of Mg2+ in patients with various types of Bartter syndromes may similarly upregulate DCT TRPM6, and hence reduce net Mg2+ wasting compared with Gitelman syndrome.55
Factors known to be associated with hypomagnesemia that could act via inhibition of the DCT basolateral Na+-K+-ATPase include hypophosphatemia, calcineurin inhibitors, and ethanol.70,71 As phosphate is required for all ATP-requiring cellular activities, it is conceivable that hypophosphatemia can lead to suboptimal Na+-K+-ATPase activity and, if proven present and significant in the DCT, basolateral Mg2+-ATPase.
While mutations involving EGF affect apical TRPM6 expression hence magnesium reabsorption, the use of anti-EGF receptor antibodies, particularly cetuximab and panitumumab, has accordingly been reported to be associated with hypomagnesemia. Other commonly used drugs associated with hypomagnesemia that have been shown to reduce EGF expression include cisplatin and cyclosporine.59,72,73
Clinical conditions and drugs that could potentially be equivalent to CaSR gain-of-function mutations to induce hypomagnesemia include hypercalcemia and aminoglycosides. Both may stimulate the CaSR and in turn reduce ROMK activity hence reduce the favorable potential difference necessary for optimal paracellular Mg2+ reabsorption at the TAL. Moreover, CaSR activation at the DCT is also thought to induce renal Mg2+ wasting via inhibition of TRPM6 expression. Nonetheless, common conditions associated with hypercalcemia including primary hyperparathyroidism and granulomatous disease, however, have not been reported to be associated with significant hypomagnesemia. In contrast, aminoglycosides including amikacin and gentamicin are well known to induce hypomagnesemia, hypothesized to occur via binding to and activation of the CaSR.74,75 Whether the CaSR agonist cinacalcet can induce hypomagnesemia is not known. Incidentally, our research into this question led to the review of a case series with reported pre- and post-cinacalcet treatment serum magnesium levels in four patients with familial hypocalciuric hypercalcemia. Notably, all four patients’ serum magnesium levels were lower at follow-up than at baseline. The serum magnesium levels before and at follow-up were 1.01±0.07 mmol/L and 0.95±0.01 mmol/L, respectively, P=0.11. Whether this observation is fortuitous, further investigation is warranted.76
Diabetes mellitus is well known to be associated with hypomagnesemia, likely via a plethora of mechanisms.53 Most recently, given reports of reduced insulin activation of TRPM6 in patients with TRPM6 polymorphisms, it may be speculated that the lack of insulin in the diabetic state may reduce TRPM6 activity and could thus contribute to hypomagnesemia.77
Other drugs associated with hypomagnesemia include amphotericin and pentamidine. The former is thought to self-insert into renal tubular membranes and act as an ionophore for urinary magnesium leak, whereas the latter is thought to induce hypomagnesemia via nonspecific, yet to be determined kidney injury.54,78
Finally, hypomagnesemia has been reported in severe burn patients. The involved mechanisms are likely multifactorial and may involve the need for aminoglycoside administration, or associated hypokalemia, among others.79
Diagnosis of hypomagnesemia
The clinical evaluation for the underlying cause of hypomagnesemia requires a thorough investigation for the presence of diabetes mellitus, alcoholism, gastrointestinal conditions involving poor absorption and/or poor nutritional intake, or a family history of hypomagnesemia without or without other electrolyte abnormalities, and a complete list of medications used. The suspected underlying etiology may be confirmed with urinary studies based on its mechanism via renal wasting or extrarenal cause. Patients with hypomagnesemia due to renal Mg2+ wasting have been suggested to present with a fractional excretion of Mg2+ greater than 4%, whereas those with extrarenal causes present with a much lower percentage, typically 2% or less. The fractional excretion of Mg2+ is defined as:
where the 0.7 factor indicates that in most circumstances, only 70% of serum magnesium is filterable.80
Management of hypomagnesemia: authors’ opinion
Magnesium metabolism and kidney handling of magnesium have been significantly elucidated over the last decade. A correlation between clinical conditions and specific mechanisms leading to hypomagnesemia will no doubt lead to better prevention and mechanism-specific therapy. Although mild-to-moderate hypomagnesemia is typically asymptomatic, long-term deficiencies have been reported to be associated with a spectrum of adverse micro- and macrovascular outcomes including increased risk of diabetes mellitus, various diabetic complications, arrhythmias particularly in association with congestive heart failure, hypertension, and more rapid progression of kidney disease, among others. Therefore, if safely tolerated, patients should receive therapy to correct the hypomagnesemic state.2–5,81,82 Currently, however, the management of hypomagnesemia still relies on relatively nonspecific management including avoidance or discontinuation of responsible agents if possible, corrections of underlying metabolic derangements, and/or magnesium supplementation.
