

Abstract
PTP1B, a validated therapeutic target for diabetes and obesity, plays a critical positive role in HER2 signaling in breast tumorigenesis. Efforts to develop therapeutic inhibitors of PTP1B have been frustrated by the chemical properties of the active site. We defined a novel mechanism of allosteric inhibition that targets the C-terminal, non-catalytic segment of PTP1B. We present the first ensemble structure of PTP1B containing this intrinsically disordered segment, within which we identified a binding site for the small molecule inhibitor, MSI-1436. We demonstrate binding to a second site close to the catalytic domain, with cooperative effects between the two sites locking PTP1B in an inactive state. MSI-1436 antagonized HER2 signaling, inhibited tumorigenesis in xenografts and abrogated metastasis in the NDL2 mouse model of breast cancer, validating inhibition of PTP1B as a therapeutic strategy in breast cancer. This new approach to inhibition of PTP1B emphasizes the potential of disordered segments of proteins as specific binding sites for therapeutic small molecules.
INTRODUCTION
Disruption of the normal patterns of protein phosphorylation results in aberrant regulation of signal transduction and has been implicated in the etiology of a variety of major human diseases. The ability to modulate signaling pathways selectively holds enormous therapeutic potential. The first drugs directed against protein tyrosine kinases (PTKs) represent breakthroughs in cancer therapy. For example, the humanized antibody Herceptin (Trastuzumab) targets the PTK HER2 (ERBB2), which is amplified and/or overexpressed in ~25% of breast tumors, where it associated with poor prognosis1,2. Although Herceptin is a treatment of choice, the overall success rate is low and patients develop resistance to the drug. Similar problems have limited the success of other PTK-directed inhibitors3,4. Therefore, it is anticipated that alternative therapies, to target simultaneously different signaling enzymes and processes, may be more effective than targeting individual PTKs alone. Consequently, a major problem remains the identification of such alternative therapies.
The focus on PTKs for drug development ignores the other major component of phosphorylation-dependent regulation of signaling. Protein phosphorylation is a reversible process, in which the coordinated and competing activities of kinases and phosphatases are important for determining signaling outcome, but the protein tyrosine phosphatases (PTPs) remain a largely untapped resource for drug development. Since its discovery 25 years ago5, PTP1B has become a highly validated therapeutic target for diabetes and obesity6. Consequently, there have been major programs in industry focused on developing small molecule inhibitors of PTP1B that followed standard procedures of targeting the active site; however, these efforts have been frustrated by technical challenges arising from the chemistry of PTP catalysis. Although it was possible to generate potent, specific and reversible inhibitors of PTP1B, such molecules were highly charged and thus of limited drug development potential7.
PTP1B function is not restricted to metabolic regulation; it is over-expressed in breast tumors together with HER28,9. Mice expressing activated alleles of HER2 in mammary glands develop multiple mammary tumors and frequent metastases to the lung; however, when such mice were crossed with PTP1B-null mice, tumor development was delayed and the incidence of lung metastases was decreased. Conversely, targeted overexpression of PTP1B alone was sufficient to drive mammary tumorigenesis8. These observations suggest that PTP1B may play a positive role in promoting signaling events associated with breast tumorigenesis. Therefore, inhibition of PTP1B function may represent a novel therapeutic strategy not only to address diabetes and obesity, but also mammary tumorigenesis and malignancy. Consequently new approaches to inhibition of PTP1B, which circumvent the problems with active site-directed small molecule inhibitors, are required to reinvigorate drug development efforts against this highly validated target.
Our approach has been to develop allosteric inhibitors of PTP1B that bind to unique sites on the enzyme distinct from the active site. PTP1B was purified originally from human placenta as a 37kDa catalytic domain comprising residues 1–3215, which has been the focus of attention to date for mechanistic analysis, as well as for drug screening. Nevertheless, PTP1B exists in vivo as a longer protein of ~50kDa, in which the C-terminal segment, which is deleted from the 37kDa protein, serves a regulatory function10. We have demonstrated that an aminosterol natural product, MSI-1436/Trodusquemine11, inhibited the full-length form of PTP1B preferentially in a reversible, selective manner. We have identified the binding sites for MSI-1436 in PTP1B and defined the mechanism of inhibition. Furthermore, we have demonstrated that by targeting PTP1B, MSI-1436 attenuated HER2 signaling, resulting in extensive inhibition of tumor growth and abrogation of metastasis to the lung in HER2-positive animal models of breast cancer. Overall, these data establish that PTP1B is a bona fide target for therapeutic intervention in HER2-positive cancer and illustrate a novel mechanism for specific inhibition of PTP1B through which such intervention may be achieved.
RESULTS
MSI-1436 was a non-competitive inhibitor of PTP1B
Using a small molecule fluorescent substrate DiFMUP, we observed no change in Km, but a significant decrease in Vmax (Supplementary Results, Supplementary Fig. 1). MSI-1436 was a more potent inhibitor of PTP1B1-405, which contains the non-catalytic C-terminal segment of the protein, than the PTP1B1-321 catalytic domain (Fig. 1a, b). It was a reversible non-competitive inhibitor of both forms of the enzyme, although with a 7-fold higher affinity for PTP1B1-405 (0.6 μM) compared to PTP1B1-321 (4 μM). Assays performed with pTyr-reduced carboxamidomethylated, maleylated lysozyme yielded a similar Ki of 600 nM for PTP1B1-405. Inhibition was independent of incubation time and was reversible (Supplementary Fig. 2). Although MSI-1436 inhibited PTP1B1-405, it was less effective against a panel of seven PTPs and two dual specificity phosphatases (Supplementary Table 1).
Figure 1. MSI-1436 was a non-competitive inhibitor of PTP1B.

a. Structure of MSI-1436/Trodusquemine.
b. Dixon plot for PTP1B1-321 (left) and PTP1B1-405 (right) showing 1/rate versus varying concentrations of MSI-1436 at 2 μM (●), 5 μM (■) and10 μM (▲) substrate concentrations. The lines intersect on the x-axis to give a Ki of 4 μM for PTP1B1-321 and 0.6 μM for PTP1B1-405. Data are representative of three independent experiments.
c. Isothermal titration calorimetry analysis. Binding thermograms for PTP1B1-321 (left), and PTP1B1-405 (right) are shown in the upper panels. Non-linear least square curves (lower panels) were fitted to enthalpies integrated from the individual titrations. The binding constants are 300 nM and 1 μM for PTP1B1-405 and 2 μM for PTP1B1-321. Data are representative of three independent experiments.
d. Binding of radiolabelled MSI-1436. PTP1B1-405 (●) or PTP1B1-321 (■) (100 nM) was incubated with varying concentrations of [3H]-1436, PTP1B:[3H]-1436 complex was separated from unbound compound and bound radioactivity (cpm) was measured. The stoichiometry was found to be 1:1 for the PTP1B1-321:MSI-1436 and 1:2 for the PTP1B1-405: MSI-1436 complex. Data represent mean ± s.e.m from three independent experiments.
e. Scatchard analysis of MSI-1436 binding to PTP1B1-405 (upper). B and F represent bound and free MSI-1436 (μmol). Hill plot (lower) where Y represents fraction of binding sites that are occupied by MSI-1436 on PTP1B1-405. The slope yields a Hill coefficient of 1.3 and Kd of 0.7 μM. Data are representative of three independent experiments.
f. Displacement titrations. PTP1B1-321 (left) or PTP1B1-405 (right) (100 nM) was incubated with 5 μM tritiated MSI-1436 for 5 minutes in assay buffer. Following the incubation, varying concentrations of BBR (0–10 μM) were added for 30 minutes. The extent of MSI-1436 displaced by BBR was determined by measuring the residual [3H]-MSI-1436 bound to the enzyme. The experiment was repeated three times and error bars represent S.E.M.
We investigated the stoichiometry of the enzyme-inhibitor complex using isothermal titration calorimetry (ITC) and demonstrated that binding of MSI-1436 to the “short form” of PTP1B1-321 was exothermic and fitted a one-site reaction with a Kd of 2 μM. In contrast, the isotherm generated by binding to PTP1B1-405 was distinct and best fitted a two-site model, with a Kd of 0.3 μM for one site and 3.0 μM for the second site (Fig. 1c). To validate the two-site model for PTP1B1-405, we used [3H]-MSI-1436 and measured inhibitor binding directly by scintillation counting, revealing a stoichiometry of 2 mol/mol for PTP1B1-405 and 1 mol/mol for PTP1B1-321 (Fig. 1d). The calculated binding constants correlated well with the enzyme kinetics data. Scatchard analysis revealed a pronounced concave curvature in the plot, indicative of positive cooperativity between the two sites in PTP1B1-405. Furthermore, a Hill plot indicated a Hill co-efficient (nH) of 1.3, also consistent with positive cooperativity (Fig. 1e). In addition, we performed a displacement titration with a previously characterized allosteric inhibitor, a derivative of benzbromarone12. We observed that this benzbromarone derivative displaced MSI-1436 completely from PTP1B1-321, whereas only 50% displacement was achieved with PTP1B1-405 (Fig. 1f). These data illustrate that MSI-1436 is a selective, non-competitive, reversible inhibitor of PTP1B, utilizing a novel mechanism of inhibition with a unique binding site in the C-terminus of the protein.
