

Abstract
Ensifer (syn. Sinorhizobium) meliloti is an important symbiotic bacterial species that fixes nitrogen. Strains BO21CC and AK58 were previously investigated for their substrate utilization and their plant-growth promoting abilities showing interesting features. Here, we describe the complete genome sequence and annotation of these strains. BO21CC and AK58 genomes are 6,985,065 and 6,974,333 bp long with 6,746 and 6,992 genes predicted, respectively.
Keywords: Aerobic, motile, Gram-negative, mesophilic, chemoorganotrophic, chemoautotrophic, soil, plant symbiont, biological nitrogen fixation, Ensifer (Sinorhizobium) meliloti, legume yield
Introduction
Strains AK58 and BO21CC belong to the species Ensifer (syn. Sinorhizobium) meliloti (Alphaproteobacteria, Rhizobiales, Rhizobiaceae, Sinorhizobium/Ensifer group) [1,2], an important symbiotic nitrogen fixing bacterial species that associates with roots of leguminous plants of several genera, mainly from Melilotus, Medicago and Trigonella [3]. These strains have been originally isolated from Medicago spp. during a long course experiment (BO21CC) and from plants collected in the north Aral sea region (Kazakhstan) (AK58). Previous analyses conducted by comparative genomic hybridization (CGH), nodulation tests and Phenotype Microarray™(Biolog, Inc.) showed that AK58 (= DSM 23808) and BO21CC (= DSM 23809) are highly diverse in both genomic and phenotypic properties. In particular, they show different symbiotic phenotypes with respect to the crop legume Medicago sativa L [4,5]. In a previous collaboration with DOE-JGI, the genomes of strains AK83 (= DSM 23913) and BL225C (= DSM 23914) were also sequenced, allowing the identification of putative genetic determinants for their different symbiotic phenotypes [6]. Consequently, interest in strains AK58 and BO21CC arose, since genomic analysis of these strains would foster a greater understanding of the E. meliloti pangenome [7], and facilitate deeper investigation of the genomic determinants responsible for differences in symbiotic performances between E. meliloti strains found in nature. These research goals may lead to improved strain selection and better inoculants of the legume crop M. sativa.
Classification and features
Representative genomic 16S rRNA sequences of strains AK58 and BO21CC were compared with those present in the Ribosomal Database by using Match Sequence module of Ribosomal Database Project [8]. Representative genomic 16S rRNA sequences of closer phylogenetic relatives of the genus Ensifer/Sinorhizobium and of Rhizobiales family (as outgroup) were then selected from IMG-ER database [Table 1] [16]. All strains from the genus Ensifer/Sinorhizobium form a close cluster, including strains AK58 and BO21CC, thus confirming the affiliation of these two strains within the species. Figure 1 shows the phylogenetic neighborhood of E. meliloti AK58 and BO21CC in a 16S rRNA based tree.
Table 1. Classification and general features of E. meliloti AK58 and BO21CC according to the MIGS recommendations [9] and the Names for Life database [10].