Minimization of renal Mg2+ wasting
Routine corrections of the hyperfiltrative state, chronic metabolic acidosis, and low organic anion state (eg, hypophosphatemia and hypoproteinemia/hypoalbuminemia) should be done whenever applicable to minimize filtration of large amounts of free ionized Mg2+. Consultation with a dietitian may be necessary to increase dietary magnesium intake as well as protein supplementation in malnourished patients. Reduction of glomerular hyperfiltration may be achieved with the addition of any renin–angiotensin inhibitors. The use of an aldosterone antagonist may be considered if safely tolerated. Diarrheal states should be evaluated by a gastrointestinal specialist and promptly treated.
Dietary intake of foods containing high levels of magnesium should be encouraged. In general, grains, and beans are good sources of magnesium. Selected food sources of magnesium may be found at ods.od.nih.gov/factsheets/Magnesium-HealthProfessional/#h3.83 A few food sources containing at least 10% of the daily value of recommended total daily dietary magnesium (400 mg for adults and children aged 4 and older) as developed by the US Food and Drug Administration are listed in Table 4. If all measures fail to achieve “normal” range or serum magnesium concentration >0.66 mmol/L (1.6 mg/dL), magnesium supplement should be added. Various magnesium formulations are available on the market and are listed in Table 5. In the authors’ experience, Mg Plus Protein (Miller Pharmacal Group, Inc., Carol Stream, IL, USA) appears to be best tolerated by patients in terms of magnesium supplement-induced diarrhea.
Table 4.
Selected food sources of magnesium
| Food | mg per serving | Percent daily value |
|---|---|---|
| Almonds, dry roasted, 1 ounce | 80 | 20 |
| Spinach, boiled, ½ cup | 78 | 20 |
| Cashews, dry roasted, 1 ounce | 74 | 19 |
| Peanuts, oil roasted, ¼ cup | 63 | 16 |
| Cereal, shredded wheat, 2 large biscuits | 61 | 15 |
| Soymilk, plain or vanilla, 1 cup | 61 | 15 |
| Black beans, cooked, ½ cup | 60 | 15 |
| Edamame, shelled, cooked, ½ cup | 50 | 13 |
| Peanut butter, smooth, 2 tablespoons | 49 | 12 |
| Bread, whole wheat, 2 slices | 46 | 12 |
| Avocado, cubed, 1 cup | 44 | 15 |
| Potato, baked with skin, 3.5 ounces | 43 | 11 |
| Rice, brown, cooked, ½ cup | 42 | 11 |
| Yogurt, plain, low fat, 8 ounces | 42 | 11 |
| Breakfast cereals, fortified with magnesium | 40 | 10 |
Note: Adapted from US Department of Health and Human Services. National Institutes of Health. Magnesium fact sheet for health professionals. Available from ods.od.nih.gov/factsheets/Magnesium-HealthProfessional/#h3.83
Table 5.