MSI-1436 induced a conformational change in PTP1B
In previous attempts to crystallize forms of PTP1B containing the C-terminal, non-catalytic segment, we noted that this segment remained largely unstructured. Consistent with this, dynamic light scattering revealed that the radius of hydration (Rh) of PTP1B1-393 (32.8 Å ± 0.2 Å) was more than 8 Å larger than that of PTP1B1-301 (24.1 Å ± 0.4 Å), demonstrating that PTP1B residues 300–393 must adopt an extended structure in solution (Supplementary Fig. 3). Although such intrinsically disordered segments in proteins have been associated with oligomerization, such as dimer formation13,14, we demonstrated that MSI-1436 did not cause the phosphatase to oligomerize. In fact, PTP1B1-405 was retained on the gel filtration column for longer in the presence, compared to the absence of MSI-1436; the calculated molecular weight of PTP1B1-405 was ~47k in the absence, and ~43k in the presence of the inhibitor (Supplementary Fig. 4). In contrast, there was no change in the elution of PTP1B1-321, which displayed an apparent Mr of 37k (Supplementary Fig. 5). Intrinsically disordered segments of proteins are also known to be sensitive to proteases. Therefore, we tested the effect of saturating concentrations of MSI-1436 on sensitivity to trypsin and observed that PTP1B1-405 adopted a more trypsin-resistant state (Fig. 2a, Supplementary Fig. 6). These data suggest that binding of inhibitor may impose a more compact structure on the flexible C-terminus in PTP1B1-405, thereby decreasing the effective hydrodynamic radius of the protein and protecting the enzyme from proteolysis.
Figure 2. MSI-1436 induced a conformational change in PTP1B.

a. SDS-PAGE following limited proteolysis of PTP1B1-405 with trypsin in the absence (left) and presence (right) of MSI-1436. Purified PTP1B1-405 (2 μM) was incubated without (left) or with (right) MSI-1436 (5 μM) for 30 min with trypsin/PTP1B (mol/mol) ratio of 1:10, 0.5:10, 0.25:10, 0.125:10, 0.06:10,0.03:10, 0.01:10, 0.005:10, 0:10 from left to right, respectively. Reactions were terminated with SDS sample buffer, and the digested products were analyzed by 20% SDS-PAGE and stained by Coomassie blue.
b. Schematic model of the PTP1B fusion protein tagged with CFP and YFP on the N- and C- terminus respectively, showing a conformational change induced by binding of the inhibitor (MSI-1436) that results in a change in FRET (upper panel). The effect of MSI-1436 binding to PTP1B1-405 was followed by FRET (lower left panel). Representative spectra from the experiment in which CFP-PTP1B1-405-YFP was titrated with MSI-1436, covering a concentration range of 0 – 10 μM revealed the inhibitor-induced decrease in CFP emission at 475 nm and concomitant increase in the YFP emission at 535 nm. The increase in YFP with addition of MSI- 1436 was plotted against the concentration of MSI-1436 to derive a Kd of 0.8 μM (lower right panel).
We generated a construct in which the two fluorophores CFP and YFP were fused to the amino and carboxy termini of PTP1B, respectively, to investigate MSI-1436-induced conformational changes by intramolecular FRET. The CFP-PTP1B-YFP fusion protein was excited at 440 nm. The emission peak of CFP is expected to be at 475 nm; however, if CFP and YFP were brought into close proximity by inhibitor binding, the energy from CFP would be transferred to YFP, causing the YFP emission peak at 535 nm, with a concomitant decrease in the emission peak for CFP at 475 nm (Fig. 2b). At low concentrations of MSI-1436 (10-fold below the Ki) a strong emission peak at 475 nm (CFP emission) and a relatively smaller peak at 535 nm (YFP emission) were observed. As the concentration of MSI-1436 was increased, a strong FRET signal between CFP and YFP was observed. A plot of the emission at 535 nm against the concentration of MSI-1436 yielded a Kd of 0.8 μM for MSI-1436 binding, in close agreement with the Ki (Fig 2b). In contrast, excitation of CFP-PTP1B1-321-YFP at 440nm did not result in FRET at any concentration of MSI-1436 tested (Supplementary Fig. 7), highlighting the importance of the C-terminal segment for the recognition of the inhibitor by PTP1B. The data suggest that binding of MSI-1436 induced a conformational change that brought the C-terminus of CFP- PTP1B1-405-YFP closer to the N-terminus, which agrees with our gel-filtration data indicating that inhibitor binding induced a more compact structure.
The structure of PTP1B1-393 defined by NMR spectroscopy
To understand the interaction of MSI-1436 with the long form of PTP1B, structural data were essential. Biomolecular NMR spectroscopy is a technique that permits structural analysis of extended, flexible segments of biomacromolecules. To overcome the technical complexities that are characteristic of NMR analysis of proteins of 35 kDa, all proteins were expressed in D2O-based medium and TROSY versions of all 2D and 3D experiments were recorded at 800/850 MHz 1H Larmor frequency15. To overcome spectral overlap, 2D [1H,15N] TROSY spectra of single amino acid labeled (15N-Leu, 15N-Tyr, 15N-Phe or 15N-Val) PTP1B were used both to verify existing assignments and to facilitate the assignment of additional residues. The chemical shift index (CSI) calculated from the deviations of Cα and Cβ chemical shifts from random coil values (RefDB16) correlated very well with secondary structures observed in the crystal structure of PTP1B (PDBid 1SUG17).
Peaks corresponding to residues 300–393 of PTP1B in the 2D [1H,15N] TROSY spectrum were on average 5-times more intense than those from PTP1B1-301 and clustered in the center of the spectrum (7.5 to 8.5 ppm 1H dimension; Fig. 3a). This is typical for flexible and mostly unstructured amino acids. Cross peaks in the 2D [1H,15N] TROSY spectrum of PTP1B1-301 overlayed almost perfectly with the corresponding peaks in the 2D [1H,15N] TROSY spectrum of PTP1B1-393 (Fig. 3a, Supplementary Fig. 8). This indicates that residues 300–393 were flexible, intrinsically disordered, did not interact with, and extended away from, PTP1B1-301.
Figure 3. PTP1B residues 300-393 were flexible and predominantly disordered.

a. Overlay of the 2D [1H,15N] TROSY spectra of PTP1B1-301 (black) and PTP1B1-393 (purple). Boxed region (7.5 - 8.5 ppm, 1H dimension) marks PTP1B residues 300-393.
b. Secondary structure propensity (SSP) scores for PTP1B residues 300–393. A score of 1 indicates a fully populated α-helix, while a score of 0.5 indicates an α-helix that is ~50% populated in solution. Helices α8′ and α9′, which are 20 % populated, are denoted with a gray cylinder and labeled above.
c. Representative ensemble of 100 conformers that are superimposed over PTP1B residues 1–284 (gold). PTP1B residues 285–398 (gray) sample a wide range of conformational space.
Secondary structure propensity (SSP) scores derived from the chemical shift index (CSI) revealed that PTP1B residues 300–393 were not entirely unstructured; instead, they contained two α-helices, composed of residues 320 – 327 (α8′) and 360 – 377 (α9′), which were, on average, ~33 and ~22% populated in solution, respectively (Fig. 3b).
To gain further insights into the structure of PTP1B1-393, we modeled an ensemble structure of PTP1B1-393, rather than a single structure, using the crystal structure of PTP1B (PDBid: 1SUG), NMR chemical shift and SAXS data (Supplementary Fig. 9a–e). Experimental chemical shifts were matched with predicted Cα, Cβ, N and HN chemical shifts for PTP1B residues 285–393. We built 15000 starting conformers and the ensemble exhibited, as expected, a Gaussian distribution of Rg values. However, this average calculated SAXS curve did not fit the experimental curve, which, again as expected, did not follow a Gaussian distribution; rather, it was composed of more than average compact and extended conformations (Supplementary Fig. 9f, g). Therefore, from this initial pool, sub-ensembles containing different numbers of conformers were selected for which the average theoretical curve reproduced the experimental SAXS curve (Supplementary Fig. 9h). Selected ensembles of 100 conformers reproduced the SAXS curve and the NMR chemical shifts for the 108 C-terminal residues remarkably well. Thus, these ensembles provide an atomic representation of PTP1B1-393 that is in excellent agreement with all experimental data (Fig. 3c).
