

Abstract
In this report, we present a rare association between Ebstein anomaly (EA) and isovaleric acidemia (IVA) in a newborn who was admitted to our cardiac center. He underwent for PDA stenting to maintain adequate pulmonary blood flow, later he developed recurrent metabolic acidosis, prominent sweaty feet odor, neutropenia and thrombocytopenia. His organic acids profile in the urine confirmed the diagnosis of IVA.
To the best of our knowledge, there is no association between these two rare diseases. We are presenting this case report to highlight this rare association.
Keywords: Ebstein anomaly, Isovaleric acidemia, Congenital heart disease, Metabolic disorder
Introduction
Ebstein’s anomaly (EA) is a congenital heart disease characterized primarily by abnormalities of the tricuspid valve (TV), right atrium (RA) and right ventricle (RV). The estimated risk of Ebstein’s anomaly in the general population is one in 200,000 live births [1]. In newborns with this anomaly, tricuspid valve regurgitation may be significant as it can result in ineffective RV contractility that is unable to generate forward enough force to open the pulmonary valve in systole, thus producing ‘functional’ pulmonary atresia [2]. Isovaleric acidemia (IVA) is known as one of the ‘classical’ organic acidemias, and is caused by a genetic deficiency of isovaleryl-CoA dehydrogenase catalyzing the third step in leucine catabolism [3].
In this report, we describe one of the rare associations between EA and IVA diagnosed in an infant admitted to our center.
Case presentation
A two-day male newborn (3.9 kg) was admitted to our Pediatric Cardiac Intensive Care Unit. Diagnosed with cyanotic congenital heart disease, he was placed on prostaglandin E1 (PGE1) for further diagnosis and management, and intubated shortly after that. His chest X-ray examination showed cardiomegaly. Trans-thoracic echocardiography (TTE) confirmed diagnosis of Ebstein’s anomaly, functional pulmonary atresia, and patent ductus arteriosus (PDA) with systemic to pulmonary shunt through PDA (Fig. 1). The patient was found to be dependent on PDA, and a trial of weaning from PGE1 lead to significant desaturation. He was hemodynamically stable with PGE1 infusion, and was maintained on 70–85% oxygen saturation.
Figure 1.
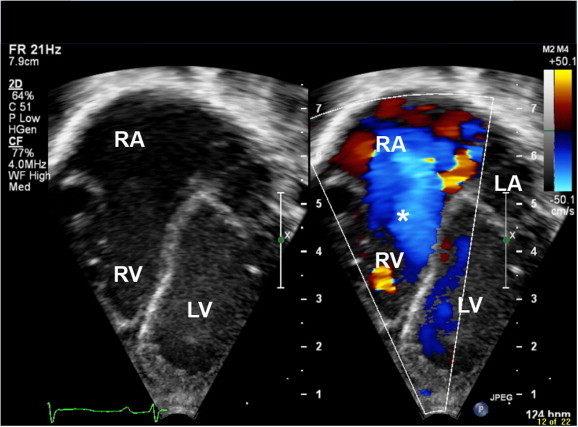
Trans thoracic echocardiography with Doppler showing tricuspid valve regurgitation. ∗ Tricuspid valve regurgitation, LV: Left ventricle, RV: Right ventricle, RA: Right atrium.
The patient underwent PDA stenting by interventional cardiac catheterization on day 10 of life in order to maintain adequate pulmonary flow through PDA and to allow liberation from PGE1 infusion. Following successful PDA stenting and liberation from PGE1, the patient was weaned from the ventilator, and PGE1 infusion was stopped completely with no hemodynamic compromise or desaturation. Saturation was maintained with normal pulmonary blood flow and normal lung vascularity (Fig. 2).
Figure 2.
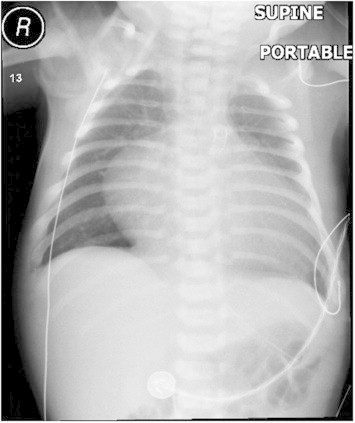
CXR after PDA stenting.
During the first 2 weeks of life, attempts to initiate feeding were unsuccessful due to both hemodynamic instability and to intolerance of enteral feeding. After extubation, feeding was established with regular baby formula. However, within 48 h of achieving full feeding, the baby started to have decreased levels of consciousness, became lethargic with episodes of jitteriness coupled with hypocalcemia, feeding intolerance, and persistent metabolic acidosis. The patient had also developed neutropenia, and thrombocytopenia (Table 1). Septic workup was completed, repeatedly demonstrating no evidence of infection during the first 4 weeks of life.