Common oral magnesium formulations
| Common formulations | Trade names | Elemental magnesium content (mg) |
|---|---|---|
| Magnesium oxide | Mag-Ox 400 tablets (400 mg) | 241 |
| Uro-Mag capsules (140 mg) | 84 | |
| Magnesium gluconate | Magnesium gluconate tablets (500 mg) | 27 |
| Magnesium chloride | Slow-Mag tablets (535 mg plus 212 mg calcium) | 64 |
| Magnesium oxide with soy protein | Mg Pro Plus tablets (133 mg plus 26 mg soy protein) | 133 |
| Magnesium L-lactate dihydrate | Mag Tab SR tablets (84 mg) | 84 |
| Magnesium-L-aspartate hydrochloride | Maginex tablets (615 mg) | 61 |
Notes: Mag-Ox 400, Uro-Mag, Blaine Pharmaceuticals, Santa Fe Springs, CA, USA; Magnesium gluconate 500 mg, eg, Mag-G, Pernix Therapeutics, Houston, TX, USA; Slow-Mag, Purdue Pharma LP, Stamford, CT, USA; Mg Plus Protein, Miller Pharmacal Group, Inc., Carol Stream, IL, CA, USA; Mag Tab SR, Niche Pharmaceuticals, Southlake, TX, USA; Maginex 615 mg, Health Care Prod/Hi-Tech, Amityville, NY, USA.
Abbreviation: SR, slow release.
In conclusion, recent research has contributed a great deal to our understanding of various congenital and acquired hypomagnesemic states. Although no specific targeted therapy is currently available, routine corrections of underlying etiologies and electrolyte and metabolic derangements, increase in dietary magnesium intake, and/or magnesium supplementation are recommended, as chronic hypomagnesemic state has been reported to be associated with various micro- and macrovascular complications.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Sanders GT, Huijen HJ, Sanders R. Magnesium in disease: a review with special emphasis on the serum ionized magnesium. Clin Chem Lab Med. 1999;37(11–12):1011–1033. doi: 10.1515/CCLM.1999.151. [DOI] [PubMed] [Google Scholar]
- 2.Saris NE, Mervaala E, Karppanen H, Khawaja JA, Lewenstam A. Magnesium: an update on physiological, clinical and analytical aspects. Clin Chim Acta. 2000;294(1–2):1–26. doi: 10.1016/s0009-8981(99)00258-2. [DOI] [PubMed] [Google Scholar]
- 3.Swaminathan R. Magnesium metabolism and its disorders. Clin Biochem Rev. 2003;24(2):47–66. [PMC free article] [PubMed] [Google Scholar]
- 4.Flink EB. Magnesium deficiency: etiology and clinical spectrum. Acta Med Scand Suppl. 1981;647:125–137. doi: 10.1111/j.0954-6820.1981.tb02648.x. [DOI] [PubMed] [Google Scholar]
- 5.Jahnen-Dechent W, Ketteler M. Magnesium basics. Clin Kidney J. 2012;5(Suppl 1):i3–i14. doi: 10.1093/ndtplus/sfr163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz R, Spencer H, Welsh JJ. Magnesium absorption in human subjects from leafy vegetables, intrinsically labeled with stable 26 Mg. Am J Clin Nutr. 1984;39(4):571–576. doi: 10.1093/ajcn/39.4.571. [DOI] [PubMed] [Google Scholar]
- 7.Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Intestinal absorption of magnesium from food and supplements. J Clin Invest. 1991;88(2):396–402. doi: 10.1172/JCI115317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quamme GA. Recent developments in intestinal magnesium absorption. Cur Opin Gastroenterol. 2008;24(2):230–235. doi: 10.1097/MOG.0b013e3282f37b59. [DOI] [PubMed] [Google Scholar]
- 9.Schweigel M, Martens H. Magnesium transport in the gastrointestinal tract. Front Biosci. 2000;5:D666–D677. doi: 10.2741/schweigel. [DOI] [PubMed] [Google Scholar]
