

Abstract
Latently infected resting CD4+ T cells are the major barrier to curing HIV. We have recently demonstrated that chemokines, which bind to the chemokine receptors CCR7, CXCR3 and CCR6, facilitate efficient HIV nuclear localisation and integration in resting CD4+ T cells, leading to latency. As latently infected cells are enriched in lymphoid tissues, where chemokines are highly concentrated, this may provide a mechanism for the generation of latently infected cells in vivo. Here we review the role of chemokines in HIV persistence; the main signalling pathways that are involved; and how these pathways may be exploited to develop novel strategies to reduce or eliminate latently infected cells.
Keywords: Human immunodeficiency virus (HIV), Chemokine, Signalling, Latency, Persistence
1. Introduction
Combination antiretroviral therapy (cART) for the treatment of human immunodeficiency virus (HIV) has resulted in a significant reduction in morbidity and mortality [1,2]. However, cART is currently unable to cure HIV, which means treatment needs to be life-long [3]. The major reason why HIV persists on treatment is the ability of HIV to establish a latent infection in long-lived CD4+ T cells [4–7]. Latently infected cells are enriched in lymphoid tissues, in particular the lymph nodes, spleen and gastrointestinal tract [8–11]. These tissue sites are rich in small chemotactic cytokines known as chemokines that, together with chemokine receptors, coordinate the highly regulated process of lymphocyte migration [12–14]. Here we review the role of chemokines in the establishment of HIV latency and how these pathways may be exploited to develop novel strategies to reduce or eliminate latently infected cells.
2. HIV latency
The first step of the HIV life cycle is binding of the viral glycoprotein 120 (gp120) to both the CD4 receptor and a chemokine co-receptor (CCR5 or CXCR4) expressed on the surface of the host cell [15]. This is followed by uncoating and reverse transcription to form double-stranded cDNA [16,17], which then enters the nucleus, accompanied by integrase, in the form of pre-integration complexes (PICs). Inside the nucleus, the viral cDNA will either integrate into the host genome or form 1-long terminal repeat (LTR) or 2-LTR circles that are made following “self-ligation” of the HIV LTR at each end of the cDNA [18]. In activated CD4+ T cells, the integrated HIV provirus undergoes transcription to form unspliced (US) and multiply-spliced (MS) RNA in the nucleus. These transcripts are then translated in the cytoplasm to produce viral proteins leading to the production of new viral particles. Latency occurs when the virus life cycle is arrested following integration.
2.1. Pre- and post-integration latency
HIV latency can occur either as pre-integration or post-integration latency. Pre-integration latency refers to unintegrated HIV DNA that is unstable and will either degrade or will integrate into the host cell genome, usually following cell activation [19]. Post-integration latency is defined by the presence of stable integrated HIV DNA in the absence of virus production [6]. In vivo, the major reservoir of cells that harbour post-integration latency consists of resting central (CD45RA−CCR7+CD27+) and transitional (CD45RA−CCR7−CD27+) memory CD4+ T cells [20,21], and to a lesser extent, naïve CD4+ T cells [22].
In activated CD4+ T cells the virus life cycle is efficient with rapid integration, virion production and death of infected cells (Fig. 1A). In contrast, direct infection of resting CD4+ T cells is difficult to establish in vitro due to multiple blocks in the viral life cycle. These blocks include low or absent expression of the HIV co-receptor CCR5, which limits the ability for CCR5-using virus to enter resting CD4+ T cells, inefficient reverse transcription [19,23] and limited nuclear import of the reverse transcription complex (RTC)/PIC [24]. However, resting CD4+ T cells are clearly infected in vivo [25,26], as well as ex vivo in tissue blocks [27,28], and contain stable integrated forms of HIV. Once integration occurs in resting CD4+ T cells, the virus remains silent due to multiple molecular mechanisms. These include transcriptional interference [29–31]; a lack of transcription factors such as nuclear factor-kappa B (NF-κB), nuclear factor of activated T cells (NFAT) and transcriptional elongation factor (pTEFb; [32]); impaired export of MS RNA from the nucleus to the cytoplasm [33,34]; and microRNAs that inhibit translation [35,36].
Fig. 1.
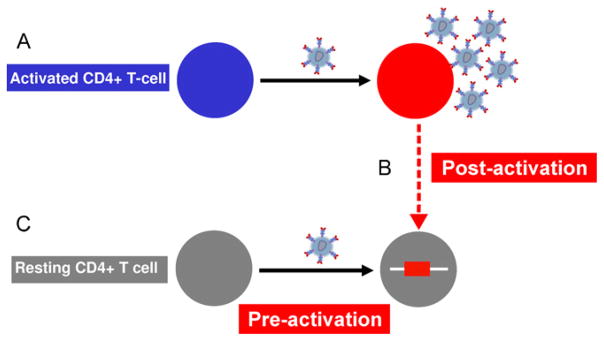
Pathways for the establishment of latent infection in resting CD4+ T cells. (A) In activated CD4+ T cells (blue) the virus life cycle is efficient, multiple virions are produced and the infected cell dies. In latently infected cells, virus enters and integrates but there is limited or no virus production. There are two potential pathways for the establishment of latent infection in resting CD4+ T cells. (B) Some activated infected cells (red) may survive and revert to become central memory T cells with an associated integrated provirus (post-activation latency). (C) The alternate pathway is the direct infection of resting CD4+ T cells (grey) in the presence of factors that modify the actin cytoskeleton, such as chemokines (pre-activation latency).
2.2. Establishment of post-integration latency in resting CD4+ T cells
There are two theories on how post-integration latency is established in resting CD4+ T cells. One suggests that latency arises from HIV-infected activated CD4+ T cells that survive and revert to a resting memory CD4+ T cell carrying integrated virus. We refer to this as post-activation latency (Fig. 1B). In vitro models that support this pathway demonstrate that latency can be established after prolonged culture of activated infected cells [30,37,38]; together with IL-7 [39]; or by inhibiting cell death of infected activated cells via over-expression of the anti-apoptotic gene, bcl-2 [40].
The alternative pathway is that HIV can directly infect a subset of resting CD4+ T cells. We refer to this as pre-activation latency (Fig. 1C). This has been demonstrated in vitro using CD4+ thymocytes [41,42] and circulating resting CD4+ T cells by some but not all groups [43–45]. We have recently demonstrated that latent infection can be established in resting memory CD4+ T cells following incubation with multiple chemokines [43].
