

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening clinical syndrome characterized by dysregulation of the immune system. Impaired function of cytotoxic T cells and natural killer cells is often seen, and T-cell malignancies represent most cases of lymphoma-associated HLH. HLH associated with B-cell lymphoma is rare. We describe a case of a 30-year-old man who presented with fever, splenomegaly, and hyperferritinemia. Bone marrow biopsy revealed T-cell/histiocyte-rich large B-cell lymphoma, a rare, aggressive B-cell malignancy. This case highlights the interplay between a pro-inflammatory cytokine microenvironment and tumor-mediated immune suppression, and addresses the importance of accurately diagnosing these entities for appropriate clinical management.
Keywords: Hemophagocytic lymphohistiocytosis (HLH), T-cell/histiocyte rich large B-cell lymphoma (THRLBCL), Chemotherapy, Stem cell transplant
Highlights
-
•
We present an uncommon case of HLH associated with TCHRBCL with cytotoxic T-cells.
-
•
The case highlights the interplay between immune hyperactivation and tumor immune evasion.
-
•
Treatment considerations for this difficult case based on updated guidelines are reviewed.
1. Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an uncommon, hyperinflammatory syndrome that is often fatal when treatment is delayed [1]. Diagnosis may be difficult due to the wide range of symptoms associated with HLH and lack of specific diagnostic tests. Genetic causes of familial HLH relate to defects in cytotoxic granule exocytosis or function, whereas the acquired or secondary form is often associated with infection, malignancy, or autoimmune/immunodeficiency conditions. Defects in cytotoxic T cells and natural killer (NK) cells are seen in nearly all cases of HLH [1]. Hematologic neoplasms account for the majority of malignancy-associated HLH, and T-cell malignancies predominate. HLH associated with underlying B-cell lymphoma is rare, and seems to occur in older patients with a low incidence of marrow involvement by lymphoma [2]. Here, we describe a case of HLH arising in a young patient with diffuse marrow involvement by T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL), a subtype representing 1–3% of diffuse large B-cell lymphomas (DLBCL) with a characteristic background of cytotoxic T cells and histiocytes.
2. Clinical presentation
A 30-year-old male with a past medical history of adult-onset Still׳s disease diagnosed 1.5 years ago was transferred from an outside hospital with fever, jaundice, weight loss, and impending respiratory failure. On physical exam, there was splenomegaly and imaging demonstrated retroperitoneal lymphadenopathy and pulmonary infiltrates. Lab findings revealed pancytopenia and liver function tests were suggestive of severe hepatitis. Significant laboratory findings included total bilirubin 10.2 mg/dL (reference range 0.3–1.2 mg/dL), alkaline phosphatase 410 IU/L (30–115 IU/L), AST 320 IU/L (10–41 IU/L), ALT 476 IU/L (10–40 IU/L), LDH 749 IU/L (110–240 IU/L), ferritin 14,298 ng/mL (23–336 ng/mL), WBC 2100/µL (4300–10,300/µL), hemoglobin 7.5 g/dL (14.0–18.0 g/dL), and platelet count 68,000/μL (140,000–440,000/μL). An extensive infectious disease workup did not reveal an etiology. Concern for hemophagocytic lymphohistiocytosis was raised and a bone marrow biopsy was performed.
3. Pathologic findings
Bone marrow aspirate smears were hemodilute but notable for frequent macrophages exhibiting erythro- and leukophagocytosis (Fig. 1A). Bone biopsy demonstrated absent normal hematopoiesis and diffuse infiltration by lymphocytes and histiocytes (Fig. 1B). CD20 highlighted scattered, large atypical cells (Fig. 1C) that were negative for CD15 and CD30. The background infiltrate was composed predominantly of T cells with a cytotoxic phenotype (CD3+, CD5+, CD7+, CD8+, and TIA-1+ [Fig. 2]), with admixed CD68+-histiocytes (Fig. 2D). Flow cytometry analysis did not show evidence of an abnormal B- or T-cell population. Polymerase chain reaction studies did not detect clonal IgH@, IgK@ or TCR gene rearrangements, although neoplastic cells were scant. in situ hybridization for Epstein Barr virus (EBV)-encoded RNA was negative.