- 10.Schlingmann KP, Gudermann T. A critical role of TRPM channel-kinase for human magnesium transport. J Physiol. 2005;566(Pt 2):301–308. doi: 10.1113/jphysiol.2004.080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlingmann KP, Weber S, Peters M, et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31(2):166–170. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- 12.Plasma and bone magnesium. Nutrition Reviews. 1960;18(7):214–216. doi: 10.1111/j.1753-4887.1960.tb01776.x. [no authors listed] [DOI] [PubMed] [Google Scholar]
- 13.Quamme GA, Dirks JH. The physiology of renal magnesium handling. Ren Physiol. 1986;9(5):257–269. doi: 10.1159/000173090. [DOI] [PubMed] [Google Scholar]
- 14.Quamme GA. Control of magnesium transport in the thick ascending limb. Am J Physiol. 1989;256(2 Pt 2):F197–F210. doi: 10.1152/ajprenal.1989.256.2.F197. [DOI] [PubMed] [Google Scholar]
- 15.Blanchard A, Jeunemaitre X, Coudol P, et al. Paracellin-1 is critical for magnesium and calcium reabsorption in the human thick ascending limb of Henle. Kidney Int. 2001;59(6):2206–2215. doi: 10.1046/j.1523-1755.2001.00736.x. [DOI] [PubMed] [Google Scholar]
- 16.Weber S, Schneider L, Peters M, et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2001;12(9):1872–1881. doi: 10.1681/ASN.V1291872. [DOI] [PubMed] [Google Scholar]
- 17.Konrad M, Schaller A, Seelow D, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79(5):949–957. doi: 10.1086/508617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou J, Goodenough DA. Claudin-16 and claudin-19 function in the thick ascending limb. Curr Opin Nephrol Hypertens. 2010;19(5):483–488. doi: 10.1097/MNH.0b013e32833b7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voets T, Nilius B, Hoefs S, et al. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem. 2004;279(1):19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 20.Konrad M, Schlingmann KP, Guderman T. Insights into the molecular nature of magnesium homeostasis. Am J Physiol Renal Physiol. 2004;286(4):F599–F605. doi: 10.1152/ajprenal.00312.2003. [DOI] [PubMed] [Google Scholar]
- 21.Reily RF, Ellison DH. Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol Rev. 2000;80(1):277–313. doi: 10.1152/physrev.2000.80.1.277. [DOI] [PubMed] [Google Scholar]
- 22.Lemmink HH, Knoers NV, Károlyi L, et al. Novel mutations in the thiazide-sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int. 1998;54(3):720–730. doi: 10.1046/j.1523-1755.1998.00070.x. [DOI] [PubMed] [Google Scholar]
- 23.Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115(6):1651–1658. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yogi A, Callera GE, O’Connor SE, et al. Dysregulation of renal transient receptor potential melastatin 6/7 but not paracellin-1 in aldosterone-induced hypertension and kidney damage in a model of hereditary hypomagnesemia. J Hypertens. 2011;29(7):1400–1410. doi: 10.1097/HJH.0b013e32834786d6. [DOI] [PubMed] [Google Scholar]
- 25.Glaudemans B, van der Wijst J, Scola RH, et al. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest. 2009;119(4):936–942. doi: 10.1172/JCI36948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groenestege WM, Thébault S, van der Wijst J, et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest. 2007;117(8):2260–2267. doi: 10.1172/JCI31680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol. 2009;20(1):78–85. doi: 10.1681/ASN.2008030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meij IC, Koenderink JB, van Bokhoven H, et al. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+), (K+)-ATPase gamma-subunit. Nat Genet. 2000;26(3):265–266. doi: 10.1038/81543. [DOI] [PubMed] [Google Scholar]