2.3. Chemokines and HIV latency
Our initial experiments focused on the chemokines CCL19 and CCL21. These chemokines bind to the chemokine receptor CCR7 that is highly expressed on both central memory and naïve CD4+ T cells [46]. We demonstrated that incubation of resting CD4+ T cells with either CCL19 or CCL21, allowed for high levels of HIV integration with only low levels of productive infection [43]. This was observed with both CXCR4-using and CCR5-using viruses and in memory but not naïve CD4+ T cells [43]. Chemokine exposure did not result in cell activation (as measured by expression of CD69, CD25 and HLA-DR) [43,47] or significant changes in gene expression [47]. This was the first evidence to support a role for chemokines both in mediating infection of resting CD4+ T cells and in the establishment of HIV latency.
We subsequently demonstrated that multiple chemokines, in addition to CCL19 and CCL21, are able to induce latency in resting CD4+ T cells [47]. These included CXCL9 and CXCL10, which bind to CXCR3, and CCL20, which binds to CCR6 [48–50]. By quantifying various viral intermediates in the virus life cycle, we showed that the major effect of these chemokines was to facilitate efficient nuclear localisation and integration. Following HIV infection of unactivated or CCL19-treated resting CD4+ T cells, as well as PHA/IL-2-activated CD4+ T cells, we found no difference in early and late reverse transcripts. However, when we compared the CCL19-treated cells to unactivated cells we observed a more rapid and significant increase in the production of 2-LTR circles and integrated HIV DNA [47]. These results demonstrated that in the presence of CCL19, HIV can efficiently enter the nucleus and integrate in resting memory CD4+ T cells.
The lack of progression to productive infection in CCL19-treated cells suggested that there were factors that blocked the virus life cycle following integration. We therefore analysed viral transcripts in the nucleus and cytoplasm, including US RNA and MS RNA. In latently infected CCL19-treated CD4+ T cells we detected expression of MS RNA in the nucleus, but not in the cytoplasm [51]. Additionally, we did not detect any US RNA in the CCL19-treated latently infected cells. As MS RNA encodes for the positive regulators Rev and Tat that are crucial for the efficient expression of US RNA [52,53], the absence of MS RNA in the cytoplasm could explain the lack of US RNA expression and viral production observed in infected CCL19-treated CD4+ T cells. These data demonstrate that in latently infected CCL19-treated CD4+ T cells, production of MS RNA occurs, but that there is a block in the transport of MS RNA from the nucleus to the cytoplasm, similar to descriptions of resting CD4+ T cells from HIV-infected patients on cART [34]. Taken together, our data support the hypothesis that latency can result from direct infection of resting memory CD4+ T cells, possibly as a result of exposure to chemokines found in lymphoid tissues, which could explain the efficient infection of resting cells observed in vivo [54] and in lymphoid tissue explants [28].
3. Chemokine signalling: a role in HIV nuclear localisation and integration
Chemokine ligation of G-coupled chemokine receptors leads to the activation of three main pathways, which include the Rho/GTPase pathway [55], the phosphoinositide-3 kinase (PI3K) pathway [56] and the phospholipase (PLC) pathway [57,58]. Activation of the Rho/GTPase pathway activates RhoA leading to cofilin dephosphorylation and changes in actin polymerisation [59]. This pathway also mediates endocytosis via cdc42 and RAC1 [60]. The PI3K pathway mediates chemotaxis via activation of the extracellular signal-regulated kinases 1 and 2 (Erk1/2) and c-Jun N-terminal kinases (JNK), as well as cell survival via activation of NF-κB (reviewed in [61,62]) (Fig. 2).
Fig. 2.

Signalling pathways activated by chemokine receptors. The binding of both gp120 to CXCR4 and exogenous chemokines to their respective chemokine receptors is associated with signalling by (A) cdc42/RAC1/RhoA (yellow); (B) phosphoinositide-3 kinase (PI3K; blue/green); and (C) phospholipase (PLC; blue). These pathways can mediate HIV replication via modification of post entry events such as (1) reverse transcription; (2) migration of the reverse transcriptase and pre-integration complex to the perinuclear region; and (3) nuclear entry. After nuclear entry, (4) the full length linear DNA may be integrated or form 2-long terminal repeat circles, a virologically dead end product. The RhoA pathway has been shown to modify cofilin activation and actin depolymerisation, which enhances nuclear localisation in chemokine treated cells. The PI3K and PLC pathways, via nuclear factors such as nuclear factor-kappa B (NF-κB) and nuclear factor of activated T cells (NFAT), mediate cellular functions, such as cell survival and chemotaxis, and can also activate viral transcription. These pathways may potentially have a role in enhancing the efficiency of HIV integration in resting CD4+ T cells.
3.1. Rho/GTPase pathway
The Rho family of GTPases, including RAC1, RhoA and Cdc42, directly modulate the cytoskeleton through effects on actin/myosin and microtubules [63,64]. Actin/myosin and microtubules are important for cell migration [63], as well as migration of HIV to the nucleus in T cell lines [65] and activated CD4+ T cells [66]. In HIV infection, the viral PIC binds to actin via the matrix protein [67].
Infection of resting CD4+ T cells with a CXCR4-using virus, results in HIV gp120-induced chemokine signalling, through the HIV entry co-receptor CXCR4, and activation of the RhoA/GTPase pathway. This pathway activates Lim kinase, the actin-binding protein cofilin and changes in cortical F-actin, which allow for nuclear localisation of HIV [45,68]. We found that a similar pathway was critical for CCL19-induced latency and showed that the addition of jasplakolinolide, which blocks F-actin polymerisation, inhibited the detection of integrated HIV DNA and 2-LTR circles [47].
It has been proposed that in unactivated resting CD4+ T cells the PIC is carried to the nucleus by a process of actin cycling that does not require microtubule involvement [69]. This is supported by recent data showing that microtubule inhibitors do not inhibit direct infection of resting T cells [70]. However, it is not yet clear if microtubule involvement will be critical for infection of resting T cells that have undergone cytoskeletal changes following exposure to exogenous chemokine [47].
These findings shed new light on how HIV can exploit cellular machinery, including the cytoskeleton in resting CD4+ T cells. However, unlike our model of chemokine-induced latency, binding of gp120 to CXCR4 in unactivated cells did not result in viral integration and post-integration latency [71,72]. Furthermore, it is unclear if a similar process occurs following infection of resting CD4+ T cells with CCR5-using virus. In unactivated cells, accumulation of pre-integrated virus occurs [19,23], which may persist for many days [73] and can induce apoptosis [74]. We therefore propose that additional signalling is required for efficient integration in resting CD4+ T cells to generate stable post-integration latency.