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [11] | |
| Phylum Proteobacteria | TAS [12] | ||
| Class Alphaproteobacteria | TAS [12] | ||
| Order Rhizobiales | TAS [12] | ||
| Family Rhizobiaceae | TAS [12] | ||
| Genus Ensifer | TAS [2,12] | ||
| Species Ensifer meliloti | TAS [13] | ||
| Strain BO21CC Strain AK58 |
TAS [4,5] | ||
| Gram stain | negative | TAS [12] | |
| Cell shape | rods | TAS [12] | |
| Motility | Motile | TAS [12] | |
| Sporulation | non-sporulating | TAS [12] | |
| Temperature range | mesophile, 20-37°C | TAS [12] | |
| Optimum temperature | 25-30°C | TAS [12] | |
| Salinity | Tolerate 1.0% NaCl | TAS [12] | |
| MIGS-22 | Oxygen requirement | Aerobe | TAS [12] |
| Carbon source | carbohydrates and salts of organic acids | TAS [12] | |
| Energy metabolism | chemoorganotroph | TAS [12] | |
| MIGS-6 | Habitat | Soil, root nodules of legumes | TAS [3,12] |
| MIGS-15 | Biotic relationship | free living, symbiont | TAS [12] |
| MIGS-14 | Pathogenicity | not reported | |
| Biosafety level | 1 | TAS [14] | |
| MIGS-23.1 | Isolation | BO21CC: root nodules of Medicago sativa cv. ‘Oneida’ AK58: root nodules of Medicago falcata |
TAS [4] |
| MIGS-4 | Geographic location | BO21CC: Lodi, Italy AK58: Kazakhstan, |
TAS [4] |
| MIGS-5 | Sample collection time | BO21CC: 1997 AK58: 2001 |
NAS |
| MIGS-4.1 | Latitude | BO21CC: 45.31 AK58: 58.75 |
NAS |
| MIGS-4.2 | Longitude | BO21CC: 9.50 AK58: 48.98 |
NAS |
| MIGS-4.3 | Depth | not reported | |
| MIGS-4.4 | Altitude | BO21CC: 70 m AK58: 305 m |
NAS |
Evidence codes - TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [15].
Figure 1.
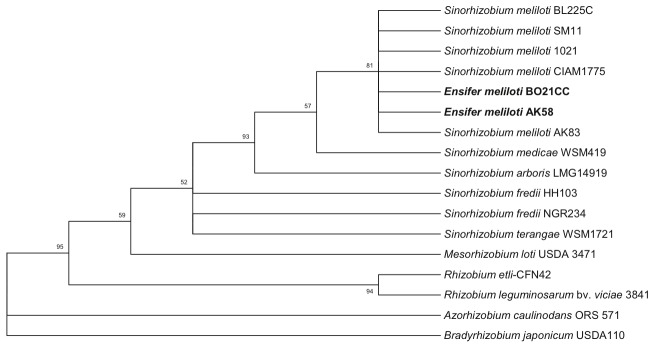
Phylogenetic consensus tree showing the position of E. meliloti AK58 and BO21CC strains in the Ensifer/Sinorhizobium genus. The phylogenetic tree was inferred by using the Maximum Likelihood method based on the Tamura 3-parameter model [17], chosen as model with the lowest BIC scores (Bayesian Information Criterion) after running a Maximum Likelihood fits of 24 different nucleotide substitution models (Model Test). The bootstrap consensus tree inferred from 500 replicates [18] is taken to represent the phylogenetic pattern of the taxa analyzed [18]. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches. The tree with the highest log likelihood (-3411.7124) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. A discrete Gamma distribution was used to model evolutionary rate differences among sites (G, parameter = 0.3439). A total of 1,284 nt positions were present in the final dataset. Model test and Maximum Likelihood inference were conducted in MEGA5 [19]. In bold E. meliloti AK58 and BO21CC strains.
E. meliloti AK58 and BO21CC show different symbiotic phenotypes with respect to the host plant Medicago sativa, as well as differences in substrates utilization [5]. Moreover E. meliloti AK58 and BO21CC present differences in cell morphology also, with AK58 being smaller than BO21CC and the other E. meliloti strains for which genome sequencing is available (Figure 2). Interestingly, BO21CC is also showing cells with a ratio between cell axes nearer 1 (more rounded cells), when compared with AK58 and with the other E. meliloti strains (Figure 2).
Figure 2.

Cell morphology and cell size analysis of E. meliloti strains. Cell size analysis with Pixcavator IA 5.1.0.0 software [20] of logarithmically grown cultures (OD600=0.6) in TY medium of AK58, BO21CC, plus other completely sequenced E. meliloti strains is reported. Cell size is expressed as cell area in µm2, while roundness is the ratio between the two main axes of the cell. Standard errors after more than 300 individual observations are reported. Different letters indicate significant differences (P<0.05) after 1-way ANOVA.