- 29.Arystarkhova E, Sweadner KJ. Splice variants of the gamma subunit (FXYD2) and their significance in regulation of the Na, K-ATPase in kidney. J Bioenerg Biomembr. 2005;37(6):381–386. doi: 10.1007/s10863-005-9475-y. [DOI] [PubMed] [Google Scholar]
- 30.Ferrè S, Veenstra GJ, Bouwmeester R, Hoenderop JG, Bindels RJ. HNF-1B specifically regulates the transcription of the gamma a-subunit of the Na+/K+-ATPase. Biochem Biophys Res Commun. 2011;404(1):284–290. doi: 10.1016/j.bbrc.2010.11.108. [DOI] [PubMed] [Google Scholar]
- 31.Kolatsi-Joannou M, Bingham C, Ellard S, et al. Hepatocyte nuclear factor-1 beta: a new kindred with renal cysts and diabetes and gene expression in normal human development. J Am Soc Nephrol. 2001;12(10):2175–2180. doi: 10.1681/ASN.V12102175. [DOI] [PubMed] [Google Scholar]
- 32.Scholl UI, Choi M, Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106(14):5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ 10 mutations. N Engl J Med. 2009;360(19):1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulais M, Bloch-Faure M, Picard N, et al. Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSame/EAST syndrome. Proc Natl Acad Sci U S A. 2011;108(25):10361–10366. doi: 10.1073/pnas.1101400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cha SK, Huang C, Ding Y, Qi X, Huang CL, Miller RT. Calcium-sensing receptor decreases cell surface expression of the inwardly rectifying K+ channel Kir4.1. J Biol Chem. 2011;286(3):1828–1835. doi: 10.1074/jbc.M110.160390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stuiver M, Lainez S, Will C, et al. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet. 2011;88(3):333–343. doi: 10.1016/j.ajhg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song Y, Hsu YH, Niu T, Manson JE, Buring JE, Liu S. Common genetic variants of the ion channel transient receptor potential membrane melastatin 6 and 7 (TRPM6 and TRPM7), magnesium intake, and risk of type 2 diabetes in women. BMC Med Genet. 2009;10:4. doi: 10.1186/1471-2350-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groenestege WM, Hoenderop JG, van den Heuvel L, Knoers N, Bindels RJ. The epithelial Mg2+ channel transient receptor potential melastatin 6 is regulated by dietary Mg2+ content and estrogens. J Am Soc Nephrol. 2006;17(4):1035–1043. doi: 10.1681/ASN.2005070700. [DOI] [PubMed] [Google Scholar]
- 39.Barr CS, Lang CC, Hanson J, Arnott M, Kennedy N, Struthers AD. Effects of adding spironolactone to an angiotensin-converting enzyme inhibitor in chronic congestive heart failure secondary to coronary artery disease. Am J Cardiol. 1995;76(17):1259–1265. doi: 10.1016/s0002-9149(99)80353-1. [DOI] [PubMed] [Google Scholar]
- 40.Gao X, Peng L, Adhikari CM, Lin J, Zuo Z. Spironolactone reduced arrhythmia and maintained magnesium homeostasis in patients with congestive heart failure. J Cardiac Fail. 2007;13(3):170–177. doi: 10.1016/j.cardfail.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Sontia B, Montezano AC, Paravicini T, Tabet F, Touyz RM. Downregulation of renal TRMP7 and increased inflammation and fibrosis in aldosterone-infused mice: effects of magnesium. Hypertension. 2008;51(4):915–921. doi: 10.1161/HYPERTENSIONAHA.107.100339. [DOI] [PubMed] [Google Scholar]