There may be mechanisms other than direct chemokine receptor signalling, that could contribute to changes in the cell cytoskeleton allowing for efficient nuclear localisation. Cell migration alone may also lead to cytoskeletal changes that will favour transport of virus to the nucleus. Support for this theory can be derived from infection of resting CD4+ T cells via spinoculation, which involves infection of cells in the presence of high centrifugal force. Spinoculation was initially conceived as a method for increasing virus exposure during infection [44], however, it has since been shown to be effective in inducing efficient integration and establishment of latency in resting memory and, to some extent, naïve T cells [75–77]. Recent work has now demonstrated that spinoculation is associated with cytoskeletal changes, in addition to the activation of the cofilin/Lim kinase pathway [78]. These changes resemble the changes observed in the cytoskeleton following both direct gp120-co-receptor signalling and exogenous chemokine signalling [45,47]. Further studies on the relationship between cytoskeletal changes and the signalling pathways downstream of the chemokine receptors will help clarify the mechanisms controlling nuclear localisation and integration in resting CD4+ T cells.
3.2. PI3K pathway
The PI3K signalling pathway and its downstream transcription factors have been intensively studied for their role in lymphocyte proliferation, survival and cycle control in cancer [79,80]. PI3K is also upstream of the critical nuclear transcription activator NF-κB, which regulates gene expression in response to inflammation and activates transcription of the HIV provirus [81]. A recent report has suggested that the generation of either latent or productive infection in T cell lines is determined by the relative amount of NF-κB [38], and that basal levels of NF-κB are required for HIV integration, even in the absence of productive infection [31,38]. Therefore, NF-κB may indeed play a role in both facilitating integration as well as viral transcription.
The PI3K pathway also activates JNK and ERK1/2, which are important in cell survival and proliferation. The Raf/Mek/ERK pathway has previously been associated with the nuclear import of the HIV reverse transcriptase complex [69]. PI3K signalling, via the transcription factor JNK and the protein PIN-1, has been shown to result in the phosphorylation of HIV integrase, leading to increased stability of integrase and enhanced HIV integration in activated CD4+ T cells [82]. Whether similar pathways are required for efficient integration in resting CD4+ T cells remains unknown.
3.3. PLC signalling
The PLC pathway is also activated by chemokine receptor ligation and participates in the regulation of NFAT through Ca2+ and the Ca2+/calmodulin-dependent serine phosphatase calcineurin. NFAT consists of a family of transcription factors that play important roles in the transcription of many cytokine genes. Reactivation of HIV from latency in some but not all models of latently infected memory CD4+ T cells has been shown to be mediated via NFAT, rather than NF-κB [38]. Furthermore, the inhibition of NFAT is associated with the inhibition of HIV gene expression [83]. The potential role of NFAT in HIV integration in resting CD4+ T cells is still unknown. However, the potency of NFAT activation of virus expression [84,85] would suggest that latency occurs in the presence of low levels of NFAT activation.
Multiple chemokine signalling pathways seem to play a role either directly or indirectly in HIV infection. It is clear from our work and others that in resting CD4+ T cells, exogenous chemokines do increase the rate of nuclear localisation and dramatically increase the efficiency of viral integration compared to gp120-mediated chemokine signalling. It is currently unknown which of the signalling pathways is critical for HIV integration and latency in resting CD4+ T cells and whether these pathways differ from activated CD4+ T cells. It is most likely that separate downstream chemokine signalling pathways are critical for establishing latent infection in resting CD4+ T cells.
4. Chemokine receptors and productive HIV infection
Specific chemokine receptors (other than the HIV co-receptors CXCR4 and CCR5) have also recently been shown to be important in productive HIV infection. Activated memory CD4+ T cells are heterogeneous in their susceptibility to infection. In particular, Th17 cells, which express the chemokine receptor CCR6 [86], have been shown to be highly permissive to HIV infection [87]. Integrated HIV was found to be significantly enriched in CCR6+ cells when compared with other memory T cell subsets in treatment naïve HIV-infected patients [50]. Furthermore, the frequency of CCR6 expressing cells was diminished in blood from HIV-infected patients [50,88], even when HIV was controlled following cART [50]. Although Th17 cells also express the α4β7 receptor, which has recently been shown to bind HIV gp120 [89], CCR6 and not α4β7 expression has been shown to account for the increased permissiveness to HIV infection. CCR6+ T cells have the ability to be recruited to both gut and lymph node tissues [87] via the CCR6 ligand, CCL20 [90,91]. Therefore, HIV infection of CCR6+ CD4+ memory T cells may provide a pathway for HIV dissemination to the T cell rich compartments of lymphoid tissues.
5. Effects of HIV infection on chemokine expression in vivo
Elevated levels of numerous chemokines have been detected in the plasma of HIV-infected patients, including CCL2, CCL19, CCL21, CCL20 [92–95], CXCL9, CXCL10 [96], CXCL16 [97] and CXCL13 [98,99], and expression of these chemokines increases with disease progression. Additionally, many of these chemokines remain elevated on cART [95,96,100] or, if reduced, fail to return to the levels observed in uninfected individuals [95,98,99]. The addition of CCL19, CCL21 or CXCL16 to PBMC isolated from HIV-infected individuals, resulted in a significant increase in pro-inflammatory cytokines, such as TNF-alpha and IFN-gamma [93,100]. Furthermore, elevated plasma CXCL10 and CXCL9 have recently been associated with elevated markers of immune activation (including IL-12 and soluble IL-2 receptor) in HIV-infected patients on cART [96]. Therefore, it is possible that elevated chemokines may potentially drive immune activation in the setting of HIV infection. Persistent immune activation may also facilitate ongoing virus replication in tissue sites where penetration of cART may be suboptimal [101]. Further studies, however, are required to establish if there is any relationship between elevated chemokine levels and the localisation of latently infected cells and/or residual virus replication on cART.
6. Blocking chemokine function – implications for clinical strategies aimed at eradicating latency
Currently, there is intense interest in finding novel strategies to eliminate latency as a potential path towards finding a cure for HIV. How might we then translate some of our findings on the role of chemokines and latent infection into novel therapeutics?
The lack of decay of long-lived latently infected resting memory CD4+ T cells may be explained by the long half-life of memory CD4+ T cells [102,103]. Alternatively, there could be proliferation or replenishment of this reservoir. There is some evidence to suggest that IL-7 may drive homeostatic proliferation of latently infected cells [20]. Ongoing infection of resting CD4+ T cells in sites, such as the gastrointestinal tract or lymphoid tissue, may be another way the pool of latently infected CD4+ T cells is replenished. The source of this virus could be intermittent release of virus from long-lived latently infected cells and/or long-lived productively infected cells, such as infected myeloid cells and dendritic cells, or their progenitors [104]. Reduced efficacy of cART in these tissues may also arise from suboptimal penetration of cART [101] and/or limited efficacy of cART in blocking cell–cell transfer of virus [105,106]. Therefore, cART alone may never be able to sufficiently limit infection of resting CD4+ T cells in these tissue sites. Strategies other than cART that specifically block infection of resting memory CD4+ T cells and, therefore, the establishment of latent infection, may be an important intervention to limit replenishment and accelerate the decay of latently infected cells.