MSI-1436 bound to the disordered C-terminus of PTP1B
To define the primary binding site within the C-terminal segment of PTP1B, we tested the interaction between PTP1B1-393 and MSI-1436 at atomic resolution using NMR spectroscopy. We recorded 2D [1H,15N] TROSY spectra of PTP1B1-301 and PTP1B1-393 with MSI-1436 (Fig. 4a, b). No chemical shift perturbations (CSPs) were detected upon addition of MSI-1436 to PTP1B1-301; however, NMR CSP mapping showed that Arg371, Arg373 and Val375 in α9′ and residues Leu299, His310, Ile311, Val334 and Ser393, were most affected by MSI-1436 binding to PTP1B1-393 (Fig. 4c, d), consistent with the importance of the C-terminal segment for the interaction with MSI-1436.
Figure 4. PTP1B residues were perturbed upon MSI-1436 binding.<.

br>a. Overlay of the 2D [1H,15N] TROSY spectra of [2H,15N]-PTP1B1-393 with (light blue) and without (dark blue) MSI-1436 (1.5x molar excess). Inset: 2D [1H,15N] TROSY spectra of 15N-Val-PTP1B1-393 (same coloring scheme). Dashed box marks the region of the spectrum shown in b.
b. Segment of the 2D [1H,15N] TROSY spectrum corresponding to PTP1B residues 300–393.
c. CSP mapping of MSI-1436 binding to PTP1B residues 300–393. Colored lines indicate one (blue) and three (red) standard deviations from the mean,<Δδ>.
d. CSPs mapped onto the structure of a single conformer of PTP1B1-393, using the same color scheme as in panel c. MSI-1436 binding site 1 and 2 are highlighted and the close proximity of binding site 2 to the exosite highlighted by an arrow.
We generated truncation mutants of PTP1B and observed that compared to PTP1B1-405 and PTP1B1-394, the inhibitory potency of MSI-1436 was lower for the two truncation mutants PTP1B (1-367) and (1-346), which were similar to that of the catalytic domain construct PTP1B1-321 (Supplementary Fig. 10). These data indicate that the primary binding site must be located between residues 367–394, which contain helix α9′ (Fig. 4c). Three point mutations, to convert residues V370, S372 and R373 to alanine did not affect enzymatic activity or inhibition by MSI-1436 (data not shown). Nevertheless, helix-destabilizing mutations, replacing the residues with Pro, increased the Ki for MSI-1436 10–20 fold. The S372P and R373P mutations, which were centered in helix α9′, had a greater effect than V370P (Fig. 5a). Taken together, these data show that the primary PTP1B binding site of MSI-1436 is helix α9′.
Figure 5. Effect of mutations in the C-terminus of PTP1B on inhibition by MSI-1436<.
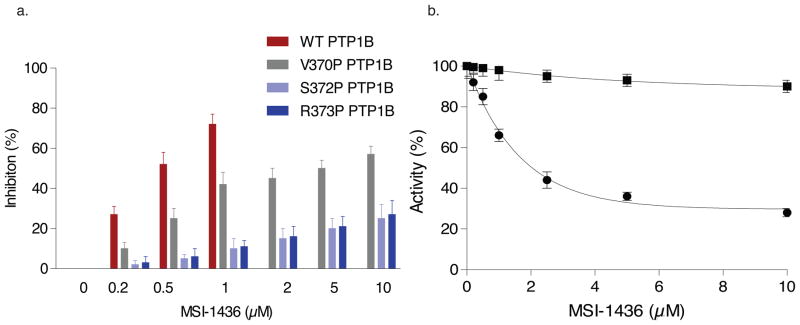
br>a. The bar graph compares the extent of inhibition of PTP1B1-405 and three point mutants located in the NMR-detected helix α9′ (residues 360 – 379 in the C-terminus). The inhibition constants were ki 0.8 μM for PTP1B1-405 and 10 μM, 15 μM and 18 μM for V370P, S372P and R373P, respectively. Data represent mean ± s.e.m from three independent experiments.
b. Introduction of two point mutations in PTP1B1-405, L192A and S372P, resulted in complete loss of inhibition by MSI-1436. Data represent mean ± s.e.m from three independent experiments.
Identification of a second binding site for MSI-1436
The ability of MSI-1436 to inhibit PTP1B1-321, albeit with reduced activity compared to PTP1B1-405, suggested the presence of a binding site within this truncated form of the phosphatase. Previously, Hansen and colleagues identified an allosteric site (exosite) within the PTP1B catalytic domain that binds a series of benzofuran compounds (benzbromarones) that bear no structural similarity to MSI-1436. These compounds were shown to stabilize an inactive state of the enzyme in which the active site cleft is locked in the open conformation12. Mutation of residues known to be critical for benzbromarone binding also impaired inhibition by MSI-1436 (Supplementary Fig. 11, Supplementary Tables 2, 3). The most significant effect on Ki was observed following mutation of Leu192 to Ala; the mutant displayed a Ki of 8 μM, >10-fold greater than wild type PTP1B1-405. This, and the results of the competition studies between benzbromarone and MSI-1436 (Fig. 1f), indicates that there is some overlap between this exosite and the second MSI-1436 binding site.
Nevertheless, there are unique features to the second MSI-1436 binding site. No interaction was detected between PTP1B1-301 and MSI-1436 by NMR spectroscopy, indicating that residues within the 20 amino acids at the C-terminus of the catalytic domain were important for this second binding site. In fact, NMR CSP mapping revealed that Leu299, His310, Ile311 were affected by MSI-1436 binding, consistent with a contribution to the modest inhibition of PTP1B1-321 by MSI-1436. Residues 302–321 in PTP1B flank helix α7, which, as shown in our ensemble PTP1B structure, can adopt numerous conformations. Mutation of residues Val287, Lys292, Leu294 to Ala impaired inhibition by MSI-1436 (Supplementary Tables 2, 3), indicating the importance of α7 helix in mediating MSI-1436 binding. This indicates that MSI-1436 targets a site that is distinct from, but incorporates features of, the already characterized allosteric site, stabilizing an inactive conformation of PTP1B by a novel mechanism that involves the C-terminal segment and a second binding site close to the catalytic segment.
The importance of the combined effects of both binding sites for MSI-1436 in the “long form” of PTP1B was revealed by the fact that a double point mutant in which one residue from each site was mutated, Ser372 to Pro and Leu192 to Ala, preserved catalytic activity but was insensitive to MSI-1436 (Fig. 5b and Supplementary Fig. 11b).
MSI-1436 attenuated HER2-mediated tumorigenesis
In order to determine whether this novel mechanism for inhibition of PTP1B has therapeutic potential, we examined the effects of MSI-1436 in models of HER2-dependent breast cancer. We used MCF10A mammary epithelial cells that express a well-characterized chimeric form of HER2/ErbB2 (10A.B2), the activity of which can be induced with a small molecule dimerizer, AP151018. Activation of HER2 by AP1510 induced characteristic changes in acinar morphogenesis, proliferation and luminal apoptosis in 3D culture, which were blocked by MSI-1436 (Supplementary Fig. 12a). Furthermore, MSI-1436 inhibited migration of 10A.B2 cells in 2D culture, to a similar extent to PTP1B-directed shRNA (Supplementary Fig. 12b). Similarly, MSI-1436 attenuated migration of BT-474 and SKBR3 cells, which are HER2-positive, but not MCF-7 and MDA-MB-231 cells, which are HER2-negative (Supplementary Fig. 12c). Finally, we observed that MSI-1436 inhibited proliferation (Supplementary Fig. 12d), but had limited effect on apoptosis (Supplementary Fig. 12e), in a panel of breast cancer cells that are sensitive to the HER2/EGF receptor dual kinase inhibitor Lapatinib19. These data illustrate that MSI-1436 is an inhibitor of HER-induced signaling in cells; however, it is possible that MSI-1436 may also affect other pathways by targeting PTP1B.
The effect of MSI-1436 was also assessed on breast tumorigenesis in animals. Using a xenograft model, in which HER2-positive BT-474 cells expressing luciferase were injected orthotopically into the mammary fat pad, we tested the effect of administering MSI-1436 both as soon as the cells were injected and following the appearance of palpable tumors. In both cases, treatment with the PTP1B inhibitor intraperitoneally reduced tumor burden (Fig. 6a). In addition, we tested the effects of MSI-1436 in the NDL2 transgenic mouse model of human breast cancer that expresses an activated mutant form of Neu, the rat homolog of human HER2, and develops multiple mammary tumors with metastasis to the lung8,20. NDL2 animals were allowed to develop palpable tumors, then randomly assigned into two groups to receive vehicle control (saline) or MSI-1436 (5 mg/kg) intraperitoneally every third day. MSI-1436-treated animals displayed a marked decrease in tumor size and tumor number relative to the control group (Fig. 6b). Even more striking was the observation that all control animals developed lung metastases, although the extent of metastasis varied within the group. In contrast, metastasis was essentially abrogated in the MSI-1436-treated animals. In two separate studies, conducted with six animals in each group, we detected only one drug-treated animal that developed a small metastasis in the lung, whereas all the animals in the untreated control group presented with a high occurrence of lung metastasis (Fig. 6c). These data highlight the potential of MSI-1436 as an effective approach to counter HER2-induced mammary tumorigenesis and malignancy.