Table 1.
Trends of arterial blood gases PH, base excess, blood WBC counts, and platelets counts during pediatric cardiac intensive care course, showing neutropenia after third week of life and persistent thrombocytopenia after second week of life.
| Day 1 | Day 5 | Day 10 | Day 15 | Day 20 | Day 25 | Day 30 | Day 35 | Day 37 | |
|---|---|---|---|---|---|---|---|---|---|
| PH | 7.35 | 7.43 | 7.37 | 7.34 | 7.33 | 7.36 | 7.32 | 7.20 | 7.09 |
| Base Excess (mmol/L) | −3.9 | 3 | −0.9 | −8.9 | −5.7 | −3.4 | −2.5 | −14 | −15.4 |
| WBC (x103/ml) | 21 | 11.3 | 9.6 | 9.2 | 3.4 | 3.2 | 1.7 | 1.3 | 1.4 |
| PLT (x103/ml) | 241 | 220 | 195 | 97 | 54 | 22 | 28 | 41 | 9 |
During his intensive care stay, the patient started to give off a distinctively foul odor, similar to ‘sweaty feet’, despite good hygiene. The odor coincided with the patient’s feeding initiation during the second week of life. Organic acidemia was suspected. Serum ammonia level was found to be within normal limits but urine organic acid profile was positive and showed highly elevated levels of isovalerylglycine and 3-hydroxyisovaleric, consistent with isovaleric acidemia. Based on these results, the patient was started on carnitine and given parenteral nutrition. Glycine was unavailable in our hospital. Subsequently, overwhelming sepsis developed after 1 month, and the patient deteriorated due to overwhelming refractory septic shock with Klebsiella pneumonia. He succumbed at day 37 of life. Pre-mortal blood sample had been obtained to analyze the gene responsible for IVA, and genetic counseling for the family of the deceased ensued.
Discussion
Tanaka and associates were the first to recognize and describe isovaleric acidemia in 1966 [4]. There is no published data about the prevalence of IVA in Saudi Arabia. Research shows that IVA is caused by mutations in the IVD gene, or isovaleryl-CoA dehydrogenase [5]. The IVD enzyme is encoded by the nuclear gene, which is located on chromosome 15q14-15 [6].
IVA may manifest in acute or chronic intermittent form. Clinical manifestations in the acute form include vomiting and severe acidosis in the first few days of life. Lethargy, convulsions, and coma may ensue. Death may follow if proper therapy is not initiated. A characteristic odor of ‘sweaty feet’ may be present [7]. This odor was a prominent sign in our patient, and led us to start investigations for IVA.
Infants who survive this acute episode will go onto have the chronic intermittent form later on in life. A milder form of the chronic intermittent disease also exists. In both forms, acute episodes of metabolic decompensation may occur during a catabolic state such as an infection [7].
We think that three factors possibly delayed clinical manifestations of IVA in our patient. These were the use of IV fluid since birth, delayed feeding, and the frequent replacements of sodium bicarbonate, attributed initially to inadequate cardiac output secondary to heart disease.
Laboratory findings during the acute attacks include ketoacidosis, neutropenia, thrombocytopenia and, occasionally, pancytopenia due to bone marrow suppression. Hypocalcaemia, hyperglycemia and moderate to severe hyperammonemia may be present in some patients [7]. All the above listed laboratory findings were found in our patient except ammonia level, which remained within normal levels.
Diagnosis of IVA is established by demonstrating marked elevations of isovaleric acid and its metabolites in body fluids, especially in urine, as first described in 1967 [8]. We based our patient diagnosis on urine tests, which repeatedly showed highly positive results.
Isovaleric acidemia is inherited as an autosomal recessive trait [6]. Our patient was the firstborn to his parents, and this disease was not previously diagnosed in either family.
Treatment of the acute attack targets hydration, reversal of the catabolic state by providing adequate calories orally or intravenously, correction of metabolic acidosis, and removal of the excess isovaleric acid.
Isovalerylglycine has a high urinary clearance, and therefore administration of glycin is recommended to enhance formation of isovalerylglycine.
l-carnitine also increases removal of isovaleric acid by forming isovalerylcarnitine, which is excreted in the urine. Our patient received l-carnitine directly after confirmation of IVA.
