

Abstract
Patients with HIV are at an increased risk of cardiovascular disease. In this study we investigated the effect of Nef, a secreted HIV protein responsible for the impairment of cholesterol efflux, on the development of atherosclerosis in two animal models. ApoE−/− mice fed a high-fat diet and C57BL/6 mice fed a high-fat, high-cholesterol diet were injected with recombinant Nef (40 ng/injection) or vehicle, and the effects of Nef on development of atherosclerosis, inflammation, and dyslipidemia were assessed. In apoE−/− mice, Nef significantly increased the size of atherosclerotic lesions and caused vessel remodeling. Nef caused elevation of total cholesterol and triglyceride levels in the plasma while reducing high-density lipoprotein cholesterol levels. These changes were accompanied by a reduction of ABCA1 abundance in the liver, but not in the vessels. In C57BL/6 mice, Nef caused a significant number of lipid-laden macrophages presented in adventitia of the vessels; these cells were absent from the vessels of control mice. Nef caused sharp elevations of plasma triglyceride levels and body weight. Taken together, our findings suggest that Nef causes dyslipidemia and accumulation of cholesterol in macrophages within the vessel wall, supporting the role of Nef in pathogenesis of atherosclerosis in HIV-infected patients.—Cui, H. L., Ditiatkovski, M., Kesani, R., Bobryshev, Y. V., Liu, Y., Geyer, M., Mukhamedova, N., Bukrinsky, M., Sviridov, D. HIV protein Nef causes dyslipidemia and formation of foam cells in mouse models of atherosclerosis.
Keywords: HIV, cholesterol, metabolic syndrome, vessels, macrophages, reverse cholesterol transport
It has been consistently demonstrated that HIV infection is associated with increased risk of development of atherosclerosis and cardiovascular disease (1, 2). The mechanism was originally ascribed to the antiretroviral regiments causing dyslipidemia; however, emerging evidence suggests that HIV infection itself also contributes significantly to dyslipidemia and the risk of atherosclerosis (3). One mechanism of how HIV infection may accelerate the development of atherosclerosis is the inhibition of cellular cholesterol efflux and, consequently, accumulation of cellular cholesterol caused by the viral protein Nef (3). Not only does Nef impair cholesterol efflux in macrophages infected by HIV, but soluble Nef, released from infected cells, has a systemic effect impairing ATP binding cassette transporter A1 (ABCA1), the key element of cholesterol efflux machinery, in cells not infected with the virus (4). However, direct demonstration of proatherogenic activity of Nef has not been provided yet. Pathogenesis of atherosclerosis is a complex process, and in addition to disturbances of cholesterol metabolism also includes inflammation, oxidation, and apoptosis. It remains unclear whether Nef protein contributes to these pathological events associated with accelerated development of atherosclerosis in HIV-infected patients.
In this study we investigated the effect of recombinant Nef injected into uninfected animals on the development of atherosclerosis. We used two dissimilar animal models of atherosclerosis. One is a widely used and extensively characterized genetic model, apoE−/− mice fed a high-fat diet (HFD). Another is a dietary model, C57BL/6 mice fed with high-fat/high-cholesterol diet. The latter model is a reflection of atherosclerosis prompted by nongenetic causes; however, atherosclerosis in this model rarely progresses beyond a very early stage of fatty streaks, restricting utility of this model to the initial stages of pathogenesis of atherosclerosis (5). On the other hand, this model provides a background with limited number of proatherogenic factors, which could be an advantage when studying an intervention that is expected to be proatherogenic.
MATERIALS AND METHODS
Recombinant Nef
Full length, myristoylated wild-type HIV-1 Nef protein, allele SF2, was prepared as described previously (6). The protein was concentrated to 2 mg/ml in 10 mM Tris/HCl, 100 mM NaCl buffer (pH 7.6), portioned into aliquots, snap-frozen in liquid nitrogen, and stored at −80°C. Prior to injection the protein was diluted in saline to a final concentration of 1 μg/ml.
Animal models of atherosclerosis
All experiments were approved by the Animal Ethics Committee of the Alfred Medical Research and Education Precinct (AMREP; Melbourne, VIC, Australia) and conformed to the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and the Weatherall Report on the use of nonhuman primates in research.