Figure 6. Effect of the allosteric inhibitor of PTP1B, MSI-1436, in cell and animal models of HER2-positive breast cancer<.

br>a. Effect MSI-1436 on tumor xenografts. BT474 cells (5×106) were injected orthotopically into the mammary fat pad of SCID-beige mice. MSI-1436 (5 mg/kg) or saline (control) was injected intraperitoneally as soon as the surgery was performed or once the tumors reached ~100 mm3 in volume. Tumor volume was assessed with a digital caliper for control (open circles) and MSI-1436-treated. Quantitation of tumor volume, measured using a digital caliper, over time (Right panel) (Student t-test, p<0.01). Representative images of three independent experiments are shown.
b. Distribution of tumor weight (left) and tumor number (right) in NDL2 animals treated with MSI-1436 (5 mg/kg) (●) or vehicle control (■) (Student t-test, p<0.001). Data are representative of three independent experiments with more than six animals in each experiment.
c. Effect of MSI-1436 on lung metastases, monitored by histopathological analysis of hematoxylin and eosin-stained lung sections taken from the animals. Mice were treated with saline, or MSI-1436 at 5 mg/kg or 10 mg/kg. Each treatment group contained 6 animals. Left panels are representative images of lung sections from control and MSI-1436-treated animals subjected to H&E staining. The right panel is a quantitation of the whole lung obtained from all the animals in each group (control and MSI-1436-treated) (Student t-test, p<0.001). Data are representative of three independent experiments with more than six animals in each experiment.
d. Tumor lysate was incubated with MSI-1436 immobilized to NHS-activated Sepharose beads for 30 minutes at 4 °C. The beads were washed and the samples were subjected to SDS-PAGE and silver staining (upper panel). Both beads and supernatant were subjected to immunoblotting using anti-PTP1B (FG6) antibody (lower panel) to demonstrate that PTP1B was cleared from the lysate by immobilized MSI-1436. Data are representative of three independent experiments.
e. Migration of control and PTP1B-depleted MCF10-NeuNT cells, in the absence and presence of doxycycline, were compared to MCF10-NeuNT cells overexpressing wild type (WT) PTP1B or the double mutant, PTP1B L192A/S372P, in the presence of doxycycline and in the absence (pale grey bars) and presence of MSI-1436 (1 μM, grey bars and 2 μM dark grey bars). Data are representative of three independent experiments done in duplicates.
f. PTP1B knock-down and re-expression of wild type (WT) and L192A/S372P mutant PTP1B in MCF10A-Neu-NT cells was analyzed by immunoblotting with anti-PTP1B (FG6) antibody. Data are representative of three independent experiments. (Full blot image Supplementary Fig. 20).
MSI-1436 specifically targeted PTP1B in breast cancer models
We took a variety of approaches to demonstrate that the mechanism by which MSI-1436 antagonized HER2 function was manifested through inhibition of PTP1B. First, we tested its effects on tyrosine phosphorylation-dependent signaling. Using the NDL2 mice, we observed a significant increase in tyrosine phosphorylation of p62DOK in tumor samples from animals treated with MSI-1436. Concomitantly, we also observed a decrease in phosphorylation of p44/42 MAP kinase upon MSI-1436 treatment (Supplementary Fig. 13). These signaling changes are consistent with PTP1B being the target of MSI-143621.
To demonstrate a direct interaction with PTP1B, we developed a binding assay in which MSI-1436 was linked to NHS-activated magnetic beads via its free amino group on the spermine tail (Supplementary Fig. 14). The structurally related inactive analog MSI-1459 (3β-spermine, 5α, cholenic acid methyl ester), which has a free amino group like MSI-1436 but does not inhibit PTP1B (Supplementary Fig. 15), was included as a negative control. Bead-immobilized MSI-1436 bound to recombinant PTP1B1-405 and PTP1B1-321 in vitro, whereas MSI-1459 did not (Supplementary Fig. 16a). Furthermore, upon mixing bead-immobilized MSI-1436 with tumor lysates from NDL2 mice a major band at 50 kDa, confirmed as PTP1B by immunoblotting, was retained by the MSI-1436 beads, but was not bound by MSI-1459 (Fig. 6d). Similar results were obtained with lysates of MCF10A cells (Supplementary Fig. 16).
To establish that inhibition of PTP1B was the critical step underlying the effects of MSI-1436, we turned to the PTP1B-L192A/S372P double mutant, which preserved catalytic activity but was insensitive to MSI-1436 (Fig. 5b). We used a transformed breast epithelial cell line, MCF10A-NeuNT, expressing a PTP1B-directed shRNA from a doxycycline-inducible lentiviral vector. In the absence of doxycycline, migration of these cells was attenuated by MSI-1436. In contrast, doxycycline treatment essentially ablated PTP1B expression, which inhibited migration (Fig. 6e). We reconstituted expression of either wild type or L192A/S372P double mutant forms of PTP1B to a similar level as endogenous PTP1B in the control cells (Fig. 6f). Reconstitution with wild type PTP1B rescued cell migration, which remained sensitive to inhibition by MSI-1436; however, although reconstitution with PTP1B-L192A/S372P double mutant rescued cell migration, this was no longer sensitive to MSI-1436 (Fig. 6e). Similarly, we tested the effects of MSI-146 on growth of orthotopic BT474 xenografts in which PTP1B had been suppressed by RNAi, then rescued by re-expressing wild type or MSI-1436-resistant forms of PTP1B. In two independent experiments, we observed that tumor xenografts from BT474 cells re-expressing wild type PTP1B were more sensitive to MSI-1436 than xenografts from BT474 cells re-expressing the mutant protein (Supplementary Fig. 17). These data illustrate that PTP1B is the primary target of MSI-1436 in these effects on HER2-induced cell migration.
DISCUSSION
Through its ability to antagonize insulin and leptin signaling, PTP1B is a highly validated metabolic regulator and therapeutic target for diabetes and obesity. Although there is also evidence to support a role in cancer, this function is less well defined, with reports suggesting both tumor suppressive and oncogenic functions22. Although there are reports linking PTP1B to the down-regulation of several growth promoting signaling pathways, such as inhibitory effects on EGFR signaling23, PTP1B-deficient mice do not display increased incidence of tumors8. Furthermore, although PTP1B has been implicated in the regulation of cadherin-dependent cell adhesion and apoptosis, the literature contains contradictions that prevent the precise definition of a tumor suppressor role for the phosphatase. In fact, the PTPN1 gene encoding PTP1B is located on chromosome 20q13.1-13.224, which is amplified in several cancers, and PTP1B is highly expressed in several tumor types25. For example, HER2 amplification has been associated with overexpression of PTP1B8,26. Furthermore, PTP1B plays a positive role in promoting the tumor phenotype in gastric cancer, where its expression is correlated with metastasis27,28, as well as in prostate29 and colon30 cancer cells. However, this oncogenic function of PTP1B also remains to be fully defined. Although PTP1B has been reported to play a positive role in breast cancer cell invasion8,31, it has also been suggested that the phosphatase is not important for breast tumor maintenance32. Nevertheless, through utilizing a PTP1B-directed allosteric inhibitor, now we provide compelling evidence for the validation of PTP1B as a therapeutic target in HER2-positive breast cancer.
Given the importance of the unmet medical needs that could be addressed by targeting PTP1B, many pharmaceutical companies and academic labs took up the challenge of generating inhibitors of the phosphatase. The current focus has been on producing active site-directed inhibitors, but this has encountered challenges due to the charged nature of the active site and the susceptibility of PTPs to irreversible inactivation due to covalent modification of an essential, catalytic cysteine. The most potent, selective inhibitors are based upon non-hydrolyzable analogues of a pTyr substrate, such as fluorophosphonates33; however, these inhibitors are highly charged, with limited membrane permeability. As industry has set a hurdle of oral bioavailability for the next generation of treatments of diabetes and obesity, these substrate analogues have limited drug development potential. A second problem, and one that also impeded the early stages of development of kinase inhibitors, is the issue of inhibitor selectivity. In the case of kinase inhibitors, most now not only target the active site, but also the ATP binding pocket within the active site, a common feature of all kinases. Furthermore, the ability of an inhibitor to act on more than one kinase has often proven to be advantageous34. This concern about inhibitor selectivity is fuelled by the structural similarities among the catalytic domains of the members of the PTP family. In particular, TCPTP, which is the closest relative of PTP1B, sharing 75% sequence identity in the catalytic domain, regulates inflammation, and ablation of the gene encoding TCPTP in mice is lethal35. Although there are data to suggest that some inhibition of TCPTP may be of benefit to promoting insulin signaling36,37, drug discovery programs have always sought inhibitors that distinguish between these two enzymes. Our approach addresses these concerns. We devised methods to overcome technical problems associated with using recombinant forms of PTP1B that contain the C-terminal segment, including susceptibility to proteolysis. By targeting the unique, non-catalytic segment that is unrelated to TCPTP, or any other member of the PTP family, such allosteric inhibitors would have the potential to be highly specific for PTP1B. Also, the inhibitors we identify in this way will have been missed in screens conducted to date in industry, which have used primarily the truncated form of the enzyme comprising only the catalytic domain.