Genome sequencing information
Genome project history
AK58 and BO21CC strains were selected for sequencing on the basis of the Community Sequencing Program 2010 of DOE Joint Genome Institute (JGI) in relation to the project entitled “Complete genome sequencing of Sinorhizobium meliloti AK58 and BO21CC strains: Improving alfalfa performances through the exploitation of Sinorhizobium genomic data”. The overall rationale for their genome sequencing was related to the identification of genomic determinants of different symbiotic performances between S. meliloti strains. The genome project is deposited in the Genomes On Line Database [21] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE-JGI. A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | High-Quality Draft |
| MIGS-28 | Libraries used | Two genomic libraries: one 454 PE library (9 kb insert size), one Illumina library |
| MIGS-29 | Sequencing platforms | Illumina GAii, 454 GS FLX Titanium |
| MIGS-31.2 | Sequencing coverage | 60 × (AK58) 910 × (BO21CC) Illumina; 8.8 × pyrosequence |
| MIGS-30 | Assemblers | Newbler version 2.3, Velvet version 1.0.13, phrap version, 1.080812, Allpaths version 39750, |
| MIGS-32 | Gene calling method | Prodigal |
| GenBank Date of Release | BO21CC: AXBL01000000 AK58: AXBM00000000 |
|
| GOLD ID | BO21CC: Gi07569 AK58: Gi07577 |
|
| NCBI project ID | BO21CC: 375171 AK58: 928722 |
|
| Database: IMG | BO21CC: 9144 AK58: 7327 |
|
| MIGS-13 | Source material identifier | BO21CC: DSM23809 AK58: DSM23808 |
| Project relevance | CSP2010, biotechnological, biodiversity |
Growth conditions and DNA isolation
E. meliloti strains AK58 and BO21CC (DSM23808 and DSM23809, respectively) were grown in DSMZ medium 98 (Rhizobium medium) [22] at 28°C. DNA was isolated from 0.5-1 g of cell paste using Jetflex Genomic DNA Purification kit (GENOMED 600100) following the standard protocol as recommended by the manufacturer with modification st/LALMP [23] for strain AK58 and additional 5 µl proteinase K incubation at 58° for 1 hour for strain BO21CC, respectively. DNA will be available on request through the DNA Bank Network [24].
Genome sequencing and assembly
The draft genomes were generated at the DOE Joint Genome Institute (JGI) using Illumina data [25]. For BO21CC genome, we constructed and sequenced an Illumina short-insert paired-end library with an average insert size of 270 bp which generated 76,033,356 reads and an Illumina long-insert paired-end library with an average insert size of 9,141.74 ± 1,934.63 bp, which generated 4,563,348 reads totaling 6,463 Mbp of Illumina data. For AK58, a combination of Illumina [25] and 454 technologies [26] was used. For the AK58 genome we constructed and sequenced an Illumina GAii shotgun library which generated 80,296,956 reads totaling 6,102.6 Mb, a 454 Titanium standard library which generated 0 reads and 1 paired end 454 library with an average insert size of 10 kb, which generated 326,569 reads totaling 96 Mb of 454 data. All general aspects of library construction and sequencing performed at the JGI can be found at the JGI website [27]. The initial draft assemblies contained 194 contigs in 16 scaffold(s) for BO21CC, and 311 contigs in 5 scaffolds for AK58.
For BO21CC the initial draft data was assembled with Allpaths and the consensus was computationally shredded into 10 Kbp overlapping fake reads (shreds). The Illumina draft data was also assembled with Velvet, version 1.1.05 [28], and the consensus sequences were computationally shredded into 1.5 Kbp overlapping fake reads (shreds). The Illumina draft data was assembled again with Velvet using the shreds from the first Velvet assembly to guide the next assembly. The consensus from the second Velvet assembly was shredded into 1.5 Kbp overlapping fake reads. The fake reads from the Allpaths assembly and both Velvet assemblies and a subset of the Illumina CLIP paired-end reads were assembled using parallel phrap, version 4.24 (High Performance Software, LLC). Possible mis-assemblies were corrected with manual editing in Consed [29-31].