- 42.MacIntyre I, Robinson CJ. Magnesium and the gut: experimental and clinical observations. Ann N Y Acad Sci. 1969;162(2):865–873. doi: 10.1111/j.1749-6632.1969.tb13016.x. [DOI] [PubMed] [Google Scholar]
- 43.Hersh T, Siddiqui DA. Magnesium and the pancreas. Am J of Clin Nutr. 1973;26(3):362–366. doi: 10.1093/ajcn/26.3.362. [DOI] [PubMed] [Google Scholar]
- 44.Agus ZS. Hypomagnesemia. J Am Soc Nephrol. 1999;10(7):1616–1622. doi: 10.1681/ASN.V1071616. [DOI] [PubMed] [Google Scholar]
- 45.Elisaf M, Merkouropoulos M, Tsianos EV, Siamopoulos KC. Pathogenetic mechanisms of hypomagnesemia in alcoholic patients. J Trace Elem Med Biol. 1995;9(4):210–214. doi: 10.1016/S0946-672X(11)80026-X. [DOI] [PubMed] [Google Scholar]
- 46.Tambyah PA, Rauff A, Lee KO. Persistent hypomagnesaemia following parathyroid surgery, hypermagnesuria as a possible cause. Ann Acad Med Singapore. 1990;19(4):536–539. [PubMed] [Google Scholar]
- 47.Weiselberg EC, Gonzalez M, Fisher M. Eating disorders in the twenty-first century. Minerva Ginecol. 2011;63(6):531–545. [PubMed] [Google Scholar]
- 48.Frazier TG, Mucha ME, Rush IH, Trull EJ, Carlson SA, O’Connor JA. Hypomagnesemia: higher risk using total parenteral nutrition in the treatment of patients with malignancies. J Surg Oncol. 1980;13(1):35–38. doi: 10.1002/jso.2930130106. [DOI] [PubMed] [Google Scholar]
- 49.Flink EB. Magnesium deficiency. Etiology and clinical spectrum. Acta Med Scand Suppl. 1981;647:125–137. doi: 10.1111/j.0954-6820.1981.tb02648.x. [DOI] [PubMed] [Google Scholar]
- 50.Ho KM, Leonard A. Risk factors and outcome associated with hypomagnesemia in massive transfusion. Transfusion. 2011;51(2):270–276. doi: 10.1111/j.1537-2995.2010.02855.x. [DOI] [PubMed] [Google Scholar]
- 51.Aglio LS, Stanford GG, Maddi R, Boyd JL, 3rd, Nussbaum S, Chernow B. Hypomagnesemia is common following cardiac surgery. J Cardiothorac Vasc Anesth. 1991;5(3):201–208. doi: 10.1016/1053-0770(91)90274-w. [DOI] [PubMed] [Google Scholar]
- 52.Gearhart MO, Sorg TB. Foscarnet-induced severe hypomagnesemia and other electrolyte disorders. Ann Pharmacother. 1993;27(3):285–289. doi: 10.1177/106002809302700304. [DOI] [PubMed] [Google Scholar]
- 53.Pham PCT, Pham PMT, Pham SV, Miller JM, Pham PTT. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol. 2007;2(2):366–373. doi: 10.2215/CJN.02960906. [DOI] [PubMed] [Google Scholar]
- 54.Shah GM, Alvarado P, Kirschenbaum MA. Symptomatic hypocalcemia and hypomagnesemia with renal magnesium wasting associated with pentamidine therapy in a patient with AIDS. Am J Med. 1990;89(3):380–382. doi: 10.1016/0002-9343(90)90354-g. [DOI] [PubMed] [Google Scholar]
- 55.Lee CT, Lien YHH, Lai LW, Ng HY, Chiou TT, Chen HC. Variations of dietary salt and fluid modulate calcium and magnesium transport in the renal distal tubule. Nephron Physiol. 2012;122(3–4):19–27. doi: 10.1159/000353199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holmes AM, Hesling CM, Wilson TM. Drug-induced secondary hyperaldosteronism in patients with pulmonary tuberculosis. Q J Med. 1970;39(154):299–315. [PubMed] [Google Scholar]