In this study, we have identified two binding sites for MSI-1436 in the long form of PTP1B, a novel site in the C-terminal segment (helix α9′) and an extended site incorporating helix α7 and residues 299, 310 and 311. Our data suggest a model in which one molecule of the cholestane MSI-1436 could be bound between α7 and the α9′ helices, in a manner analogous to the cholesterol-binding motif that has been identified in the crystal structure of the β-adrenergic receptor38. We noted a similar spatial arrangement and identity of residues in the α7 and α9′ helices in the C-terminal segment of PTP1B to those engaged by cholesterol in two critical α-helices in the β-adrenergic receptor (Supplementary Fig. 18). PTP1B helix α7, together with helix α3 and α6, also forms a previously identified allosteric inhibition site in PTP1B. Benzofuran compounds have been shown to bind to this site and change the ability of helix α3 and the WPD loop to adopt the catalytic competent conformation of PTP1B12. This suggests a mechanism by which the two binding sites for MSI-1436 may communicate to yield the positive cooperativity we observed. Thus, a first molecule of MSI-1436 sandwiched between α7 and the C-terminal α9′ helix may bring the α7 helix close to α3 and α6, forming the second site where another molecule of MSI-1436 would bind. Molecular dynamic simulations revealed that the best conformation with least binding energy between MSI-1436 and PTP1B was consistent with this proposed mechanism of inhibition (Supplementary Table 4 and Supplementary Fig. 18).
The approach of generating allosteric inhibitors has also been applied to several protein kinases with the goal of producing cancer therapeutics. For example, GNF-5 engages a myristate-binding pocket in the ABL PTK, which alters the ATP binding site and may represent an approach to overcome resistance to ATP-competitive inhibitors of BCR-ABL39. However, another unique feature of our approach is that we target a segment of PTP1B that is intrinsically disordered, rather than addressing defined binding pockets. Intrinsically disordered segments of proteins do not have stable secondary or tertiary structure; instead, as we have noted for the C-terminal segment of PTP1B, there is an ensemble of rapidly interconverting structures. It is interesting to note that disordered proteins are particularly prevalent in signaling and disease, where the potential for specificity in the interactions is stimulating efforts to exploit them for therapeutic development13. Attempts have been made to target Myc-Max interactions by this approach40, but our definition of the effects of MSI-1436 on PTP1B is one of the first examples of a therapeutic candidate that works by this mechanism.
In conclusion, inhibition of PTP1B may offer a new approach to attenuating HER2-dependent signaling in a clinical setting. As has been reported for PTK inhibitors, such as lapatinib41,42 and gefitinib43, the effects of MSI-1436 appear to be primarily cytostatic, rather than cytotoxic. This suggests that inhibition of PTP1B may perhaps be most effective in combinatorial therapies, together with other signal transduction-based drugs and cytotoxic agents. Herceptin is the treatment of choice for HER2-dependent cancer, although there are problems with de novo and acquired resistance. It will be of interest to test whether inclusion of MSI-1436 together with Herceptin, to target simultaneously the PTK and its downstream signaling, will prolong the time to development of resistance, or even overcome resistant states. In addition, unlike PTK inhibitors, which have significant side effects, MSI-1436 is well tolerated in patients11. Thus, we are poised for a clinical trial to test this novel therapeutic strategy in HER2-positive cancer.
ONLINE METHODS
Reagents
All chemicals and reagents were purchased from Sigma-Aldrich unless otherwise specified. Global tyrosyl phosphorylation was detected by anti-phosphotyrosine antibody 4G10 (#05-321, Millipore). Anti-p62DOK, anti–phospho-Erk and total ERK antibodies were obtained Cell Signaling Technology. For loading controls for corresponding phosphotyrosine proteins, the membrane was stripped by Restore Stripping Buffer (#21059, Thermo Scientific) and reprobed with antibodies against the individual protein. For the FRET reporter, ECFP and EYFP cDNA was PCR-amplified and cloned on either side of PTP1B in pET28b vector. A spacer containing residues GSGSG was employed between the protein and the fluorophores on each side to provide a flexible connection and also to allow rotational mobility as the individual domains of the protein will be folded separately. CFP-PTP1B-YFP was expressed under IPTG induction in BL21 cells (E. coli) and purified with Ni-NTA matrix exploiting the C-terminal His tag.
Enzyme Kinetics
PTP assays were performed in black polystyrol 96-well plate using DiFMUP as substrate. DiFMUP (10 μM) was added to assay buffer (50 mM Hepes, 100 mM NaCl, 0.1 % BSA, 2 mM DTT, 2 mM EDTA pH 6.5) containing 10 nM purified PTP1B in a final volume of 100 μl. The fluorescence emitted at 450 nm was monitored continuously for 20 minutes using a Gemini XPS fluorescence plate reader. For assays using radiolabelled substrate, reduced carboxamidomethylated and maleylated lysozyme (RCML) was phosphorylated on tyrosine to a stoichiometry up to 0.8 mol 32P/mol of protein, by recombinant GST-FER kinase and [γ32P] ATP, and activity measured as described in44.
Binding assay with [3H]-MSI-1436
Direct binding assays were performed using tritiated MSI-1436. Histidine-tagged PTP1B (100 nM) was incubated with varying concentrations of [3H]-MSI-1436 for 30 minutes in assay buffer (50 mM Hepes, 100 mM NaCl, 0.1 % BSA, 2 mM DTT, 2 mM EDTA pH 6.5) at 25°C. Protein-bound and free [3H]-MSI-1436 were separated by incubating the protein-inhibitor mixture with 50 μl of 50% Ni-NTA beads for 10 minutes at 25°C. The beads were washed with assay buffer containing 150 mM NaCl three times and the inhibitor bound to protein was determined by scintillation counting. The amount of bead-bound protein was estimated by BCA protein quantitation. For Scatchard analysis the number of moles of [3H]-MSI-1436 bound to PTP1B was obtained directly from scintillation counting and number of moles of free (F) [3H]-MSI-1436 was calculated by subtracting the bound from the total number of moles of [3H]-MSI-1436 used in each assay. The ratio of B/F was plotted against B. The extent of positive cooperativity was estimated using a Hill plot, where Y represents the fraction of binding sites that are occupied on the protein and 1 - Y represents the fraction of binding sites that are not occupied. Using the equation , the Kd and the Hill co-efficient (n) were obtained. For displacement titrations PTP1B (100 nM) was saturated with [3H]-MSI-1436, the unbound ligand was separated as mentioned above. The enzyme saturated with [3H]-MSI-1436 was titrated against sulphanamido-benzbromarone (0–10 μM). The residual MSI-1436 bound to the enzyme was measured by scintillation counting.
Isothermal titration calorimetry
The studies were performed using a VP-ITC isothermal titration calorimetry system (GE Healthcare). The titrations where performed at 25°C, in degassed buffer (50 mM HEPES pH 7.0, 100 mM NaCl, 1 mM DTT and 2% DMSO). Protein concentration in the calorimeter cell was 50–100 μM. All the protein samples used in the titration were dialyzed completely against the buffer. Inhibitor (10 mM) in DMSO was diluted to make a stock solution of 500 μM in degassed buffer. The instrument was calibrated by using the heat of dilution of NaCl in water. By titrating buffer into protein solution the heat generated due to protein dilution was estimated and was found to be negligible. The heat of ligand dilution was corrected by subtracting the average heat of injection after saturation. Origin software was used to analyze and fit the data.