Gap closure was accomplished using repeat resolution software (Wei Gu, unpublished), and sequencing of bridging PCR fragments with Sanger and/or PacBio (unpublished, Cliff Han) technologies. For improved high quality draft and noncontiguous finished projects, one round of manual/wet lab finishing may have been completed. Primer walks, shatter libraries, and/or subsequent PCR reads may also be included for a finished project. A total of 128 additional sequencing reactions and 126 PCR PacBio consensus sequences were completed to close gaps and to raise the quality of the final sequence. The total ("estimated size" for unfinished) size of the BO21CC genome is 7.1 Mb and the final assembly is based on 6,463 Mbp of Illumina draft data, which provides an average 910 × coverage of the genome.
For AK58, the 454 Titanium standard data and the 454 paired end data were assembled together with Newbler, version 2.6 (20110517_1502). The Newbler consensus sequences were computationally shredded into 2 kb overlapping fake reads (shreds). Illumina sequencing data was assembled with Velvet, version 1.1.05 [28], and the consensus sequence was computationally shredded into 1.5 kb overlapping fake reads (shreds). We integrated the 454 Newbler consensus shreds, the Illumina Velvet consensus shreds and the read pairs in the 454 paired end library using parallel phrap, version SPS - 4.24 (High Performance Software, LLC). The software Consed [29-31] was used in the following finishing process. Illumina data was used to correct potential base errors and increase consensus quality using the software Polisher developed at JGI (Alla Lapidus, unpublished). Possible mis-assemblies were corrected using gapResolution (Cliff Han, unpublished), Dupfinisher [32], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR (J-F Cheng, unpublished) primer walks. A total of 0 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. The estimated genome size of AK58 is 7 Mb and the final assembly is based on 61.5 Mb of 454 draft data which provides an average 8.8 × coverage of the genome and 420 Mb of Illumina draft data which provides an average 60 × coverage of the genome.
Genome annotation
Genes were identified using Prodigal [33] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [34]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [16].
Genome properties
The High-Quality draft assemblies of the genomes consist of 41 scaffolds for BO21CC and 9 scaffolds for AK58 representing overall 6,985,065 and 6,974,333 bp, respectively. The overall G+C content was 62.12% and 62.04% for BO21CC and AK58, respectively (Table 3a and Table 3b). Of the 6,746 and 6,992 genes predicted, 5,357 and 5,549 were protein-coding genes, and 105 and 79 RNAs were present in BO21CC and AK58, respectively. The large majority of the protein-coding genes (79.32% and 78.03%, BO21CC and AK58, respectively) were assigned a putative function as COGs. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3a. Genome Statistics for strain BO21CC.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 6,985,065 | 100.00% |
| DNA coding region (bp) | 6,011,953 | 86.07% |
| DNA G+C content (bp) | 4,339,356 | 62.12% |
| Number of scaffolds | 41 | |
| Total genes | 6,746 | 100.00% |
| RNA genes | 105 | 1.72% |
| rRNA operons | 3 | |
| tRNA genes | 58 | 0.86% |
| Protein-coding genes | ||
| Genes with function prediction (proteins) | 5,357 | 79.41% |
| Genes in paralog clusters | 3,275 | 48.55% |
| Genes assigned to COGs | 5,351 | 79.32% |
| Genes assigned Pfam domains | 5,318 | 78.83% |
| Genes with signal peptides | 1,427 | 21.15% |
| Genes with transmembrane helices | 1,521 | 22.55% |
Table 3b. Genome statistics for strain AK58.
| Attribute | Value | %age |
|---|---|---|
| Genome size (bp) | 6,974,333 | 100.00% |
| DNA coding region (bp) | 5,914,246 | 84.80% |
| DNA G+C content (bp) | 4,315,694 | 62.04% |
| Number of scaffolds | 9 | |
| Total genes | 6,992 | 100.00% |
| RNA genes | 79 | 1.13% |
| rRNA operons | 1* | |
| tRNA genes | 49 | 0.70% |
| Protein-coding genes | 6,934 | 98.87% |
| Genes with function prediction (proteins) | 5,459 | 77.84% |
| Genes in paralog clusters | 2,912 | 41.52% |
| Genes assigned to COGs | 5,472 | 78.03% |
| Genes assigned Pfam domains | 5,420 | 77.29% |
| Genes with signal peptides | 1,432 | 20.42% % |
| Genes with transmembrane helices | 1,465 | 20.89% |
*only one rRNA operon appears to be complete.