Male Apoe−/− (C57BL/6J-apoetm1Unc) mice were obtained from the AMREP animal center. At 10 wk of age, the animals were fed an HFD containing 21% fat and 0.15% cholesterol (SF00-219; Specialty Feeds, Glen Forrest, WA, Australia), available ad libitum for 12 wk. Recombinant Nef was resuspended in saline and administered to the animals intraperitoneally at a dose of 40 ng/injection 2×/wk (n=8); a control group (n=9) received the same volume of saline at the same frequency as the treatment group. This dose of Nef was chosen to roughly correspond to the concentration of Nef in plasma of HIV-infected patients (10 ng/ml; ref. 7), concentration of Nef active in the in vitro tests (4) and EC50 of a Nef peptide in an apoptosis assay (8). Blood samples were collected by submandibular puncture. At the end of the treatment, mice were euthanized by CO2 inhalation, and blood was collected by cardiac puncture. The aorta, up to the point of renal arteries, was removed for subsequent analysis. The heart was removed, allowing for analysis of plaques within the aortic sinus region. The liver was removed for analysis of ABCA1 abundance and lipid content.
Male C57BL/6 mice were obtained from the AMREP animal center. At 10 wk of age the animals were fed a high-fat/high-cholesterol diet (HFHCD) containing 15% fat, 1% cholesterol and 0.5% cholate (SF00-245; Specialty Feeds) available ad libitum for 20 wk. Recombinant Nef was resuspended in saline and administered to the animals intraperitoneally at a dose of 40 ng/injection 2×/wk (n=10); a control group (n=9) received the same volume of saline at the same frequency as the treatment group. Blood and tissue samples were obtained as described above.
Histology
Aortas were perfused with PBS containing 2 mM EDTA and excised from the animal. Aortas were cleaned of periaortic fat and stained for lipids with Sudan IV. Within each aorta, the stained area was quantified using Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Bethesda, MD, USA) and expressed as a proportion of the aorta surface occupied by plaques.
Segments of aorta or heart tissue containing the aortic sinus were embedded in optimal cutting temperature (OCT) compound (Sakura, Torrance, CA, USA) and frozen for assessment of plaques within the aortic regions. Consecutive 10 μm sections spanning 240 μm of the aortic segments or aortic sinus were cut and collected. Three sections per segment per mouse, 80 μm apart, were stained with Oil Red O to determine lipid content within the lesion or with Masson's trichrome stain to detect collagen. The cross-sectional size of the lesions (depth) was expressed as absolute area in sections stained with Oil Red O. Cellularity and inflammation in the plaques was assessed by immunohistochemistry. Briefly, following fixation in ice-cold acetone, the sections were incubated with 3% H2O2, followed by 10% serum and avidin/biotin blocking solution. The sections were then incubated with a primary antibody (anti-CD68, AbD Serotec, Kidlington, UK; anti-CD3, and anti-ABCA1, Abcam, Cambridge, MA, USA; anti-eNOS, BD Biosciences, San Jose, CA, USA; anti-VCAM-1, BD Pharmingen, San Diego, CA, USA; anti-nitrotyrosine, Merck, Darmstadt, Germany); followed by corresponding biotinylated secondary antibodies. Images were collected on an Olympus FSX100 microscope (Olympus, Tokyo, Japan) and quantified using ImagePro plus 6.0 software. Stained areas were expressed as a percentage of the total area of atheromatous plaque stained with each of the antibodies. For coimmunofluorescence staining, CD68 was detected with Alexa633-labeled anti-rat IgG, and ABCA1 was detected with Alexa488-labeled anti-rabbit IgG antibodies. Staining was visualized using a BX61 fluorescence microscope (Olympus). Images were stitched and analyzed using ImageJ (NIH, Bethesda, MD, USA).
Analysis of plasma lipids, lipoproteins, and cytokines
Blood was collected from animals by submandibular bleeding; at end point, blood was collected by cardiac puncture. Blood was centrifuged and plasma collected. Plasma was analyzed for total cholesterol (TC) and total triglyceride (TG) contents using colorimetric kits (Wako, Osaka, Japan) according to the manufacturer's instructions. To quantify high-density lipoprotein (HDL), apoB-containing particles were precipitated from the plasma by the dextran sulfate and magnesium chloride method (9) and the remaining plasma analyzed for total cholesterol using a colorimetric kit. Size distribution of HDL subfractions has been assessed by nondenaturing gradient gel electrophoresis using 4–20% Tris-glycine gels (Life Technologies, Carlsbad, CA, USA) as described previously (10). Plasma lecithin cholesterol acyltransferase (LCAT) activity was assessed using fluorometric LCAT activity assay kit (Merck) according to the manufacturer's instructions. For fast protein liquid chromatography (FPLC) analysis, 25 μl of plasma was separated using Superose 6 PC3.2/30 column (GE Healthcare, Piscataway, NJ, USA) and 100 μl fractions were collected for 2 h. Total and free cholesterol, triglyceride, and phospholipid concentrations were evaluated in each fraction using commercially available kits (T-Chol E, F-Chol E, TG E, PL E; Wako). The cytokines levels in the plasma were assessed using the CBA Mouse Inflammation Kit (BD Biosciences) according to the manufacturer's instructions.