Fig. 1.
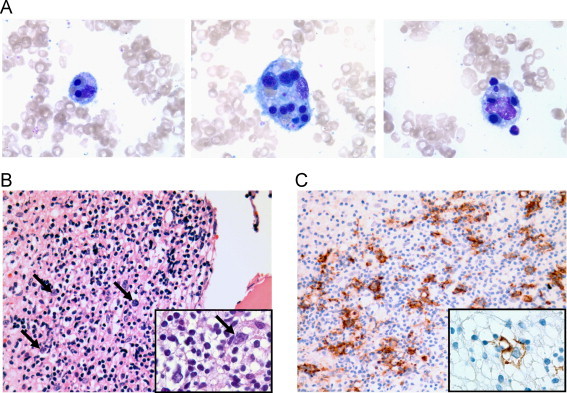
Aspirate smear showing examples of hemophagocytosis, Wright Geimsa stain, ×1000 (A). Bone marrow core biopsy showing diffuse lymphohistiocytic infiltration effacing the normal marrow architecture with scattered large atypical cells (arrows), H&E stain, ×400 (B), inset ×1000. CD20 immunostain highlights large neoplastic B cells, ×400 (C), inset ×1000.
Fig. 2.
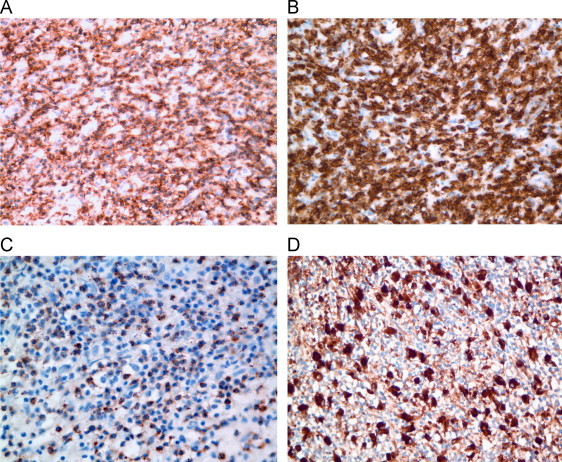
Bone marrow core biopsy stained with antibody for CD3 (A) and CD8 (B) showing diffuse lymphocytic infiltrate. TIA-1 (C) demonstrates punctate staining of cytotoxic T cells. CD68 (D) highlights admixed histiocytes. (A, B, D: ×400, C: ×600).
A diagnosis of hemophagocytic lymphohistiocytosis arising in a background of T-cell/histiocyte-rich large B-cell lymphoma was made. This case was sent for expert review at an outside institution, and the diagnosis was upheld. Biopsies of the retroperitoneal lymph node and lung infiltrate also demonstrated involvement by THRLBCL.
4. Clinical management
Diagnostic criteria for HLH were updated in 2004 and rely on a combination of clinical, laboratory, pathologic, and genetic results (Table 1) [3,4]. Further laboratory workup for the patient revealed hypertriglyceridemia (1040 mg/dL; reference range <150 mg/dL), hypofibrinogenemia (113 mg/dL; 150–400 mg/dL), and elevated soluble IL-2 receptor (7469 U/mL; 45–1105 U/mL). NK cell function was within normal limits. Thus, this patient fulfilled 7 out of 8 criteria for the diagnosis of HLH. Primary HLH was considered unlikely given the patient׳s age, and studies on peripheral blood did not show abnormalities in expression of perforin, granzyme B, SLAM-associated protein, or X-linked inhibitor of apoptosis [3,4]. The patient did show a very low titer of EBV DNA (278 IU/mL) of uncertain significance, which was undetectable 3 weeks into treatment.