Gel filtration
PTP1B (5 μM) was incubated with 10 μM MSI-1436 in a final volume of 200 μl buffer (50 mM HEPES pH7.0, 100 mM NaCl, 0.2 mM EDTA, 2 mM DTT) at 4 C. The reaction mixture was then subjected to gel filtration on a Superdex 200 column (HR30/10; Pharmacia Biotech Inc.). Prior to loading the MSI-1436-saturated protein sample, the column was equilibrated with 50 mM HEPES pH 7.0, 100 mM NaCl, 0.2 mM EDTA, 2 mM DTT containing 100 μM MSI-1436 to ensure the protein is bound to the inhibitor throughout the run. The void volume (Vo) and total volume (Vt) were measured using blue dextran and Commassie blue dye respectively. Fractions of 0.5 ml were collected and the protein concentration in each sample was determined by Bradford assay or recording the UV absorption spectrum at 280 nm. Alternatively, the fractions were precipitated with 10% trichloroacetic acid, and subjected to SDS-PAGE followed by staining of the gel with Coomassie blue. Relative retention (Rt) was determined from the absolute retention volumes (Vabs) according to the equation . The column was calibrated with ferritin, catalase, cytochrome C and BSA as molecular weight standards.
Limited Proteolysis
PTP1B (2 μM) was incubated with MSI-1436 (5 μM) for 10 minutes at 4 °C prior to adding varying amounts of trypsin for 30 minutes at room temperature. The trypsin/PTP1B (mol/mol) ratios were 1:10, 0.5:10, 0.25:10, 0.125:10, 0.06:10,0.03:10, 0.01:10, 0.005:10, 0:10. Reactions were terminated after 5 min by adding phenylmethylsulfonyl fluoride (PMSF) (2 mM final concentration) and hot SDS sample buffer at a 3:2 (v/v) ratio of SDS sample buffer/sample and analysed by SDS-PAGE using 10 % polyacrylamide separating gels.
FRET
Fluorescence spectra were recorded from 400 to 600 nm with excitation at 435 nm using a Gemini XPS fluorescence microplate reader. Excitation and emission bandwidths were set to be 0.5 nm. The emission of 10 nM CFP-PTP1B-YFP in 50 mM HEPES pH7.0, 100 mM NaCl, 2 mM DTT and 0.1% BSA was measured in the absence of MSI-1436, for which a very low FRET signal was observed. MSI-1436 was added to CFP-PTP1B-YFP and FRET was monitored as the concentration of MSI-1436 was increased from 1 nM to 10 μM. The Forster radius (R0), FRET efficiency (E), and distance (R) were calculated using the software PTI Felix32 Analysis, version 1.2. Errors for E and R were estimated to be ~5%.
Protein Expression for NMR studies
A human PTP1B catalytic domain construct (residues 1–301; PTP1B1-301) and PTP1B1-393 (residues 1–393) were subcloned into pRP1B. The plasmid was transformed into E. coli BL21 (DE3) RIL cells (Agilent) and protein expression was induced using 1 mM IPTG at 18 °C for 18h. To prepare uniformly [2H,13C,15N]-labeled and [2H,15N]-labeled protein, cultures were grown in M9 minimal media containing selective antibiotics and [13C]-D-d7-glucose (4 g/l) and/or 15NH4Cl (1 g/l) in 99% D2O (Cambridge Isotope Laboratories or Isotec). A single round of 50% D2O adaptation was necessary to increase the yield of PTP1B. To incorporate single 15N-isotopically labeled amino acids into PTP1B, cultures were grown in minimal media, replacing an unlabeled amino acid with the corresponding 15N-labeled amino acid where appropriate. PTP1B1-301 was labeled with 15N-L-Valine, 15N-L-Tyrosine, 15N-L-Phenylalanine or 15N-L-Leucine, whereas PTP1B1-393 was labeled with 15N-L-Valine or 15N-L-Phenylalanine. Cells were harvested by centrifugation and stored at -80 °C.
Protein Purification
Cell pellets were resuspended in ice-cold lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 5 mM imidazole, 0.1% Triton X-100, and EDTA-free protease inhibitor tablets [Roche]) and lysed by high-pressure cell homogenization (Avestin C3 Emulsiflex). The bacterial lysate was clarified by centrifugation at 45,000 x g for 60 min at 4 °C. After filtration, the supernatant was loaded onto a HisTrap HP column (GE Healthcare) quilibrated with 50 mM Tris pH 7.5, 5 mM imidazole, and 500 mM NaCl, and the His6-tagged protein was eluted using a 5–500 mM imidazole gradient. Fractions containing PTP1B were pooled and cleaved with tobacco etch virus (TEV) protease overnight at 4 °C while being dialyzed against 50 mM Tris pH 8.0, 500 mM NaCl. Cleaved protein was further purified using Ni2+-NTA immobilized metal affinity chromatography followed by size exclusion chromatography (SEC; Superdex 75 26/60; GE Healthcare) equilibrated in NMR buffer (50 mM HEPES pH 6.8, 150 mM NaCl, 0.5 mM TCEP) to a purity of >98% and a final yield of 40 mg PTP1B1-301 or 20 mg PTP1B1-393 per liter of LB cell culture. Purified PTP1B1-301 and PTP1B1-393 were concentrated to 1 mM and 0.3 mM, respectively. For long-term storage, the protein was flash frozen in liquid nitrogen and stored at −80 °C.
NMR Spectroscopy
NMR data of PTP1Bcat and PTP1B1-393 were collected at 298 K on Bruker Avance 500 and 800 MHz spectrometers and a Bruker AvanceIIIHD 850 MHz spectrometer all equipped with a TCI HCN z-gradient cryoprobe. All NMR spectra were processed with Topspin 2.1/3.0/3.1 (Bruker) and analyzed using Cara (http://cara.nmr.ch). TROSY versions of a 3D HNCA, 3D HN(CO)CA, 3D HNCACB and 3D HN(CO)CACB were recorded on 1 mM [2H,15N,13C]-PTP1Bcat and 0.3 mM [2H,15N,13C]-PTP1B1-393 samples. A TROSY version of a 3D 15N-resolved [1H,1H] NOESY (Tm = 120 ms) was also recorded on 1 mM [2H,15N]-PTP1Bcat to verify assignments using sequential backbone 1HN-1HN NOEs. Additionally, 2D [1H,15N] TROSY spectra of single 15N-isotopically labeled amino acid samples (15N-Leu, 15N-Tyr, 15N-Phe or 15N-Val) were recorded.
Small Angle X-ray Scattering (SAXS)
All data were collected on a Rigaku BioSAXS-1000 (Anode: FRE+-SuperBright) equipped with a Dectris Pilatus 100K detector at 10°C using 1 hour experiments. Immediately prior to data collection, proteins were verified to be monomeric and exchanged into NMR buffer using SEC. Buffer scattering curves were subtracted from sample scattering curves using the Rigaku SAXSLab Software. PTP1B1-301 was recorded at 2.0 mg/ml and 3.5 mg/ml concentrations, whereas PTP1B1-393 was recorded at 3.6 mg/ml and 5.0 mg/ml. Guinier approximation, I(q) = I(0)exp(−q2Rg2/3), was performed on each independent scattering experiment and averaged to determine the radius of gyration. The linearity of the Guinier region and the forward scattering intensity, I(0), which is proportional to the molecular weight of the sample, were used to verify that samples were monodisperse in solution.
Dynamic Light Scattering (DLS)
Dynamic light scattering (DLS) data for PTP1B1-301 and PTP1B1-393 were acquired using a Viscotek 802 DLS instrument. All proteins were measured in triplicate at a concentration of 20 μM in 50 mM HEPES pH 6.8, 150 mM NaCl, 0.5 mM TCEP. All data were analyzed using the OmniSIZE software, and each experiment reflects the average of ten measurements, each of which was acquired for 10 seconds at 25 °C.
Cell migration and invasion assays
10A.B2 cells expressing an inducible HER2 chimera53 were used to quantitate cell invasion with BD BioCoat Matrigel Invasion Chambers, 8.0μm pore size. 10A.B2 cells (1 × 106) were grown in the insert. After 24-48 h, the cells retained inside the insert were removed, and those that migrated to the other side of the insert were fixed and stained with KARYOMAX Giemsa Stain (GIBCO, Invitrogen) and counted. Suppression of PTP1B by RNAi in MCF-10AN cells was achieved using a retroviral expression vector (pMLP) that utilizes a Murine Stem Cell Virus (MSCV) backbone53. The pMLP vector containing the validated PTP1B shRNA (-CTTTGACCATAGTCGGATT-) and non-target control shRNA targeting firefly luciferase were used to create stable cell lines. MCF10AN-cells, ~30% confluent, were infected with retrovirus encoding shRNA for ~15 hours using polybrene (8 μg/ml). The infection medium was then replaced with fresh growth medium and cultured for 24 hours, following which cells were selected with puromycin (2 μg/ml) for 7 days. MCF10ANeuNT cells expressing doxycycline inducible control and PTP1B shRNA miRs were obtained from Mohamed Bentires-Alj (Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland)32. For experiments with inducible miRs, 500 ng/mL of doxycycline was added to the medium 12 h after seeding the cells and refreshed every 12h. For experiments overexpressing wild type and mutant forms of PTP1B, cells were transfected with PMT2-PTP1B WT or PMT2-PTP1BL192A/S372P for 24 h using X-tremeGene HP DNA transfection reagent (Roche), following which cells were untreated or treated with MSI-1436 (1 or 2 μM) and cell invasion was studied as described above.