Liver ABCA1 content, lipid composition, and gene expression
Frozen samples of mouse liver were homogenized with lysis buffer (150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, and 50 mM Tris, pH 7.4) supplemented with complete protease inhibitor cocktail (Roche, Basel, Switzerland). A total of 15 μg protein were loaded onto the SDS-polyacrylamide gel and subjected to electrophoresis. The samples were then transferred onto a PVDF membrane and probed with anti-ABCA1 antibody (Abcam); the antibody were validated by staining ABCA1 in HeLa cells and HeLa cells stably transfected with ABCA1 and RAW 264.7 cells treated with siRNA against ABCA1. For loading control, blots were probed with monoclonal anti-GAPDH antibody. Bands were quantified by densitometry.
Gene expression in the frozen liver samples was assessed by real-time RT-PCR using predesigned primers purchased from Life Technologies.
To analyze liver lipid composition, tissues were homogenized in PBS using the Gentle MACS dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). A total of 2.5 mg protein from each sample was used for lipid extraction. Lipids were extracted with a chloroform:methanol (2:1, v/v) solution and resuspended in 2% (v/v) Triton X-100. Total cholesterol, free cholesterol, triglyceride, and phospholipid contents were measured using colorimetric kits (Wako) according to the manufacturer's instructions.
In vitro assays
SVEC4/VCAM-1 cells were used to assess VCAM-1 expression as described previously (11). Briefly, cells were treated with indicated concentrations of Nef for 24 h. TNF-α was added in serum-free medium to the final concentration of 10 ng/ml to cells and incubated for 5 h, and luciferase activity was measured.
NO level released from SVEC4 endothelial cells were determined by measuring the accumulation of by-products (nitrate and nitrite) using a nitrate/nitrite colorimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA). Cells were treated with indicated concentrations of Nef for 24 h prior the assay.
Reactive oxygen species (ROS) production was measured in SVEC4 cells treated with Nef for 24 h. Cells were collected and incubated with 3 μM DHE for 30 min at 37°C. ROS levels were analyzed by flow cytometry.
Statistics
Data are shown as means ± sem. Statistical significance of the differences was assessed by t test or Mann-Whitney U test on ranks when data did not follow a normal distribution.
RESULTS
Effect of Nef on development of atherosclerosis in apoE−/− mice
ApoE−/− mice were fed an HFD for 12 wk and injected intraperitoneally 2×/wk with either recombinant Nef (40 ng/injection) or a vehicle. When aortas were analyzed en face after staining with Sudan IV, there was a trend for greater abundance of atherosclerosis in Nef-injected mice, but the difference did not reach statistical significance (Fig. 1A). When different regions of the aorta, aortic arch, thoracic aorta, and abdominal aorta were analyzed separately, again no statistically significant differences were found in the abundance of atherosclerosis between Nef and vehicle groups (not shown).
Figure 1.

Effect of Nef on development of atherosclerosis in apoE−/− mice. A) Percentage of atherosclerotic lesions in the total aorta. En face analysis, percentages calculated as an area stained with Sudan IV divided by total area. B) Quantitation of the area occupied by lesions in cross-sections of the aortic sinus region after staining with Oil Red O. *P < 0.03 vs. vehicle. C) Representative section of an aortic sinus from a control mouse stained with Oil Red O. D) Representative section of an aortic sinus from a mouse treated with Nef stained with Oil Red O. E) Analysis of the abundance of collagen within the aortic sinus region after staining with Masson's trichrome stain. F) Analysis of macrophage infiltration within the aortic sinus region after staining with anti-CD68, G) Analysis of T-cell infiltration within the aortic sinus region after staining with anti-CD3. H) Analysis of the abundance of VCAM-1 in sections from the aortic sinus region. I) Analysis of the abundance of the nitrotyrosine (NT) in the aortic sinus region. J) Analysis of the abundance of the eNOS in the aortic sinus region. *P < 0.01 vs. vehicle. K) Analysis of the abundance of ABCA1 within the aortic sinus region. **P < 0.03 vs. vehicle. Values represent positive staining as a percentage of total lesion area. L) Costaining of ABCA1 and CD68. *P < 0.05.