Table 4. Number of genes associated with the general COG functional categories.
| BO21CC | AK58 | ||||
|---|---|---|---|---|---|
| Code | Value | %age | Value | %age | Description |
| J | 195 | 3.27 | 201 | 3.29 | Translation, ribosomal structure and biogenesis |
| A | - | - | - | - | RNA processing and modification |
| K | 524 | 8.79 | 551 | 9.01 | Transcription |
| L | 273 | 4.58 | 327 | 5.35 | Replication, recombination and repair |
| B | 1 | 0.02 | 3 | 0.05 | Chromatin structure and dynamics |
| D | 45 | 0.76 | 53 | 0.87 | Cell cycle control, cell division, chromosome partitioning |
| Y | - | - | - | - | Nuclear structure |
| V | 64 | 1.17 | 62 | 1.01 | Defense mechanisms |
| T | 247 | 4.14 | 249 | 4.07 | Signal transduction mechanisms |
| M | 305 | 5.12 | 298 | 4.87 | Cell wall/membrane biogenesis |
| N | 69 | 1.16 | 68 | 1.11 | Cell motility |
| Z | - | - | - | - | Cytoskeleton |
| W | 1 | 0.02 | 1 | 0.02 | Extracellular structures |
| U | 104 | 1.75 | 102 | 1.67 | Intracellular trafficking and secretion, and vesicular transport |
| O | 185 | 3.10 | 189 | 3.09 | Posttranslational modification, protein turnover, chaperones |
| C | 365 | 6.13 | 356 | 5.82 | Energy production and conversion |
| G | 604 | 10.14 | 596 | 9.75 | Carbohydrate transport and metabolism |
| E | 637 | 10.69 | 685 | 11.20 | Amino acid transport and metabolism |
| F | 107 | 1.80 | 114 | 1.86 | Nucleotide transport and metabolism |
| H | 202 | 3.39 | 205 | 3.35 | Coenzyme transport and metabolism |
| I | 210 | 3.52 | 217 | 3.55 | Lipid transport and metabolism |
| P | 320 | 5.17 | 294 | 4.81 | Inorganic ion transport and metabolism |
| Q | 163 | 2.74 | 159 | 2.60 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 730 | 12.25 | 767 | 12.54 | General function prediction only |
| S | 608 | 10.20 | 617 | 10.09 | Function unknown |
Acknowledgements
We are grateful to Dr. M.L. Roumiantseva and Dr. B. Simarov (Research Institute for Agricultural Microbiology, St-Petersburg-Puskin, Russia) for original isolation and the permission to use strain AK58 in this work. The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy Under Contract No. DE-AC02-05CH11231.
References
- 1.Martens M, Delaere M, Coopman R, De Vos P, Gillis M, Willems A. Multilocus sequence analysis of Ensifer and related taxa. Int J Syst Evol Microbiol 2007; 57:489-503 10.1099/ijs.0.64344-0 [DOI] [PubMed] [Google Scholar]
- 2.Young JM. The genus name Ensifer Casida 1982 takes priority over Sinorhizobium Chen et al. 1988, and Sinorhizobium morelense Wang et al. 2002 is a later synonym of Ensifer adhaerens Casida 1982. Is the combination ˜Sinorhizobium adhaerens" (Casida 1982) Willems et al. 2003 legitimate? Request for an Opinion. Int J Syst Evol Microbiol 2003; 53:2107-2110 10.1099/ijs.0.02665-0 [DOI] [PubMed] [Google Scholar]
- 3.Sprent JI. Nodulation in legumes. London: Royal Botanic Gardens, Kew.; 2001. [Google Scholar]
- 4.Giuntini E, Mengoni A, De Filippo C, Cavalieri D, Aubin-Horth N, Landry CR, Becker A, Bazzicalupo M. Large-scale genetic variation of the symbiosis-required megaplasmid pSymA revealed by comparative genomic analysis of Sinorhizobium meliloti natural strains. BMC Genomics 2005; 6:158 10.1186/1471-2164-6-158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biondi EG, Tatti E, Comparini D, Giuntini E, Mocali S, Giovannetti L, Bazzicalupo M, Mengoni A, Viti C. Metabolic capacity of Sinorhizobium ( Ensifer) meliloti strains as determined by phenotype microarray analysis. Appl Environ Microbiol 2009; 75:5396-5404 10.1128/AEM.00196-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galardini M, Mengoni A, Brilli M, Pini F, Fioravanti A, Lucas S, Lapidus A, Cheng JF, Goodwin L, Pitluck S, et al. Exploring the symbiotic pangenome of the nitrogen-fixing bacterium Sinorhizobium meliloti. BMC Genomics 2011; 12:235 10.1186/1471-2164-12-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev 2005; 15:589-594 10.1016/j.gde.2005.09.006 [DOI] [PubMed] [Google Scholar]