- 57.Chou CL, Chen YH, Chau T, Lin SH. Acquired Bartter-like syndrome associated with gentamicin administration. Am J Med Sci. 2005;329(3):144–149. doi: 10.1097/00000441-200503000-00007. [DOI] [PubMed] [Google Scholar]
- 58.Zietse R, Zoutendijk R, Hoorn EJ. Fluid, electrolyte and acid-base disorders associated with antibiotic therapy. Nat Rev Nephrol. 2009;5(4):193–202. doi: 10.1038/nrneph.2009.17. [DOI] [PubMed] [Google Scholar]
- 59.Ledeganck KJ, Boulet GA, Horvath CA, et al. Effects of renal distal tubule transporters TRPM6 and NCC in a rat model of cyclosporine nephrotoxicity and effect of EGF treatment. Am J Physiol Renal Physiol. 2011;301(3):F486–F493. doi: 10.1152/ajprenal.00116.2011. [DOI] [PubMed] [Google Scholar]
- 60.Nijenhuis T, Hoenderop JG, Bindels RJ. Downregulation of Ca(2+) and Mg(2+) transport proteins in the kidney explains tacrolimus (FK506)-induced hypercalciuria and hypomagnesemia. J Am Soc Nephrol. 2004;15(3):549–557. doi: 10.1097/01.asn.0000113318.56023.b6. [DOI] [PubMed] [Google Scholar]
- 61.Ikari A, Okude C, Sawada H, Takahashi T, Sugatani J, Miwa M. Down-regulation of TRPM6-mediated magnesium influx by cyclosporin A. Naunyn Schmiedebergs Arch Pharmacol. 2008;377(4–6):333–343. doi: 10.1007/s00210-007-0212-4. [DOI] [PubMed] [Google Scholar]
- 62.Aisa Y, Mori T, Nakazato T, et al. Effects of immunosuppressive agents on magnesium metabolism early after allogeneic hematopoietic stem cell transplantation. Transplantation. 2005;80(8):1046–1050. doi: 10.1097/01.tp.0000174340.40585.d4. [DOI] [PubMed] [Google Scholar]
- 63.Ikari A, Sanada A, Sawada H, Okude C, Tonegawa C, Sugatani J. Decrease in transient receptor potential melastatin 6 mRNA stability caused by rapamycin in renal tubular eptiherlial cells. Biochim Biophys Acta. 2011;1808(6):1502–1508. doi: 10.1016/j.bbamem.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 64.da Silva CA, de Bragança AC, Shimizu MH, et al. Rosiglitazone prevents sirolimus-induced hypomagnesemia, hypokalemia, and downregulation of NKCC2 protein expression. Am J Physiol Renal Physiol. 2009;297(4):F916–F922. doi: 10.1152/ajprenal.90256.2008. [DOI] [PubMed] [Google Scholar]
- 65.Sánchez-Fructuoso AI, Santín Cantero JM, Pérez Flores I, Valero San Cecilio R, Calvo Romero N, Vilalta Casas R. Changes in magnesium and potassium homeostasis after conversion from a calcineurin inhibitor regimen to an mTOR inhibitor-based regimen. Transplant Proc. 2010;42(8):3047–3049. doi: 10.1016/j.transproceed.2010.07.081. [DOI] [PubMed] [Google Scholar]
- 66.Nijenhuis T, Renkema KY, Hoenderop JG, Bindels RJ. Acid-base status determines the renal expression of Ca2+ and Mg2+ transport proteins. J Am Soc Nephrol. 2006;17(3):617–626. doi: 10.1681/ASN.2005070732. [DOI] [PubMed] [Google Scholar]
- 67.Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. Am J Physiol Renal Physiol. 2013;305(9):F1277–F1287. doi: 10.1152/ajprenal.00363.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Srivastava R, Aslam M, Kalluri SR, et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367(2):115–123. doi: 10.1056/NEJMoa1110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loffing J, Loffing-Cueni D, Hegyl I, et al. Thiazide treatment of rats provokes apoptosis in distal tubule cells. Kidney Int. 1996;50(4):1180–1190. doi: 10.1038/ki.1996.426. [DOI] [PubMed] [Google Scholar]