The detailed analysis of atherosclerotic plaques was undertaken on sections of the aortic sinus region. We analyzed the region with most pronounced atherosclerosis because this provided a better opportunity to discover differences in the morphology of the plaque; also, this region can be precisely mapped, ensuring that exactly the same parts of the vessel were compared. When cross-sections of the aortic sinus were analyzed after staining with Oil Red O, the cross-sectional area of lesions in the aortas of animals injected with Nef was 60% greater compared to the vehicle group (P<0.03; Fig. 1B–D). These findings indicate that although Nef had limited effect on the number of atherosclerotic plaques, it significantly increased their cross-sectional size. Alternatively, it may reflect a different effect of Nef on development of atherosclerosis in aortic sinus vs. other areas of the aorta. To assess the possible effect of Nef on stability of the plaque, we also analyzed plaque collagen content, but no statistically significant difference between the two groups was found (Fig. 1E).
We then analyzed elements of inflammation and oxidation in the sections of the aortic sinus from the two groups of animals. We found no difference between the groups in the area of the plaque occupied by macrophages (CD68-positive cells; Fig. 1F). Nef has been shown to reduce abundance of CD4-positive T cells (12), and these cells play an important role in development of atherosclerosis (13). Therefore, we also analyzed the abundance of T cells in plaques, but no difference was found between Nef and control groups (Fig. 1G). Further, no difference was found between the groups in the abundance of VCAM-1-positive areas (Fig. 1H). When abundance of a marker of protein oxidation and NO availability, nitrotyrosine, was assessed, again, no difference between the two groups was found (Fig. 1I). We also assessed the abundance of eNOS in the atherosclerotic plaques and found it to be reduced in the Nef group (Fig. 1J). It must be recognized that the abundance of markers of inflammation and oxidation was expressed as a percentage of positive staining over the plaque area; the absolute abundance of these markers was higher in bigger plaques.
Next, we assessed the abundance of ABCA1 in the plaques; ABCA1 was shown to be inhibited by Nef in vitro (4). Unexpectedly, the abundance of ABCA1 was elevated in atherosclerotic plaques of mice treated with Nef (Fig. 1K; P=0.0028). When we analyzed costaining of ABCA1 and CD68, both CD68-positive and CD68-negative cells were positive for ABCA1; however, almost 60% of ABCA1 in the plaque was colocalized with macrophages in control group (Fig. 1L). This proportion significantly increased in mice treated with Nef, indicating that macrophage ABCA1 is predominantly responsible for the effects of Nef on ABCA1 content in the vessel wall. Our attempts to detect Nef immunohistochemically in the sections of the vessel wall or liver were unsuccessful.
We also assessed the effect of Nef on the expansive remodeling of the vessel as described by Bentzon et al. (14). Vessel area and the length of internal elastic lamina (IEL) were measured in the aortic sinus and were both increased in the Nef treated group (Fig. 2A, B). There was a positive correlation between plaque area and IEL in Nef-treated (r2=0.5, P < 0.01), but not in control animals (Fig. 2C, D). Thus, Nef caused expansive remodeling of the vessels.
Figure 2.

Effect of Nef on vessel remodeling. A) Vessel area assessed in sections of the aortic sinus of two groups of mice. *P < 0.05. B) Length of IEL in aortic sinus sections from two groups of mice. *P < 0.05. C, D) Correlation between length of IEL and plaque area in control mice (C) and mice treated with Nef (D).