Our data raise the possibility of an entirely new approach to eliminating latent infection – via inhibition of specific chemokine receptors, including CCR7, CXCR3 and CCR6 [47]. Chemokine receptor antagonists, which are active against CXCR3, have undergone phase II trials for efficacy in psoriasis (T-487; Tularik and AMG-487, Amgen; reviewed in [107,108]). Additionally, the CCR5 antagonist, TAK-779, has activity as both a CCR5 and CXCR3 antagonist [109]. There are currently no available antagonists that block CCR7 and CCR6.
In addition to chemokine receptor antagonists, chemokine function can also be inhibited using synthetic chemokine antagonists [110–112]. Studies in murine models have demonstrated that truncation of the N-terminus of some chemokines, including CCL19, CXCL11 and CCL20 can antagonise specific functions of the corresponding wild-type chemokine [110–114]. Therefore, inhibition of chemokines that are important for the establishment of latency via either chemokine antagonists or chemokine receptor antagonists may be a novel strategy to be tested in the future that could potentially block ongoing infection of resting CD4+ T cells.
7. Conclusion
Chemokines and their receptors clearly play an important role in HIV infection. In addition to their role in productive infection, numerous chemokines have now been shown to induce post-integration latency in resting memory CD4+ T cells. Chemokine signalling can induce changes in the actin cytoskeleton of resting CD4+ T cells that facilitate efficient nuclear localisation. Other signalling pathways such as PI3K may also be important for efficient integration. Increases in the concentration of specific chemokines in either tissue or blood in the setting of HIV infection may potentially provide a unique environment for ongoing infection of resting CD4+ T cells in tissue sites. Blocking ongoing infection of resting CD4+ T cells by inhibiting host targets may present a novel strategy to inhibit the establishment of latent HIV infection and accelerate the decay of latently infected cells in patients on cART.
Acknowledgments
SRL is an Australian National Health and Medical Research Council (NHMRC) practitioner fellow. This work was funded by grants from the National Institutes of Health U19 AI096109 and the American Foundation for AIDS Research 108237-51-RGRL and 108023-49-RFRL.
Biographies
 Vanessa Evans obtained her Bachelor’s degree in Science and her PhD in Immunology from Monash University, Australia. Her PhD explored the role of dendritic cells in the spread and persistence of HIV. In 2011, Dr. Evans began her post-doctoral training with Professor Sharon Lewin within the Department of Infectious Diseases at Monash University. Her research focus is on dendritic cells and the development of physiologically relevant in vitro models for the study of HIV latency.
Vanessa Evans obtained her Bachelor’s degree in Science and her PhD in Immunology from Monash University, Australia. Her PhD explored the role of dendritic cells in the spread and persistence of HIV. In 2011, Dr. Evans began her post-doctoral training with Professor Sharon Lewin within the Department of Infectious Diseases at Monash University. Her research focus is on dendritic cells and the development of physiologically relevant in vitro models for the study of HIV latency.
 Gabriela Khoury is currently completing her PhD at the Department of Medicine, Monash University, Australia. She is a successful recipient of a competitive post-graduate scholarship from the Australian National Health and Medical Research Council. Prior to starting her PhD, Gabriela had 5 years of HIV basic science research experience including the development of a new class of antiretroviral compounds. Ms. Khoury’s PhD focuses on the role of naïve T cells in HIV pathogenesis, in particular how these cells remain persistently infected during long-term antiretroviral therapy.
Gabriela Khoury is currently completing her PhD at the Department of Medicine, Monash University, Australia. She is a successful recipient of a competitive post-graduate scholarship from the Australian National Health and Medical Research Council. Prior to starting her PhD, Gabriela had 5 years of HIV basic science research experience including the development of a new class of antiretroviral compounds. Ms. Khoury’s PhD focuses on the role of naïve T cells in HIV pathogenesis, in particular how these cells remain persistently infected during long-term antiretroviral therapy.
 Suha Saleh obtained her Bachelor’s degree in Veterinary Science from Baghdad University, Iraq, and her PhD in Molecular Virology from The University of Queensland, Australia. She then joined the Department of Microbiology at Monash University, Australia, where she conducted her post-doctoral training as part of the malaria vaccine initiative team project. Dr. Saleh continued her vaccine work with Viral Holding Limited, an Australian biotech company, where she was involved in the development of new recombinant vaccine vectors for HIV, prostate cancer, Hepatitis B and other infectious and autoimmune diseases. In 2005, she joined the research team of Professor Sharon Lewin within the Department of Medicine at Monash University. Dr. Saleh is an amFAR fellow and her major research interests include chemokines and their role in the establishment of HIV latency.
Suha Saleh obtained her Bachelor’s degree in Veterinary Science from Baghdad University, Iraq, and her PhD in Molecular Virology from The University of Queensland, Australia. She then joined the Department of Microbiology at Monash University, Australia, where she conducted her post-doctoral training as part of the malaria vaccine initiative team project. Dr. Saleh continued her vaccine work with Viral Holding Limited, an Australian biotech company, where she was involved in the development of new recombinant vaccine vectors for HIV, prostate cancer, Hepatitis B and other infectious and autoimmune diseases. In 2005, she joined the research team of Professor Sharon Lewin within the Department of Medicine at Monash University. Dr. Saleh is an amFAR fellow and her major research interests include chemokines and their role in the establishment of HIV latency.
 Paul Cameron trained as a clinician in clinical immunology and immunopathology at the Royal Perth Hospital and University of Western Australia. After completing a PhD in immunogenetics he spent a post-doc with Ralph Steinman at Rockefeller University where he defined the ability of dendritic cells (DC) to efficiently infect CD4+ T cells during DC-T cells interactions. He has subsequently had research positions at the Burnet Institute and the University of Melbourne, Australia where he defined the infection of DC and T cells in ex vivo models of HIV infection. More recently, he has joined the Alfred Hospital as a Clinical Immunologist and immunopathologist and co-heads a research laboratory with Prof Sharon Lewin in the Departments of Medicine and Immunology of Monash University. Dr. Cameron is continuing his research in HIV immunopathology with a particular interest in the mechanisms of latency in T cells and during DC-T cell interactions.