- 8.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM and others. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucl. Acids Res. 2009;37(suppl_1):D141-145. [DOI] [PMC free article] [PubMed]
- 9.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9 [Google Scholar]
- 11.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner DJ, Krieg NR, Staley JT. Bergeys’ Manual of Systematic Bacteriology. Volume 2 The Proteobacteria Part C The Alpha-, Beta-, Delta-, and Epsilonproteobacteria Garrity GM, editor: Springer; 2005. [Google Scholar]
- 13.De Lajudie P, Willems A, Pot B, Dewettinck D, Maestrojuan G, Neyra M, Collins MD, Dreyfus B, Kersters K, Gillis M. Polyphasic Taxonomy of Rhizobia: Emendation of the Genus Sinorhizobium and Description of Sinorhizobium meliloti comb. nov., Sinorhizobium saheli sp. nov., and Sinorhizobium teranga sp. nov. Int J Syst Bacteriol 1994; 44:715-733 10.1099/00207713-44-4-715 [DOI] [Google Scholar]
- 14.BAuA Classification of Bacteria and Archaea in risk groups. TRBA 2010; 466:80 [Google Scholar]
- 15.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markowitz VM, Mavromatis K, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]
- 17.Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol 1992; 9:678-687 [DOI] [PubMed] [Google Scholar]
- 18.Felsenstein J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985; 39:783-791 10.2307/2408678 [DOI] [PubMed] [Google Scholar]
- 19.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol 2011; 28:2731-2739 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pixcavator IA. 5.1.0.0 Intelligent Perception <http://inperc.com >.
- 21.Pagani I, Liolios K, Jansson J, Chen IMA, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40(D1):D571-D579 10.1093/nar/gkr1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.List of growth media used at DSMZ <http://www.dsmz.de/catalogues/catalogue-microorganisms/culture-technology/list-of-media-for-microorganisms.html>.
- 23.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056-1060 10.1038/nature08656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network. The Start from a German Initiative Biopreservation and Biobanking 2011; 9:51-55 10.1089/bio.2010.0029 [DOI] [PubMed] [Google Scholar]
- 25.Bennett S. Solexa Ltd. Pharmacogenomics 2004; 5:433-438 10.1517/14622416.5.4.433 [DOI] [PubMed] [Google Scholar]
- 26.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437:376-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DOE joint Genome Institute. The Regents of the University of California <http://www.jgi.doe.gov/>.
- 28.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ewing B, Green P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res 1998; 8:186-194 10.1101/gr.8.3.175 [DOI] [PubMed] [Google Scholar]
- 30.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res 1998; 8:175-185 10.1101/gr.8.3.175 [DOI] [PubMed] [Google Scholar]
- 31.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res 1998; 8:195-202 10.1101/gr.8.3.195 [DOI] [PubMed] [Google Scholar]
- 32.Han CPC. Finishing Repetitive Regions Automatically with Dupfinisher. 2006; Las Vegas, Nevada, USA. CSREA Press. [Google Scholar]
- 33.Hyatt D, Chen GL, LoCascio P, Land M, Larimer F, Hauser L. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]