- 70.Younes-Ibrahim M, Barnese M, Burth P, Castro-Faria MV. Inhibition of purified human kidney Na+-K+-ATPase by cyclosporine A: a possible mechanism for drug human nephrotoxicity. Ann N Y Acad Sci. 2003;986:633–635. doi: 10.1111/j.1749-6632.2003.tb07270.x. [DOI] [PubMed] [Google Scholar]
- 71.Rodrigo R, Thielemann L. Effects of chronic and acute ethanol exposure on renal (Na+K)-ATPase in the rat. Gen Pharmacol. 1997;29(5):719–723. doi: 10.1016/s0306-3623(97)00009-8. [DOI] [PubMed] [Google Scholar]
- 72.do Pazo-Oubiña F, Estefanell-Tejero A, Riu-Viladoms G, Anglada-Martínez H, Molas-Ferrer G, Creus-Baró N. Magnesium monitoring practice in monoclonal anti-epidermal growth factor receptor antibodies therapy. J Clin Pharma Ther. 2013;38(2):101–103. doi: 10.1111/jcpt.12028. [DOI] [PubMed] [Google Scholar]
- 73.Ledeganck KJ, Boulet GA, Bogers JJ, Verpooten GA, De Winter BY. The TRPM6/EGF pathway is downregulated in a rat model of cisplatin nephrotoxicity. PLoS One. 2013;8(2):e57016. doi: 10.1371/journal.pone.0057016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chrispal A, Boorugu H, Prabhakar AT, Moses V. Amikacin-induced type 5 Bartter-like syndrome with severe hypocalcemia. J Postgrad Med. 2009;55(3):208–210. doi: 10.4103/0022-3859.57407. [DOI] [PubMed] [Google Scholar]
- 75.Chou CL, Chen YH, Chau T, Lin SH. Acquired Bartter-like syndrome associated with gentamicin administration. Am J Med Sci. 2005;329(3):144–149. doi: 10.1097/00000441-200503000-00007. [DOI] [PubMed] [Google Scholar]
- 76.Rasmussen AQ, Jørgensen NR, Schwarz P. Clinical and biochemical outcomes of cinacalcet treatment of familial hypocalciuric hypercalcemia: a case series. J Med Case Rep. 2011;5(1):564. doi: 10.1186/1752-1947-5-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nair AV, Hocher B, Verkaart S, et al. Loss of insulin-induced activation of TRPM6 magnesium channels results in impaired glucose tolerance during pregnancy. Proc Natl Acad Sci U S A. 2012;109(28):11324–11329. doi: 10.1073/pnas.1113811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Finkelstein A, Holz R. Aqueous pores created in thin lipid membranes by the polyene antibiotics nystatin and amphotericin B. Membranes. 1973;2:377–408. [PubMed] [Google Scholar]
- 79.Cunningham JJ, Anbar RD, Crawford JD. Hypomagnesemia: a multifactorial complication of treatment of patients with severe burn trauma. JPEN J Parenter Enteral Nutr. 1987;11(4):364–367. doi: 10.1177/0148607187011004364. [DOI] [PubMed] [Google Scholar]
- 80.Elisaf M, Panteli K, Theodorou J, Siamopoulos KC. Fractional excretion of magnesium in normal subjects and in patients with hypomagnesemia. Magnes Res. 1997;10(4):315–320. [PubMed] [Google Scholar]
- 81.Pham PC, Pham PM, Pham PA, et al. Lower serum magnesium levels are associated with more rapid decline of renal function in patients with diabetes mellitus type 2. Clin Nephrol. 2005;63(6):429–436. doi: 10.5414/cnp63429. [DOI] [PubMed] [Google Scholar]
- 82.Van Laecke S, Van Biesen W, Vanholder R. Hypomagnesaemia, the kidney and the vessels. Nephrol Dial Transplant. 2012;27(11):4003–4010. doi: 10.1093/ndt/gfs126. [DOI] [PubMed] [Google Scholar]
- 83.US Department of Health and Human Services. National Institutes of Health Magnesium fact sheet for health professionals. [Accessed March 27, 2014]. Available from ods.od.nih.gov/factsheets/Magnesium-HealthProfessional/#h3.