Paul Cameron trained as a clinician in clinical immunology and immunopathology at the Royal Perth Hospital and University of Western Australia. After completing a PhD in immunogenetics he spent a post-doc with Ralph Steinman at Rockefeller University where he defined the ability of dendritic cells (DC) to efficiently infect CD4+ T cells during DC-T cells interactions. He has subsequently had research positions at the Burnet Institute and the University of Melbourne, Australia where he defined the infection of DC and T cells in ex vivo models of HIV infection. More recently, he has joined the Alfred Hospital as a Clinical Immunologist and immunopathologist and co-heads a research laboratory with Prof Sharon Lewin in the Departments of Medicine and Immunology of Monash University. Dr. Cameron is continuing his research in HIV immunopathology with a particular interest in the mechanisms of latency in T cells and during DC-T cell interactions.
 Sharon Lewin is an infectious diseases physician and basic scientist. She is Director of the Department of Infectious Diseases The Alfred Hospital and Monash University; co-head of the Centre for Virology, Burnet Institute, Melbourne, Australia and a practitioner fellow of the National Health and Medical Research Council of Australia. Professor Lewin completed her specialist and PhD training in Melbourne and her post-doctoral fellowship at the Aaron Diamond AIDS Research Centre, The Rockefeller University. She is a past president of the Australasian Society for HIV Medicine. Her laboratory focuses on understanding how HIV persists in patients on antiviral therapy, strategies to cure HIV infection and biological determinants of immune recovery following antiviral therapy.
Sharon Lewin is an infectious diseases physician and basic scientist. She is Director of the Department of Infectious Diseases The Alfred Hospital and Monash University; co-head of the Centre for Virology, Burnet Institute, Melbourne, Australia and a practitioner fellow of the National Health and Medical Research Council of Australia. Professor Lewin completed her specialist and PhD training in Melbourne and her post-doctoral fellowship at the Aaron Diamond AIDS Research Centre, The Rockefeller University. She is a past president of the Australasian Society for HIV Medicine. Her laboratory focuses on understanding how HIV persists in patients on antiviral therapy, strategies to cure HIV infection and biological determinants of immune recovery following antiviral therapy.
References
- 1.Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. New England Journal of Medicine. 1998;338:853–60. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 2.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. New England Journal of Medicine. 1997;337:734–9. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 3.Chun TW, Davey RT, Jr, Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. 1999;401:874–5. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- 4.Lambotte O, Taoufik Y, de Goer MG, Wallon C, Goujard C, Delfraissy JF. Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. Journal of Acquired Immune Deficiency Syndromes. 2000;23:114–9. doi: 10.1097/00126334-200002010-00002. [DOI] [PubMed] [Google Scholar]
- 5.McElrath MJ, Steinman RM, Cohn ZA. Latent HIV-1 infection in enriched populations of blood monocytes and T cells from seropositive patients. Journal of Clinical Investigation. 1991;87:27–30. doi: 10.1172/JCI114981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–8. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 7.Haase AT, Henry K, Zupancic M, Sedgewick G, Faust RA, Melroe H, et al. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science. 1996;274:985–9. doi: 10.1126/science.274.5289.985. [DOI] [PubMed] [Google Scholar]
- 8.North TW, Higgins J, Deere JD, Hayes TL, Villalobos A, Adamson L, et al. Viral sanctuaries during highly active antiretroviral therapy in a nonhuman primate model for AIDS. Journal of Virology. 2010;84:2913–22. doi: 10.1128/JVI.02356-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. Journal of Infectious Diseases. 2008;197:714–20. doi: 10.1086/527324. [DOI] [PubMed] [Google Scholar]
- 10.Guadalupe M, Sankaran S, George MD, Reay E, Verhoeven D, Shacklett BL, et al. Viral suppression and immune restoration in the gastrointestinal mucosa of human immunodeficiency virus type 1-infected patients initiating therapy during primary or chronic infection. Journal of Virology. 2006;80:8236–47. doi: 10.1128/JVI.00120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz L, van Lunzen J, Arno A, Stellbrink HJ, Schneider C, Rull M, et al. Protease inhibitor-containing regimens compared with nucleoside analogues alone in the suppression of persistent HIV-1 replication in lymphoid tissue. Aids. 1999;13:F1–8. doi: 10.1097/00002030-199901140-00001. [DOI] [PubMed] [Google Scholar]
- 12.Loetscher P, Seitz M, Baggiolini M, Moser B. Interleukin-2 regulates CC chemokine receptor expression and chemotactic responsiveness in T lymphocytes. Journal of Experimental Medicine. 1996;184:569–77. doi: 10.1084/jem.184.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277:2005–7. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 14.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. Journal of Experimental Medicine. 1998;187:875–83. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–6. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 16.Karageorgos L, Li P, Burrell C. Characterization of HIV replication complexes early after cell-to-cell infection. AIDS Research and Human Retroviruses. 1993;9:817–23. doi: 10.1089/aid.1993.9.817. [DOI] [PubMed] [Google Scholar]
- 17.Miller MD, Farnet CM, Bushman FD. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. Journal of Virology. 1997;71:5382–90. doi: 10.1128/jvi.71.7.5382-5390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farnet CM, Haseltine WA. Circularization of human immunodeficiency virus type 1 DNA in vitro. Journal of Virology. 1991;65:6942–52. doi: 10.1128/jvi.65.12.6942-6952.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zack JA, Haislip AM, Krogstad P, Chen IS. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. Journal of Virology. 1992;66:1717–25. doi: 10.1128/jvi.66.3.1717-1725.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nature Medicine. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 22.Wightman F, Solomon A, Khoury G, Green JA, Gray L, Gorry PR, et al. Both CD31(+) and CD31 naive CD4(+) T cells are persistent HIV type 1-infected reservoirs in individuals receiving antiretroviral therapy. Journal of Infectious Diseases. 2010;202:1738–48. doi: 10.1086/656721. [DOI] [PubMed] [Google Scholar]
- 23.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell. 1990;61:213–22. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]