Table 1.
Revised diagnostic guidelines for HLH [3].
| Molecular diagnosis consistent with HLH |
| (PRF1, UNC13D, STXBP2, RAB27A, STX11, SH2D1A, or XIAP) |
| OR |
| ≥5 of the following 8 diagnostic criteria: |
| Fever |
| Splenomegaly |
| Cytopenias (affecting≥2 of 3 lineages) |
| Hemoglobin<9 g/dL |
| Platelets<100,000/μL |
| Neutrophils<1000/μL |
| Hypertriglyceridemia and/or hypofibrinogenemia |
| Fasting triglycerides≥265 mg/dL |
| Fibrinogen≤150 mg/dL |
| Hemophagocytosis in bone marrow, spleen, or lymph nodes |
| Ferritin≥500 ng/mL |
| Decreased or absent NK-cell activity |
| Soluble CD25≥2400 U/mL |
Upon diagnosis, the patient׳s lymphoma and HLH represented a therapeutic dilemma. Effective early therapy reduces mortality of HLH from 95% to 30–35% [4]. The HLH-2004 treatment protocol consists of T-cell targeted immunosupression with dexamethasone and cyclosporine, macrophage targeted etoposide with intrathecal methotrexate, if indicated, and monthly IV immunoglobulin [4]. In addition, patients with THRLBCL should be treated similarly to DLBCL with anthracycline-based chemotherapy and an anti-CD20 monoclonal antibody. Complicating this scenario was the patient׳s multiorgan failure with severe hepatitis and high total bilirubin levels, which represent contraindications to many chemotherapy agents. As etoposide has significantly impacted survival outcome in HLH and induced sustained remissions of HLH, a full dose was administered per HLH-2004 despite being contraindicated in liver failure [4]. Three weeks later, when the patient׳s HLH had responded (ferritin decreased from 14,298 to 3056 ng/mL), a treatment with dose-adjusted EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin) with rituximab was started. This regimen was selected as it contains etoposide, and doses for subsequent cycles can be adjusted based on cytopenias, avoiding the use of G-CSF which can be catastrophic in HLH [5].
The patient achieved remission as confirmed by clinical and laboratory workup, including follow-up bone marrow biopsy, and will be further consolidated with an autologous stem cell transplant.
5. Discussion
Recent studies of genetic HLH have shed light on the pathophysiology of the disease. Defects in cytotoxic pathways of T cells and NK cells are thought to lead to an inability to clear antigenic stimuli, resulting in a “cytokine storm” and perpetuation of the inflammatory response [1]. Unchecked macrophage activation, hemophagocytosis, and tissue infiltration ensue.
Acquired HLH is most often associated with infections (49%), of which EBV is the most common, followed by malignancies (27%), rheumatologic disorders (7%) and immune deficiency syndromes (6%) [1]. Still׳s disease, also known as systemic-onset juvenile idiopathic arthritis, is a form of juvenile rheumatoid arthritis that carries an overall risk of HLH of 10% [6]. In our particular case, the relationship between the patient׳s hematologic malignancy and underlying autoimmune disorder, diagnosed as adult-onset Still׳s disease, is unclear.
When HLH is associated with malignancy, T- and NK-cell leukemias/lymphomas predominate, although HLH has also been seen with anaplastic large cell lymphoma, other leukemias, and solid tumors. HLH association with non-Hodgkin B-cell lymphoma is relatively rare, involving predominantly older patients (>60 years), with sparing of the bone marrow, as compared with T- or NK-cell lymphomas [2].