Dynamic changes in plasma lipids after initiation of HFD are shown in Fig. 3A–D. As expected, TC and TG content gradually rose in both groups after initiation of HFD; however, this dynamic was exaggerated in animals injected with Nef. After 12 wk, TC level in the plasma was 40% higher (Fig. 3A) and TG level was increased 2-fold (Fig. 3B) in mice that were treated with Nef as compared to the control group. Changes in TC were closely followed by changes in non-HDL cholesterol (HDL-C; Fig. 3C). In contrast, while the HDL-C levels in the plasma declined after initiation of HFD in both groups, HDL-C levels in the Nef-treated group were declining at a faster rate than in the control group, resulting in an almost 2-fold difference after 8 wk; HDL-C concentrations plateaued at a very low level in both groups after 12 wk on HFD (Fig. 3D). We then analyzed the distribution of apoA-I between HDL subfractions of different sizes after 12 wk on HFD. While we found no effect of Nef on relative abundance of large, presumably mature, HDL particles, the proportion of smaller HDL particles, presumably immature preβ-HDL, was significantly increased (Fig. 3E). This change is consistent with sharp elevation of the proportion of preβ-HDL in patients with Tangier disease (15) and with changes observed in rhesus macaques infected with SIV (4). One factor potentially responsible for a higher abundance of small HDL particles could be reduced activity of LCAT. However, no difference in plasma LCAT activity between the two groups of animals was found (Fig. 3F).
Figure 3.

Effect of Nef on plasma lipoproteins and cytokines in apoE−/− mice. A) TC. B) Triglycerides; C) non-HDL-C. D) HDL-C. E) Distribution of plasma HDL among subfractions of different sizes. F) Plasma LCAT activity. G) Analysis of distribution of TC among lipoprotein fractions after FPLC. H) Analysis of distribution of triglyceride among lipoprotein fractions after FPLC. Solid symbols, control; open symbols, Nef. I–M) IL-6 (I); IL-10 (J);IL-12p70 (K); MCP-1 (K); TNF-α (M). Values for 12 wk on HFD are shown, n = 7 for each point or group. *P < 0.05, **P < 0.02, ***P < 0.001 vs. vehicle.
Analysis of plasma lipoproteins with FPLC after 12 wk on HFD confirmed the increase of total cholesterol in the very low density lipoprotein (VLDL) fraction (Fig. 3G). There was also an increase in triglyceride (Fig. 3H) and cholesteryl esters content (not shown) in the same fractions, confirming that increased abundance of VLDL-TG is responsible for hypertriglyceridemia.
Changes in plasma inflammation markers are shown in Fig. 3I–M. Treatment with Nef resulted in elevation of plasma levels of IL-6 (Fig. 3I), IL-10 (Fig. 3J), and IL-12p70 (Fig. 3K). Level of MCP-1 was not affected by Nef (Fig. 3L), and there was a marginal reduction of TNF-α levels (Fig. 3M). Thus, treatment with Nef resulted in changes in plasma levels of selected pro- and anti-inflammatory cytokines; however these changes are consistent with only a moderate systemic inflammatory response.
Most HDL is produced by the liver and HDL production is predominantly regulated by the abundance of liver ABCA1 (16); deficiency of liver ABCA1 also increases HDL fractional catabolic rate (17). We therefore assessed the abundance of ABCA1 in the livers from mice that were treated with Nef or vehicle. As shown in Fig. 4A, B, the abundance of ABCA1 in the liver samples obtained from mice that were treated with Nef was reduced by ∼60% after 12 wk on HFD. Abundance of ABCA1 in livers after 8 wk on HFD was analyzed in a limited number of animals (n=3) and was also reduced in Nef treated mice (not shown). We also assessed the expression of genes related to cholesterol metabolism in the livers of the two groups of animals. We found no effect of Nef on the expression of Abca1, lcat, abcg1, srebf1, srebf2, apoa1, apob, ldlr, scarb1, hmgcr, and lipg in the liver (Fig. 4C). When lipid composition of liver tissue was analyzed, no effect of Nef infusion on total and free cholesterol (Fig. 4D, E), phospholipid (Fig. 4F) and triglyceride (Fig. 4G) content was found.
Figure 4.

Effect of Nef on expression of genes related to lipid metabolism in liver of apoE−/− mice. A) Western blot of ABCA1 abundance in the liver of 4 control and 4 Nef-treated mice. B) Quantitation of ABCA1 abundance in the liver of control and Nef-treated mice. *P < 0.05, n = 7/group. C) Expression of genes related to lipid metabolism in liver of apoE−/− mice. D–G) lipid composition of liver: TC (D), free cholesterol (E), phospholipid (F), and triglyceride (G).
Finally, while the body weight had increased after feeding mice with HFD, no difference was found in the body weight between mice injected with Nef and vehicle (not shown).