- 24.Bukrinsky MI, Sharova N, Dempsey MP, Stanwick TL, Bukrinskaya AG, Haggerty S, et al. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:6580–4. doi: 10.1073/pnas.89.14.6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musey LK, Krieger JN, Hughes JP, Schacker TW, Corey L, McElrath MJ. Early and persistent human immunodeficiency virus type 1 (HIV-1)-specific T helper dysfunction in blood and lymph nodes following acute HIV-1 infection. Journal of Infectious Diseases. 1999;180:278–84. doi: 10.1086/314868. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 1999;286:1353–7. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- 27.Audige A, Schlaepfer E, Bonanomi A, Joller H, Knuchel MC, Weber M, et al. HIV-1 does not provoke alteration of cytokine gene expression in lymphoid tissue after acute infection ex vivo. Journal of Immunology. 2004;172:2687–96. doi: 10.4049/jimmunol.172.4.2687. [DOI] [PubMed] [Google Scholar]
- 28.Eckstein DA, Penn ML, Korin YD, Scripture-Adams DD, Zack JA, Kreisberg JF, et al. HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity. 2001;15:671–82. doi: 10.1016/s1074-7613(01)00217-5. [DOI] [PubMed] [Google Scholar]
- 29.Han Y, Lin YB, An W, Xu J, Yang HC, O’Connell K, et al. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional read-through. Cell Host and Microbe. 2008;4:134–46. doi: 10.1016/j.chom.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lenasi T, Contreras X, Peterlin BM. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host and Microbe. 2008;4:123–33. doi: 10.1016/j.chom.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duverger A, Jones J, May J, Bibollet-Ruche F, Wagner FA, Cron RQ, et al. Determinants of the establishment of human immunodeficiency virus type 1 latency. Journal of Virology. 2009;83:3078–93. doi: 10.1128/JVI.02058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. Journal of Virology. 2010;84:6425–37. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bachelerie F, Rodriguez MS, Dargemont C, Rousset D, Thomas D, Virelizier JL, et al. Nuclear export signal of IkappaBalpha interferes with the Rev-dependent posttranscriptional regulation of human immunodeficiency virus type I. Journal of Cell Science. 1997;110(Pt 22):2883–93. doi: 10.1242/jcs.110.22.2883. [DOI] [PubMed] [Google Scholar]
- 34.Lassen KG, Ramyar KX, Bailey JR, Zhou Y, Siliciano RF. Nuclear retention of multiply spliced HIV-1 RNA in resting CD4+ T cells. PLoS Pathogens. 2006;2:e68. doi: 10.1371/journal.ppat.0020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klase Z, Kale P, Winograd R, Gupta MV, Heydarian M, Berro R, et al. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Molecular Biology. 2007;8:63. doi: 10.1186/1471-2199-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, et al. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nature Medicine. 2007;13:1241–7. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- 37.Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J, et al. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. The Journal of Biological Chemistry. 2009;284:6782–9. doi: 10.1074/jbc.M807898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marini A, Harper JM, Romerio F. An in vitro system to model the establishment and reactivation of HIV-1 latency. Journal of Immunology. 2008;181:7713–20. doi: 10.4049/jimmunol.181.11.7713. [DOI] [PubMed] [Google Scholar]
- 40.Yang HC, Xing S, Shan L, O’Connell K, Dinoso J, Shen A, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. Journal of Clinical Investigation. 2009;119:3473–86. doi: 10.1172/JCI39199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brooks D, Kitchen S, Kitchen C, Scripture-Adams D, Zack J. Generation of HIV latency during thymopoiesis. Nature Medicine. 2001;7:459–64. doi: 10.1038/86531. [DOI] [PubMed] [Google Scholar]
- 42.Burke B, Brown HJ, Marsden MD, Bristol G, Vatakis DN, Zack JA. Primary cell model for activation-inducible human immunodeficiency virus. Journal of Virology. 2007;81:7424–34. doi: 10.1128/JVI.02838-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood. 2007;110:4161–4. doi: 10.1182/blood-2007-06-097907. [DOI] [PubMed] [Google Scholar]
- 44.Swiggard WJ, Baytop C, Yu JJ, Dai J, Li C, Schretzenmair R, et al. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. Journal of Virology. 2005;79:14179–88. doi: 10.1128/JVI.79.22.14179-14188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoder A, Yu D, Dong L, Iyer SR, Xu X, Kelly J, et al. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell. 2008;134:782–92. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 47.Cameron PU, Saleh S, Sallmann G, Solomon A, Wightman F, Evans VA, et al. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16934–39. doi: 10.1073/pnas.1002894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rivino L, Messi M, Jarrossay D, Lanzavecchia A, Sallusto F, Geginat J. Chemokine receptor expression identifies Pre-T helper (Th)1, Pre-Th2, and non-polarized cells among human CD4+ central memory T cells. Journal of Experimental Medicine. 2004;200:725–35. doi: 10.1084/jem.20040774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–8. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- 50.Gosselin A, Monteiro P, Chomont N, Diaz-Griffero F, Said EA, Fonseca S, et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. Journal of Immunology. 2010;184:1604–16. doi: 10.4049/jimmunol.0903058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saleh S, Wightman F, Ramanayake S, Alexander M, Kumar N, Khoury G, et al. Expression and reactivation of HIV in a chemokine induced model of HIV latency in primary resting CD4+ T cells. Retrovirology. 2011;8:80. doi: 10.1186/1742-4690-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laspia MF, Rice AP, Mathews MB. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell. 1989;59:283–92. doi: 10.1016/0092-8674(89)90290-0. [DOI] [PubMed] [Google Scholar]
- 53.Sodroski J, Patarca R, Rosen C, Wong-Staal F, Haseltine W. Location of the trans-activating region on the genome of human T-cell lymphotropic virus type III. Science. 1985;229:74–7. doi: 10.1126/science.2990041. [DOI] [PubMed] [Google Scholar]
- 54.Zhang L, Ramratnam B, Tenner-Racz K, He Y, Vesanen M, Lewin S, et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. New England Journal of Medicine. 1999;340:1605–13. doi: 10.1056/NEJM199905273402101. [DOI] [PubMed] [Google Scholar]
- 55.Ishizaki H, Togawa A, Tanaka-Okamoto M, Hori K, Nishimura M, Hamaguchi A, et al. Defective chemokine-directed lymphocyte migration and development in the absence of Rho guanosine diphosphate-dissociation inhibitors alpha and beta. Journal of Immunology. 2006;177:8512–21. doi: 10.4049/jimmunol.177.12.8512. [DOI] [PubMed] [Google Scholar]
- 56.Sotsios Y, Whittaker GC, Westwick J, Ward SG. The CXC chemokine stromal cell-derived factor activates a Gi-coupled phosphoinositide 3-kinase in T lymphocytes. Journal of Immunology. 1999;163:5954–63. [PubMed] [Google Scholar]