3D-morphogenesis assays
For 3D cultures, ~5,000 cells were plated on chamber slides coated with growth factor-reduced Matrigel (BD Biosciences) as described. To activate chimeric ErBB2 protein, 1 μM AP1510 was added to the growth medium. For studies with MSI-1436, 2.5 μM of the inhibitor was used. At least 100 acini were imaged every 4 days using a Zeiss Axiovert 200M microscope and AxioVision 4.5 software (Zeiss).
Cell proliferation assays
Cells (1 × 104) were plated in 96-well plates for 12 - 96 hours in growth medium in the absence and presence of MSI-1436 (2 and 5 μM). Cell proliferation was measured using the CellTiter-Glo (CTG; Promega) assay according to the manufacturer’s instructions using a luminescence plate reader.
Annexin/PI staining
Cells (1 × 105) were plated in 6-well plates in growth medium. The cells were untreated or treated with MSI-1436 (2 and 5 μM), stained with annexin and PI according to the manufacturer’s instructions (Invitrogen) and analyzed by flow cytometry (Becton Dickinson LSRII Cell Analyzer).
Use of animals in the research
The animal protocol application was reviewed and approved by the Cold Spring Harbor Laboratory Animal Care and Use committee and was designated protocol #10-4. The Institution has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare (number A3280-01).
PTP1B inhibitor treatment in NDL2 animals
NDL2 animals with palpable tumors where randomly grouped to receive saline (control) or MSI-1436. MSI-1436 was administered (5 or 10 mg/kg body weight, with an injection volume of 100 μl) every third day intraperitoneally for 45 days. Control animals received an injection at the same time of 100 μl of saline. Solutions were freshly prepared before each administration. Mice were sacrificed 45 d after the treatment and individual tumors were removed to measure the weight.
Xenograft studies
Luciferase-expressing BT474 cells (5×106) were injected orthotopically into mammary fat pads of SCID-beige mice (Tectonic lab) in 15 μl of a 1:1 mixture with DMEM and growth factor-reduced Matrigel (BD Biosciences). The animals were randomly assigned to two groups to receive saline (control) or MSI-1436 (5 mg/kg) (treated) intraperitoneally every third day. In another study, animals injected with BT474 cells (5×106) were allowed to develop tumors. Once the tumors reached ~100 mm3 in volume, treatment with MSI-1436 (5 mg/kg) was initiated. Tumor growth was monitored periodically by injecting 100 μl of d-luciferin (15 mg/kg) and imaging the animals using a Xenogen Imager (Xenogen IVIS-200 Optical In Vivo Imaging System). Tumor volume in mm3 was measured three times weekly by palpation using the formula: volume = width2 × length/2.
Mammary tissue harvesting for immunoblotting
After harvesting, connective tissue was carefully dissected from the tumor to exclude stromal cells from whole-tumor cell lysate to be used for immunoblotting. Tissue samples were ground into a powder under liquid nitrogen and lysed for 30 min on ice in lysis buffer (50 mM HEPES, pH 7.4; 10% glycerol; 1% Triton X-100, 150 mM NaCl; 1 mM EGTA, pH 8.0; 1 mM sodium orthovanadate; 20 mM NaF; complete EDTA-free protease cocktail inhibitors). The cell lysates were cleared by centrifugation at 12,000 x g for 15 min at 4 °C. Lysates (20 μg) were used for immunoblotting.
MSI-1436 binding assay in cell and tumor lysates
MSI-1436 was immobilized on magnetic beads through covalent linkage using its primary amine group. NHS-activated magnetic beads equilibrated in DMSO were incubated with 1 mM MSI-1436 and 100 mM triethylamine for 8h at 4°C. The reaction was terminated by pelleting the magnetic beads and gently aspirating the supernatant. Free NHS groups were blocked with 0.8 M aminoethanol. The beads were washed twice sequentially with 100 mM Tris-HCl, pH 8.0, and 100 mM acetate buffer pH 4.5 containing 150 mM NaCl.
The coupled magnetic beads were incubated with purified recombinant PTP1B (0.5 μg) for 6–8h at 4 °C in 100 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM DTT and 0.1% (w/v) BSA. Subsequently the beads were washed in 100 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM DTT, 0.01% Nonidet P-40, 1% glycerol and 0.1% BSA and eluted by heat-denaturation in SDS-PAGE sample buffer. The bound protein was separated by 10 % SDS-PAGE and subjected to Commassie or silver staining.
For experiments with cell or tumor lysates, 0–1 mg lysate protein was used. Preliminary analysis of derivatives of MSI-1436 suggested that the cholestane moiety of the compound is critical for its binding to PTP1B. Considering the hydrophobic nature of the cholestane core, we anticipated that the interaction would be stabilized predominantly by hydrophobic forces. Hence, we eliminated detergent in the wash buffer. We also observed that the interaction was resistant to salt concentration up to 200 mM (NaCl). Consequently, we chose a final buffer composition of 50 mM HEPES pH 7.0, 150 mM NaCl, 2 mM TCEP, 5% (v/v) glycerol. MSI-1459 (3β-spermine, 5α, cholenic acid methyl ester), a structurally related inactive analog of MSI-1436, was included as a negative control for binding specificity. Beads coupled to another related compound (MSI-1459) with a primary amine group but poor inhibitory potency against PTP1B was used as a negative control.
Histology
To evaluate the presence and absence of tumors in a secondary site, the entire lungs from untreated and treated animals were sectioned and stained with H&E. Whole-slide, digitized images of H&E-stained tissue were captured using the Aperio ScanScope XT automated scanning system (Vista, CA). Images can be viewed from a public database maintained by CSHL. For volumetric measurement of total lung metastasis the slides stained with H&E were analyzed by Aperio software.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software and two-tailed Student t tests were applied.
Supplementary Material
Acknowledgments
This research was supported by NIH grants CA53840 and GM55989, and the CSHL Cancer Centre Support Grant CA45508, to N.K.T.; by NIH grant GM100910, GM098482, American Diabetes Association grant 1-14-ACN-31 and a Brown University Research Seed Fund grant by the Vice President for Research to W.P.; and with financial support from the French Agence Nationale de la Recherche through ANR JCJC ProteinDisorder to M.R.J. and ANR MALZ TAUSTRUCT to M.B. The 800 MHz NMR data were recorded at Brandeis University; the instrument was purchased with support from NIH S10-RR017269. NMR data (500 and 850 MHz) were recorded in the Brown University Structural Biology Facility, which is generously supported by Brown University. N.K.T. is also grateful for support from the following foundations; The Gladowsky Breast Cancer Foundation, The Don Monti Memorial Research Foundation, Hansen Memorial Foundation, West Islip Breast Cancer Coalition for Long Island, Glen Cove CARES, Find a Cure Today (FACT), Constance Silveri, Robertson Research Fund and the Masthead Cove Yacht Club Carol Marcincuk Fund. PTP1B inhibitor MSI-1436 was provided by Ohr Pharmaceuticals and Genaera Corporation.
Footnotes
AUTHOR CONTRIBUTIONS
N.K. performed the biochemical characterization of PTP1B inhibition by MSI-1436 and tested the effects of the inhibitor in cell and animal models; D.K., D.H.M, J.K., M.R.J., R.W.P, M.B. and W.P. designed, performed and analyzed NMR-based structural work. C.M.G performed the homology modeling, molecular docking and dynamics simulation. B.X., S.D.A. and S.K.M. helped with the breast cancer studies. N.K., W.P. and N.K.T. analyzed the data and wrote the manuscript, which was reviewed by all authors. N.K.T. directed the study.
COMPETING FINANCIAL INTERESTS
A company called DepYmed Inc was founded on 3 March 2014 with the primary purpose of taking MSI-1436 into the clinic in patients with HER2-positive breast cancer. It is owned 50-50 by Ohr Pharmaceuticals and Cold Spring Harbor Lab. Authors Krishnan and Tonks would be entitled to a share based upon the conditions of the Invention Agreement signed by all scientists at Cold Spring Harbor Lab.
Accession Numbers
All chemical shifts for PTP1B1-393 were deposited in the BioMagResBank under accession number 19224.