T-cell/histiocyte-rich large B cell lymphoma accounts for 1–3% of DLBCL, presents more commonly in males with a median age of 30 years, and typically pursues an aggressive clinical course. Diagnosis is based on the identification of <10% large neoplastic B cells in a background of cytotoxic T cells and histiocytes [7]. Immunohistochemistry can assist with differentiation from nodular lymphocyte-predominant Hodgkin lymphoma and lymphocyte-rich classical Hodgkin lymphoma by demonstrating expression of pan-B cell markers and absence of CD15 and CD30 in large cells, along with a cytotoxic phenotype of the background lymphocytes. Molecular analysis for IGH@/IGK@ gene rearrangements can assist in demonstrating clonality; however, the paucity of neoplastic cells in THRLBCL can diminish the sensitivity of this PCR based assay leading to false-negative results [8].
From a pathophysiologic standpoint, it is of interest that HLH arose in the setting of THRLBCL in this patient. Although THRLBCL is a B-cell malignancy, it is unique in that the predominant cell population consists of cytotoxic T cells and histiocytes. Recent gene expression profiling studies have suggested that the tumor microenvironment of THRLBCL in fact favors a tolerogenic host immune response despite the abundance of apparently ineffective cytotoxic T cells [9]. In addition, both the tumor cells as well as the infiltrating histiocytes have been shown to express the immune suppressive molecule PD-L1, presumably as a mechanism for tumor immune evasion, representing a potential target for therapy [10]. Interestingly, in 1999, Mitterer, et al. [11], reported one other case of HLH-associated THRLBCL, which was associated with reactivated EBV infection, again raising the possibility that the overall host immune environment contributes to disease manifestation. Further studies examining the interplay between systemic immune hyperactivation, tumor-mediated immune suppression, and autoimmunity in cases of malignancy-associated HLH are warranted to uncover novel and directed therapeutic approaches.
References
- 1.Usmani G.N., Woda B.A., Newburger P.E. Advances in understanding the pathogenesis of HLH. Br J Haematol. 2013;161:609–622. doi: 10.1111/bjh.12293. [DOI] [PubMed] [Google Scholar]
- 2.Han A., Lee H.R., Park B. Lymphoma-associated hemophagocytic syndrome: clinical features and treatment outcome. Ann Hematol. 2007;86:493–498. doi: 10.1007/s00277-007-0278-6. [DOI] [PubMed] [Google Scholar]
- 3.Henter J., Horne A., Arico M. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 4.Jordan M.B., Allen C.E., Weitzman S. How I treat hemophagocytic lymphohistiocytosis. Blood. 2009;118:4041–4052. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson W.H., Grossbard M.L., Pittaluga S. Dose-adjusted EPOCH chemotherapy for untreated large B-cell lymphomas: a pharmacodynamic approach with high efficacy. Blood. 2002;99:2685–2693. doi: 10.1182/blood.v99.8.2685. [DOI] [PubMed] [Google Scholar]
- 6.Kelly A., Ramanan A.V. Recognition and management of macrophage activation syndrome in juvenile arthritis. Curr Opin Rheum. 2007;19:477–481. doi: 10.1097/BOR.0b013e32825a6a79. [DOI] [PubMed] [Google Scholar]
- 7.De Wolf-Peeters C., Delabie J., Campo E. T cell/histiocyte-rich large B-cell lymphoma. In: Swerdlow S.H., editor. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. IARC Press; Lyon: 2008. pp. 238–239. [Google Scholar]
- 8.van Dongen J.J.M., Langerak A.W., Brüggemann M. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia. 2003;17:2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 9.Van Loo P., Tousseyn T., Vanhentenrijk V. T-cell/histiocyte-rich large B-cell lymphoma shows transcriptional features suggestive of a tolerogenic host immune response. Haematologica. 2010;95:440–448. doi: 10.3324/haematol.2009.009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen B.J., Chapuy B., Ouyang J. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19:3462–3473. doi: 10.1158/1078-0432.CCR-13-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitterer M., Pescosta N., McQuain C. Epstein–Barr virus related hemophagocytic syndrome in a T-cell rich B-cell lymphoma. Ann Oncol. 1999;10:231–234. doi: 10.1023/a:1008212516595. [DOI] [PubMed] [Google Scholar]