- 57.Shannon LA, Calloway PA, Welch TP, Vines CM. CCR7/CCL21 migration on fibronectin is mediated by phospholipase Cgamma1 and ERK1/2 in primary T lymphocytes. Journal of Biological Chemistry. 2010;285:38781–87. doi: 10.1074/jbc.M110.152173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cronshaw DG, Kouroumalis A, Parry R, Webb A, Brown Z, Ward SG. Evidence that phospholipase-C-dependent, calcium-independent mechanisms are required for directional migration of T-lymphocytes in response to the CCR4 ligands CCL17 and CCL22. Journal of Leukocyte Biology. 2006;79:1369–80. doi: 10.1189/jlb.0106035. [DOI] [PubMed] [Google Scholar]
- 59.Nishita M, Aizawa H, Mizuno K. Stromal cell-derived factor 1alpha activates LIM kinase 1 and induces cofilin phosphorylation for T-cell chemotaxis. Molecular and Cellular Biology. 2002;22:774–83. doi: 10.1128/MCB.22.3.774-783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGavin MK, Badour K, Hardy LA, Kubiseski TJ, Zhang J, Siminovitch KA. The intersectin 2 adaptor links Wiskott Aldrich Syndrome protein (WASp)-mediated actin polymerization to T cell antigen receptor endocytosis. Journal of Experimental Medicine. 2001;194:1777–87. doi: 10.1084/jem.194.12.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annual Review of Immunology. 2008;26:233–59. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- 62.Wu Y, Yoder A. Chemokine coreceptor signaling in HIV-1 infection and pathogenesis. PLoS Pathogens. 2009;5:e1000520. doi: 10.1371/journal.ppat.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takesono A, Heasman SJ, Wojciak-Stothard B, Garg R, Ridley AJ. Microtubules regulate migratory polarity through Rho/ROCK signaling in T cells. PLoS One. 2010;5:e8774. doi: 10.1371/journal.pone.0008774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samaniego R, Sanchez-Martin L, Estecha A, Sanchez-Mateos P. Rho/ROCK and myosin II control the polarized distribution of endocytic clathrin structures at the uropod of moving T lymphocytes. Journal of Cell Science. 2007;120:3534–43. doi: 10.1242/jcs.006296. [DOI] [PubMed] [Google Scholar]
- 65.McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, et al. Visualization of the intracellular behavior of HIV in living cells. Journal of Cell Biology. 2002;159:441–52. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jolly C, Mitar I, Sattentau QJ. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. Journal of Virology. 2007;81:5547–60. doi: 10.1128/JVI.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu B, Dai R, Tian CJ, Dawson L, Gorelick R, Yu XF. Interaction of the human immunodeficiency virus type 1 nucleocapsid with actin. Journal of Virology. 1999;73:2901–8. doi: 10.1128/jvi.73.4.2901-2908.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vorster PJ, Guo J, Yoder A, Wang W, Zheng Y, Xu X, et al. LIM kinase 1 modulates cortical actin and CXCR4 cycling and is activated by HIV-1 to initiate viral infection. Journal of Biological Chemistry. 2011;286:12554–64. doi: 10.1074/jbc.M110.182238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bukrinskaya A, Brichacek B, Mann A, Stevenson M. Establishment of a functional human immunodeficiency virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. Journal of Experimental Medicine. 1998;188:2113–25. doi: 10.1084/jem.188.11.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoder A, Guo J, Yu D, Cui Z, Zhang XE, Wu Y. Effects of microtubule modulators on HIV-1 infection of transformed and resting CD4 T cells. Journal of Virology. 2011;85:3020–4. doi: 10.1128/JVI.02462-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dolei A, Biolchini A, Serra C, Curreli S, Gomes E, Dianzani F. Increased replication of T-cell-tropic HIV strains and CXC-chemokine receptor-4 induction in T cells treated with macrophage inflammatory protein (MIP)-1alpha, MIP-1beta and RANTES beta-chemokines. Aids. 1998;12:183–90. doi: 10.1097/00002030-199802000-00008. [DOI] [PubMed] [Google Scholar]
- 72.Nagira M, Sato A, Miki S, Imai T, Yoshie O. Enhanced HIV-1 replication by chemokines constitutively expressed in secondary lymphoid tissues. Virology. 1999;264:422–6. doi: 10.1006/viro.1999.0011. [DOI] [PubMed] [Google Scholar]
- 73.Zamborlini A, Lehmann-Che J, Clave E, Giron ML, Tobaly-Tapiero J, Roingeard P, et al. Centrosomal pre-integration latency of HIV-1 in quiescent cells. Retrovirology. 2007;4:63. doi: 10.1186/1742-4690-4-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, et al. infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell. 2010;143:789–801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dai J, Agosto LM, Baytop C, Yu JJ, Pace MJ, Liszewski MK, et al. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. Journal of Virology. 2009;83:4528–37. doi: 10.1128/JVI.01910-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Agosto LM, Yu JJ, Dai J, Kaletsky R, Monie D, O’Doherty U. HIV-1 integrates into resting CD4+ T cells even at low inoculums as demonstrated with an improved assay for HIV-1 integration. Virology. 2007;368:60–72. doi: 10.1016/j.virol.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA, Greene WC. A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One. 2012;7:e30176. doi: 10.1371/journal.pone.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo J, Wang W, Yu D, Wu Y. Spinoculation triggers dynamic actin and cofilin activity that facilitates HIV-1 infection of transformed and resting CD4 T cells. Journal of Virology. 2011;85:9824–33. doi: 10.1128/JVI.05170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising pre-clinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116:2078–88. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chiarini F, Fala F, Tazzari PL, Ricci F, Astolfi A, Pession A, et al. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Research. 2009;69:3520–8. doi: 10.1158/0008-5472.CAN-08-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nabel G, Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–3. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 82.Manganaro L, Lusic M, Gutierrez MI, Cereseto A, Del Sal G, Giacca M. Concerted action of cellular JNK and Pin1 restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nature Medicine. 2010;16:329–33. doi: 10.1038/nm.2102. [DOI] [PubMed] [Google Scholar]
- 83.Helms WS, Jeffrey JL, Holmes DA, Townsend MB, Clipstone NA, Su L. Modulation of NFAT-dependent gene expression by the RhoA signaling pathway in T cells. Journal of Leukocyte Biology. 2007;82:361–9. doi: 10.1189/jlb.0206120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cron RQ, Bartz SR, Clausell A, Bort SJ, Klebanoff SJ, Lewis DB. NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clinical Immunology. 2000;94:179–91. doi: 10.1006/clim.1999.4831. [DOI] [PubMed] [Google Scholar]
- 85.Kinoshita S, Chen BK, Kaneshima H, Nolan GP. Host control of HIV-1 parasitism in T cells by the nuclear factor of activated T cells. Cell. 1998;95:595–604. doi: 10.1016/s0092-8674(00)81630-x. [DOI] [PubMed] [Google Scholar]