Effect of Nef on development of atherosclerosis in C57BL/6 mice
C57BL/6 mice were fed an HFHCD for 20 wk and injected intraperitoneally with either recombinant Nef (40 ng/injection) or vehicle 2×/wk. Mice are resistant to the development of atherosclerosis, and after en face examination, we found no signs of atherosclerosis in aortas from either group of animals (not shown). No atherosclerotic lesions were found in cross-sections of aortic sinus in either group (Fig. 5A, B). When we assessed the expansive remodeling of the vessel, no effect of Nef was found (not shown). However, detailed analysis of aorta sections stained with Oil Red O yielded an unusual observation: distinctive lipid inclusions were found in the vessels of animals treated with Nef, with very few found in the vehicle-treated animals (Fig. 5A–D). Surprisingly, most of these lipid inclusions were found in the adventitia, with very few inclusions in the media or intima (Fig. 5A–D). These inclusions were evident in both the aortic sinus (Fig. 5A, B) and thoracic aorta (Fig. 5C, D). Analysis of the distribution of lipids in the adventitia suggested that they were located both extracellularly and intracellularly (Fig. 5E). In some local areas of the adventitia with fewer lipid accumulations, it was possible to see that, indeed, some lipids were present intracellularly (Fig. 5F). The cells containing lipids showed a typical macrophage appearance; immunohistochemical analysis utilizing a specific macrophage marker, CD68, confirmed that these cells were macrophages (Fig. 5G). Quantitation of the abundance of the lipid inclusions showed that 10 times more lipid inclusions were present in aortic arch of Nef-treated animals compared to vehicle-treated animals (Fig. 5H). To ensure that this phenomenon was not specific for aortic arch only, we also analyzed sections of thoracic aorta and found 5 times more lipid-laden cells in the thoracic aorta of Nef-treated animals compared to vehicle-treated animals (Fig. 5I). When data for aortic arch and thoracic aorta were combined, 7 times more lipid-laden cells were found in aorta of Nef-treated animals (Fig. 5J). Finally, we repeated these experiments on a limited number of mice fed HFHCD for a shorter period of time, 12 wk. We found that accumulation of lipid-laden cells is a dynamic process: while these cells were present after 12 wk of feeding with HFHCD in Nef-treated, but not in vehicle-treated, animals, the abundance of lipid-laden cells was 75% less compared with animals that were treated with Nef and HFCD for 20 wk (Fig. 5K).
Figure 5.

Effect of Nef on lipid accumulation in aorta of C57BL/6 mice. A, B) Representative micrographs of sections of aortic sinus from mice treated with vehicle (A) or Nef (B) and stained with Oil Red O. C, D) Representative micrographs of sections of thoracic aorta from mice treated with vehicle (C) or Nef (D) and stained with Oil Red O. E) Representative micrograph, high magnification, of a section of aorta of Nef-treated mice after staining with Oil Red O. F) Representative micrograph, high magnification, of a section of aorta with low abundance of lipid staining of Nef-treated mice after staining with Oil Red O. Inset: detail of G) Representative micrograph, high magnification, of a section of aorta with low abundance of lipid staining of Nef-treated mice after staining with anti-CD68. Inset: detail of cell indicated by arrow in panel. Scale bars = 50 μm. H) Quantitation of the area occupied by lipid inclusions in aortic arch region after staining of sections with Oil Red O. *P < 0.01 vs. vehicle. I) Quantitation of the area occupied by lipid inclusions within the thoracic aorta after staining of sections with Oil Red O. *P < 0.001 vs. vehicle. J) Quantitation of the area occupied by lipid inclusions within the whole aorta after staining of sections with Oil Red O. *P < 0.01 vs. vehicle. K) Comparison of the area occupied by lipid inclusions within the whole aorta after 12 and 20 wk after staining of sections with Oil Red O. Circles, control; squares, Nef. *P < 0.05, ***P < 0.01.
When we analyzed elements of inflammation in aorta cross-sections from the two groups of animals, macrophages (CD68-positive cells) were predominantly found in adventitia, with very few found in other layers of the vessel wall (Fig. 6A). Quantitation of CD68-positive staining revealed no difference between the groups in the abundance of macrophages in the aortic arch (Fig. 6B) or thoracic aorta (Fig. 6C). We also found no difference in the abundance of VCAM-1-positive areas in these regions of the aorta (Fig. 6D, E).
Figure 6.