References
- 1.Slamon DJ, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 2.Tiwari RK, Borgen PI, Wong GY, Cordon-Cardo C, Osborne MP. HER-2/neu amplification and overexpression in primary human breast cancer is associated with early metastasis. Anticancer Research. 1992;12:419–25. [PubMed] [Google Scholar]
- 3.Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Current Opinion in Genetics & Development. 2008;18:73–9. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Critical Reviews in Oncogenesis. 2012;17:1–16. doi: 10.1615/critrevoncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tonks NK, Diltz CD, Fischer EH. Purification of the major protein-tyrosine-phosphatases of human placenta. The Journal of Biological Chemistry. 1988;263:6722–30. [PubMed] [Google Scholar]
- 6.Tonks NK. Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction. The FEBS Journal. 2013;280:346–78. doi: 10.1111/febs.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen JN, Tonks NK. Protein tyrosine phosphatase-based therapeutics: lessons from PTP1B. Topics in Current Genetics. 2004;5:201–230. [Google Scholar]
- 8.Julien SG, et al. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nature Genetics. 2007;39:338–46. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
- 9.Bentires-Alj M, Neel BG. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Research. 2007;67:2420–4. doi: 10.1158/0008-5472.CAN-06-4610. [DOI] [PubMed] [Google Scholar]
- 10.Hao L, Tiganis T, Tonks NK, Charbonneau H. The noncatalytic C-terminal segment of the T cell protein tyrosine phosphatase regulates activity via an intramolecular mechanism. The Journal of Biological Chemistry. 1997;272:29322–9. doi: 10.1074/jbc.272.46.29322. [DOI] [PubMed] [Google Scholar]
- 11.Lantz KA, et al. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity. 2010;18:1516–23. doi: 10.1038/oby.2009.444. [DOI] [PubMed] [Google Scholar]
- 12.Wiesmann C, et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nature Structural & Molecular Biology. 2004;11:730–7. doi: 10.1038/nsmb803. [DOI] [PubMed] [Google Scholar]
- 13.Metallo SJ. Intrinsically disordered proteins are potential drug targets. Current Opinion in Chemical Biology. 2010;14:481–8. doi: 10.1016/j.cbpa.2010.06.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuchillo R, Michel J. Mechanisms of small-molecule binding to intrinsically disordered proteins. Biochemical Society Transactions. 2012;40:1004–8. doi: 10.1042/BST20120086. [DOI] [PubMed] [Google Scholar]
- 15.Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:12366–71. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Neal S, Wishart DS. RefDB: a database of uniformly referenced protein chemical shifts. Journal of Biomolecular NMR. 2003;25:173–95. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 17.Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263:1397–404. [PubMed] [Google Scholar]
- 18.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nature Cell Biology. 2001;3:785–92. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konecny GE, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Research. 2006;66:1630–9. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 20.Siegel PM, Dankort DL, Hardy WR, Muller WJ. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Molecular and Cellular Biology. 1994;14:7068–77. doi: 10.1128/mcb.14.11.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dube N, Cheng A, Tremblay ML. The role of protein tyrosine phosphatase 1B in Ras signaling. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1834–9. doi: 10.1073/pnas.0304242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Julien SG, Dube N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nature Reviews: Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 23.Sangwan V, et al. Protein-tyrosine phosphatase 1B modulates early endosome fusion and trafficking of Met and epidermal growth factor receptors. The Journal of Biological Chemistry. 2011;286:45000–13. doi: 10.1074/jbc.M111.270934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown-Shimer S, et al. Molecular cloning and chromosome mapping of the human gene encoding protein phosphotyrosyl phosphatase 1B. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:5148–52. doi: 10.1073/pnas.87.13.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tonks NK, Muthuswamy SK. A brake becomes an accelerator: PTP1B--a new therapeutic target for breast cancer. Cancer Cell. 2007;11:214–6. doi: 10.1016/j.ccr.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 26.Wiener JR, et al. Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: association with p185c-erbB-2 protein expression. Journal of the National Cancer Institute. 1994;86:372–8. doi: 10.1093/jnci/86.5.372. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Chen X, Liu B, Zhu Z. Suppression of PTP1B in gastric cancer cells in vitro induces a change in the genome-wide expression profile and inhibits gastric cancer cell growth. Cell Biology International. 2010;34:747–53. doi: 10.1042/CBI20090447. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, et al. PTP1B expression contributes to gastric cancer progression. Medical Oncology. 2012;29:948–56. doi: 10.1007/s12032-011-9911-2. [DOI] [PubMed] [Google Scholar]
- 29.Lessard L, et al. PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Research. 2012;72:1529–37. doi: 10.1158/0008-5472.CAN-11-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu S, Bjorge JD, Fujita DJ. PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Research. 2007;67:10129–37. doi: 10.1158/0008-5472.CAN-06-4338. [DOI] [PubMed] [Google Scholar]
- 31.Johnson KJ, et al. PTP1B suppresses prolactin activation of Stat5 in breast cancer cells. The American Journal of Pathology. 2010;177:2971–83. doi: 10.2353/ajpath.2010.090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balavenkatraman KK, et al. Epithelial protein-tyrosine phosphatase 1B contributes to the induction of mammary tumors by HER2/Neu but is not essential for tumor maintenance. Molecular Cancer Research: MCR. 2011;9:1377–84. doi: 10.1158/1541-7786.MCR-11-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He R, Zeng LF, He Y, Zhang S, Zhang ZY. Small molecule tools for functional interrogation of protein tyrosine phosphatases. The FEBS Journal. 2013;280:731–50. doi: 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kane RC, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clinical Cancer Research. 2006;12:7271–8. doi: 10.1158/1078-0432.CCR-06-1249. [DOI] [PubMed] [Google Scholar]
- 35.Wiede F, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. The Journal of Clinical Investigation. 2011;121:4758–74. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiganis T. PTP1B and TCPTP--nonredundant phosphatases in insulin signaling and glucose homeostasis. The FEBS Journal. 2013;280:445–58. doi: 10.1111/j.1742-4658.2012.08563.x. [DOI] [PubMed] [Google Scholar]
- 37.Galic S, et al. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Molecular and Cellular Biology. 2005;25:819–29. doi: 10.1128/MCB.25.2.819-829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanson MA, et al. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–6. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. Journal of the American Chemical Society. 2009;131:7390–401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]
- 41.Agulnik M, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. Journal of Clinical Oncology. 2007;25:3978–84. doi: 10.1200/JCO.2007.11.8612. [DOI] [PubMed] [Google Scholar]
- 42.Hinow P, Wang SE, Arteaga CL, Webb GF. relocating job wise? A mathematical model separates quantitatively the cytostatic and cytotoxic effects of a HER2 tyrosine kinase inhibitor. Theoretical Biology & Medical Modelling. 2007;4:14. doi: 10.1186/1742-4682-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janne PA, Taffaro ML, Salgia R, Johnson BE. Inhibition of epidermal growth factor receptor signaling in malignant pleural mesothelioma. Cancer Research. 2002;62:5242–7. [PubMed] [Google Scholar]
- 44.Meng TC, Hsu JSF, Tonks NK. Development of a modified in-gel assay to identify protein tyrosine phosphatases that are oxidized and inactivated in vivo. Methods. 2005;35:28–36. doi: 10.1016/j.ymeth.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Bernado P, et al. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ozenne V, et al. Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics. 2012;28:1463–1470. doi: 10.1093/bioinformatics/bts172. [DOI] [PubMed] [Google Scholar]
- 47.Eyal E, Najmanovich R, McConkey BJ, Edelman M, Sobolev V. Importance of solvent accessibility and contact surfaces in modeling side-chain conformations in proteins. J Comput Chem. 2004;25:712–724. doi: 10.1002/jcc.10420. [DOI] [PubMed] [Google Scholar]
- 48.Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. Journal of Biomolecular NMR. 2007;38:289–302. doi: 10.1007/s10858-007-9166-6. [DOI] [PubMed] [Google Scholar]
- 49.Nodet G, et al. Quantitative description of backbone conformational sampling of unfolded proteins at amino acid resolution from NMR residual dipolar couplings. Journal of the American Chemical Society. 2009;131:17908–17918. doi: 10.1021/ja9069024. [DOI] [PubMed] [Google Scholar]
- 50.Jensen MR, Salmon L, Nodet G, Blackledge M. Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. Journal of the American Chemical Society. 2010;132:1270–1272. doi: 10.1021/ja909973n. [DOI] [PubMed] [Google Scholar]
- 51.Ozenne V, et al. Mapping the potential energy landscape of intrinsically disordered proteins at amino acid resolution. Journal of the American Chemical Society. 2012;134:15138–15148. doi: 10.1021/ja306905s. [DOI] [PubMed] [Google Scholar]
- 52.Svergun D, Barberato C, Koch MHJ. CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- 53.Lin G, Aranda V, Muthuswamy SK, Tonks NK. Identification of PTPN23 as a novel regulator of cell invasion in mammary epithelial cells from a loss-of-function screen of the ‘PTP-ome’. Genes Dev. 2011;25:1412–1425. doi: 10.1101/gad.2018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.