- 86.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nature Immunology. 2007;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 87.Monteiro P, Gosselin A, Wacleche VS, El-Far M, Said EA, Kared H, et al. Memory CCR6+CD4+ T cells are preferential targets for productive HIV type 1 infection regardless of their expression of integrin beta7. Journal of Immunology. 2011;186:4618–30. doi: 10.4049/jimmunol.1004151. [DOI] [PubMed] [Google Scholar]
- 88.Lecureuil C, Combadiere B, Mazoyer E, Bonduelle O, Samri A, Autran B, et al. Trapping and apoptosis of novel subsets of memory T lymphocytes expressing CCR6 in the spleen of HIV-infected patients. Blood. 2007;109:3649–57. doi: 10.1182/blood-2006-01-035717. [DOI] [PubMed] [Google Scholar]
- 89.Arthos J, Cicala C, Martinelli E, Macleod K, Van Ryk D, Wei D, et al. HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nature Immunology. 2008;9:301–9. doi: 10.1038/ni1566. [DOI] [PubMed] [Google Scholar]
- 90.Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458:1034–8. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang C, Kang SG, Lee J, Sun Z, Kim CH. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunology. 2009;2:173–83. doi: 10.1038/mi.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Damas JK, Oktedalen O, Ueland T, Landro L, Barstad J, Muller F, et al. Enhanced levels of CCL19 in patients with advanced acquired immune deficiency syndrome (AIDS) Clinical and Experimental Immunology. 2012;167:492–8. doi: 10.1111/j.1365-2249.2011.04524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Damas JK, Landro L, Fevang B, Heggelund L, Tjonnfjord GE, Floisand Y, et al. Homeostatic chemokines CCL19 and CCL21 promote inflammation in human immunodeficiency virus-infected patients with ongoing viral replication. Clinical and Experimental Immunology. 2009;157:400–7. doi: 10.1111/j.1365-2249.2009.03976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Damas JK, Landro L, Fevang B, Heggelund L, Froland SS, Aukrust P. Enhanced levels of the CCR7 ligands CCL19 and CCL21 in HIV infection: correlation with viral load, disease progression and response to highly active antiretroviral therapy. Aids. 2009;23:135–8. doi: 10.1097/QAD.0b013e32831cf595. [DOI] [PubMed] [Google Scholar]
- 95.Fontaine J, Poudrier J, Roger M. Short communication: persistence of high blood levels of the chemokines CCL2, CCL19, and CCL20 during the course of HIV infection. AIDS Research and Human Retroviruses. 2011;27:655–7. doi: 10.1089/aid.2010.0261. [DOI] [PubMed] [Google Scholar]
- 96.Kamat A, Misra V, Cassol E, Ancuta P, Yan Z, Li C, et al. A plasma biomarker signature of immune activation in HIV patients on antiretroviral therapy. PLoS One. 2012;7:e30881. doi: 10.1371/journal.pone.0030881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, et al. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. Journal of Immunology. 2004;172:6362–72. doi: 10.4049/jimmunol.172.10.6362. [DOI] [PubMed] [Google Scholar]
- 98.Widney DP, Breen EC, Boscardin WJ, Kitchen SG, Alcantar JM, Smith JB, et al. Serum levels of the homeostatic B cell chemokine, CXCL13, are elevated during HIV infection. Journal of Interferon and Cytokine Research. 2005;25:702–6. doi: 10.1089/jir.2005.25.702. [DOI] [PubMed] [Google Scholar]
- 99.Regidor DL, Detels R, Breen EC, Widney DP, Jacobson LP, Palella F, et al. Effect of highly active antiretroviral therapy on biomarkers of B-lymphocyte activation and inflammation. Aids. 2011;25:303–14. doi: 10.1097/QAD.0b013e32834273ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Landro L, Damas JK, Halvorsen B, Fevang B, Ueland T, Otterdal K, et al. CXCL16 in HIV infection – a link between inflammation and viral replication. European Journal of Clinical Investigation. 2009;39:1017–24. doi: 10.1111/j.1365-2362.2009.02207.x. [DOI] [PubMed] [Google Scholar]
- 101.Fletcher C. Overcoming pharmacological sanctuaries 18th conference on retroviruses and opportunistic infections; 2012; p. abstract 108. [Google Scholar]
- 102.Chun TW, Justement JS, Moir S, Hallahan CW, Maenza J, Mullins JI, et al. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: implications for eradication of virus. Journal of Infectious Diseases. 2007;195:1762–4. doi: 10.1086/518250. [DOI] [PubMed] [Google Scholar]
- 103.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nature Medicine. 2003;9:727–8. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 104.Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell JT, Bixby D, Savona MR, et al. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nature Medicine. 2010;16:446–51. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Felts RL, Narayan K, Estes JD, Shi D, Trubey CM, Fu J, et al. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13336–41. doi: 10.1073/pnas.1003040107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, et al. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature. 2011;477:95–8. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 107.Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nature Reviews Drug Discovery. 2009;8:23–33. doi: 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- 108.Wijtmans M, Verzijl D, Leurs R, de Esch IJ, Smit MJ. Towards small-molecule CXCR3 ligands with clinical potential. ChemMedChem. 2008;3:861–72. doi: 10.1002/cmdc.200700365. [DOI] [PubMed] [Google Scholar]
- 109.Xu H, Firdawes S, Yamamoto A, Zhao Y, Ihara Y, Uehara S, et al. Effects of blocking the chemokine receptors, CCR5 and CXCR3, With TAK-779 in a rat small intestinal transplantation model. Transplantation. 2008;86:1810–7. doi: 10.1097/TP.0b013e31818fe7fe. [DOI] [PubMed] [Google Scholar]
- 110.Kohler RE, Comerford I, Townley S, Haylock-Jacobs S, Clark-Lewis I, McColl SR. Antagonism of the chemokine receptors CXCR3 and CXCR4 reduces the pathology of experimental autoimmune encephalomyelitis. Brain Pathology. 2008;18:504–16. doi: 10.1111/j.1750-3639.2008.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liston A, Kohler RE, Townley S, Haylock-Jacobs S, Comerford I, Caon AC, et al. Inhibition of CCR6 function reduces the severity of experimental autoimmune encephalomyelitis via effects on the priming phase of the immune response. Journal of Immunology. 2009;182:3121–30. doi: 10.4049/jimmunol.0713169. [DOI] [PubMed] [Google Scholar]
- 112.Pilkington KR, Clark-Lewis I, McColl SR. Inhibition of generation of cytotoxic T lymphocyte activity by a CCL19/macrophage inflammatory protein (MIP)-3beta antagonist. Journal of Biological Chemistry. 2004;279:40276–82. doi: 10.1074/jbc.M405808200. [DOI] [PubMed] [Google Scholar]
- 113.Proost P, Mortier A, Loos T, Vandercappellen J, Gouwy M, Ronsse I, et al. Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood. 2007;110:37–44. doi: 10.1182/blood-2006-10-049072. [DOI] [PubMed] [Google Scholar]
- 114.Proost P, Schutyser E, Menten P, Struyf S, Wuyts A, Opdenakker G, et al. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood. 2001;98:3554–61. doi: 10.1182/blood.v98.13.3554. [DOI] [PubMed] [Google Scholar]