Effect of Nef on inflammation in aorta of C57BL/6 mice. A) Representative micrographs of sections of aortic arch from mice treated with vehicle or Nef and stained with anti-CD68. Arrows indicate CD68-positive cells. B) Analysis of macrophage infiltration of aorta arch region after staining with anti-CD68. C) Analysis of macrophage infiltration within the thoracic aorta after staining with anti-CD68. D) Analysis of the abundance of VCAM-1 in sections from the aortic arch region. E) Analysis of the abundance of VCAM-1 in sections from the thoracic aorta.
Dynamic changes in plasma lipids after initiation of HFHCD are shown in Fig. 7. As expected, TC content rose in both groups after initiation of HFHCD, and there was no statistically significant difference between the two groups (Fig. 7A). Plasma TG initially also rose in both groups of animals after initiation of HFHCD; however, in control mice TG levels began to subside after 2 wk, while TG remained consistently high in mice injected with Nef (Fig. 7B). As a result, after 20 wk the TG level in Nef-injected mice was 2.5-fold higher than in control mice. Changes in HDL-C followed the changes in TC (Fig. 7C), with no statistically significant difference observed between the groups in plasma HDL-C concentrations. Since in the apoE−/− model the changes in HDL-C were most likely related to changes in liver ABCA1 abundance, we investigated whether Nef had the same effect on liver ABCA1 in C57BL/6 mice fed the HFHCD. As shown in Fig. 7D, the abundance of ABCA1 in livers of Nef-treated animals was reduced similar to that in apoE−/− mice.
Figure 7.

Effect of Nef on plasma lipoproteins and body weight in C57BL/6 mice. A) TC. B) Triglycerides. C) HDL-C. D) Western blot of ABCA1 abundance in the liver of 2 control and 2 Nef-treated mice and quantitation of ABCA1 abundance in the liver of control and Nef-treated mice (n=9). E) Body weight. Solid symbols, control (n=9); open symbols, Nef (n=10). *P < 0.01, ***P < 0.001 vs. vehicle.
Finally, the body weight increased significantly during the 20 wk period in mice that were injected with Nef (Fig. 7E), while remaining unchanged in control mice.
Effects of Nef on inflammation in vitro
We have previously demonstrated that recombinant Nef inhibits cholesterol efflux in macrophages and hepatocytes in vitro (4). Here, we assessed the effects of Nef on inflammation and oxidation in in vitro assays.
Mouse endothelial cells stably transfected with the reporter gene under control of VCAM-1 promoter were used to assesses the effect of Nef on VCAM-1 expression. No statistically significant effect was found across a wide range of Nef concentrations both with and without activation of cells with TNF-α (Fig. 8A). When ROS production in the same cell line was assessed, Nef had no effect at the concentrations used in this study; however, higher concentrations of Nef statistically significantly increased ROS production (Fig. 8B). When NO production was measured in the same cells, it was significantly reduced by Nef (Fig. 8C), a finding consistent with that of Duffy et al. (18).
Figure 8.
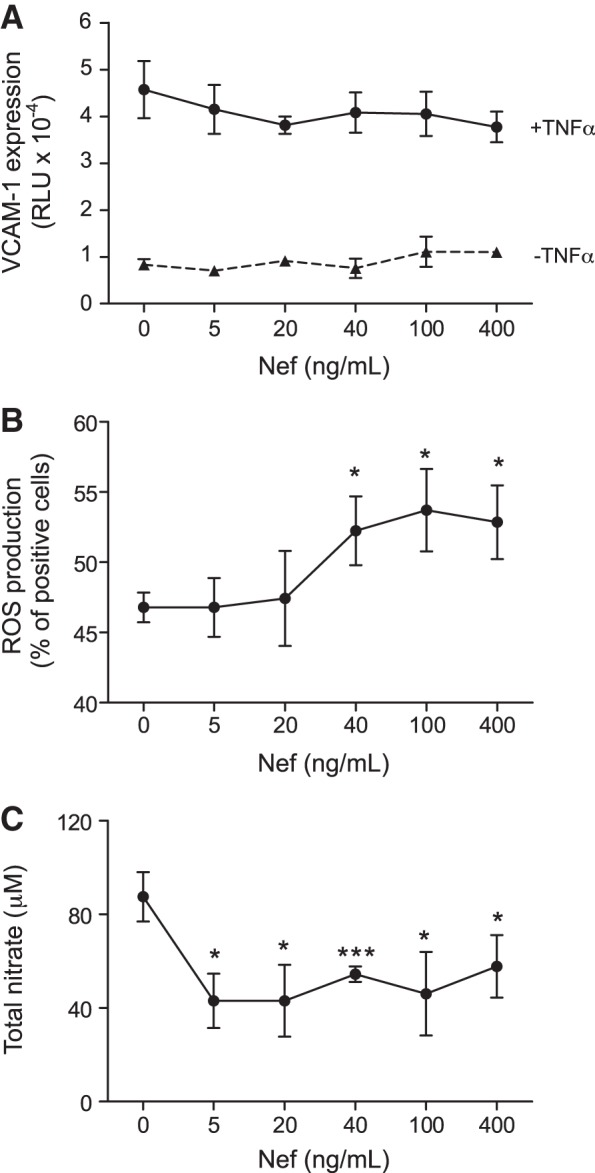
Effect of Nef on elements of inflammation in vitro. A) Effect of Nef on VCAM-1 expression in SVEC4/VCAM-1 cells with or without activation with TNF-α. B) Effect of Nef on ROS production in SVEC4 cells. C) Effect of Nef on nitrate production in SVEC4 cells. *P < 0.01, ***P < 0.001 vs. vehicle.