The etiology of Ebstein’s anomaly is unknown. However, in rare cases, genetic factors such as mutations in the transcription factor NKX2.5, 10p13-p14 deletion and 1p34.3-p36.11 deletion are described [1]. Newborns with severe forms of Ebstein’s anomaly have marked cyanosis and cardiomegaly, while the majority are dependent on a PDA patency for pulmonary blood flow [9].
Our patient was PDA dependent, and needed PGE1 to maintain ductus arteriosus patency.
Metabolic disorders are reported in association with some cardiac anomalies, mainly myopathy and cardiomyopathy. These may include glycogen storage diseases, disorders of lipid metabolism such as carnitine deficiency, disorders of mitochondrial metabolism and other storage disorders such mucopolysaccharidosis. Organic acidemias are rarely reported with cardiac abnormality [10]. In methylmalonic acidemia, hypertrophic and dilated cardiomyopathy have been described [11].
Furthermore, dilated cardiomyopathy is also a frequent complication reported in association with propionic acidemia (PA) [12].
To the best of our knowledge, there is no reported association between isovaleric acidemia and congenital heart disease in medical literature.
In conclusion, congenital heart disease may occur in association with metabolic disorder. Although this association is rare, it should not be overlooked, particularly in cases of unexplained acidemia or other investigations that cannot be attributed to congenital heart problems. Further genetic studies are needed to explore the possible association of specific heart lesions with certain metabolic diseases.
Footnotes
Peer review under responsibility of King Saud University.

References
- 1.Krieger E.V., Valente A.M. Diagnosis and management of Ebstein anomaly of the tricuspid valve. Curr Treat Options Cardiovasc Med. 2012;14(6):594–607. doi: 10.1007/s11936-012-0209-2. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki H., Nakasato M., Sato S., Komatsu H., Hayasaka K. Management of functional pulmonary atresia with isoproterenol in a neonate with Ebstein’s anomaly. Tohoku J Exp Med. 1997;181(4):459–465. doi: 10.1620/tjem.181.459. [DOI] [PubMed] [Google Scholar]
- 3.Ensenauer R., Vockley J., Willard J.M., Huey J.C., Sass J.O., Edland S.D. A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. Am J Hum Genet. 2004;75(6):1136–1142. doi: 10.1086/426318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanaka K., Budd M.A., Efron M.L., Isselbacher K.J. Isovaleric acidemia: a new genetic defect of leucine metabolism. Proc Natl Acad Sci U S A. 1966;56(1):236–242. doi: 10.1073/pnas.56.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ensenauer R., Fingerhut R., Maier E.M., Polanetz R., Olgemöller B., Röschinger W. Newborn screening for isovaleric acidemia using tandem mass spectrometry: data from 1.6 million newborns. Clin Chem. 2011;57(4):623–626. doi: 10.1373/clinchem.2010.151134. [DOI] [PubMed] [Google Scholar]
- 6.Hertecant J.L., Ben-Rebeh I., Marah M.A., Abbas T., Ayadi L., Ben Salem S. Clinical and molecular analysis of isovaleric acidemia patients in the United Arab Emirates reveals remarkable phenotypes and four novel mutations in the IVD gene. Eur J Med Genet. 2012;55(12):671–676. doi: 10.1016/j.ejmg.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Dercksen M., Duran M., Ijlst L., Mienie L.J., Reinecke C.J., Ruiter J.P. Clinical variability of isovaleric acidemia in a genetically homogeneous population. J Inherit Metab Dis. 2012;35(6):1021–1029. doi: 10.1007/s10545-012-9457-2. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka K., Isselbacher K.J. The isolation and identification of N-isovalerylglycine from urine of patients with isovaleric acidemia. J Biol Chem. 1967;242(12):2966–2972. [PubMed] [Google Scholar]
- 9.Bove E.L., Hirsch J.C., Ohye R.G., Devaney E.J. How I manage neonatal Ebstein’s anomaly. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2009;12(1):63–65. doi: 10.1053/j.pcsu.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 10.Gilbert-Barness E. Review: metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci. 2004;34(1):15–34. [PubMed] [Google Scholar]
- 11.Prada C.E., Al Jasmi F., Kirk E.P., Hopp M., Jones O., Leslie N.D. Cardiac disease in methylmalonic acidemia. J Pediatr. 2011;159(5):862–864. doi: 10.1016/j.jpeds.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Romano S., Valayannopoulos V., Touati G., Jais J.P., Rabier D., de Keyzer Y. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr. 2010;156(1):128–134. doi: 10.1016/j.jpeds.2009.07.002. [DOI] [PubMed] [Google Scholar]