DISCUSSION
There is compelling evidence that HIV infection is a strong risk factor for the development of atherosclerosis (19), but the mechanisms connecting these two diseases remain unclear and are most likely multifactorial. Systemically, HIV is associated with chronic inflammation, which is proatherogenic, and immunodeficiency, which may be pro- or antiatherogenic (20, 21). HIV infection also causes dyslipidemia characterized by hypo-β-lipoproteinemia and hypo-α-cholesterolemia, which are antiatherogenic and proatherogenic, respectively (4, 22). Locally, HIV infection inhibits cholesterol efflux in macrophages promoting formation of foam cells, a hallmark of atherosclerosis (3). Notably, HIV infection affects not only infected cells. Viral proteins released from infected cells can modify functions of uninfected bystander cells. In particular, Nef, presumably released from infected cells, may mimic the effect of the virus on cholesterol efflux locally, and on HDL metabolism systemically (4). However, studies in animal models of HIV infection make it difficult to separate contribution of Nef from the effects of other virus- and disease-related factors. A study of mice expressing transgenic Nef in cells of monocytic lineage showed significant cardiac dysfunction accompanied by cardiomyocyte apoptosis and vascular dysfunction of coronary arteries (23), but did not investigate atherogenesis. Here, we studied the effect of Nef on development of atherosclerosis by injecting recombinant Nef protein into two mouse models of this disease.
The main finding of this study is that in apoE−/− mice fed HFD, recombinant Nef increased accumulation of cholesterol in macrophages and caused formation of foam cells, while having only a limited effect on other aspects of atherosclerosis, such as local inflammation (with the exception of the effect on eNOS), oxidation, and accumulation of extracellular matrix. It must be recognized, however, that atherosclerotic plaques in apoE−/− mice are characterized by substantial inflammation and oxidation and, unless the proinflammatory effects of the treatment are very strong, their manifestation may not be apparent on such a background. In a genetic model, Nef had a limited effect on the number of plaques, as assessed by en face analysis, but enhanced the severity of atherosclerosis, as determined through a cross-sectional size of the plaques; it also caused expansive remodeling of the vessels. In a dietary model of atherosclerosis, Nef did not cause formation of plaques, instead causing the appearance of foam cells in adventitia of the vessels. Both findings are consistent with the suggestion that Nef has a limited effect on the early events of atherosclerosis, but may contribute to the later stages of plaque development and to the risk of aneurysm formation. Combination of the effects of Nef with specific and/or nonspecific inflammatory effects of the infection may result in atherosclerosis.
An intriguing finding of this study is the presence of foam cells in the adventitia layer in the Nef-treated group in the dietary model of atherosclerosis. Morphological appearance of these cells and the immunohistochemical analysis indicated that they most likely were macrophages. Several studies have indicated that adventitia may be involved in pathogenesis of atherosclerosis. First, accumulation of various subsets of lymphocytes along with dendritic cells and macrophages were observed in adventitia in regions adjacent to the atherosclerotic plaque (for review, see ref. 24); these cells play an important role in remodeling the atherosclerotic vessel (25). Second, areas of vessels predisposed to atherosclerosis often also accumulate macrophages in adventitia (26) indicating that some macrophages in the intima may have entered through vasa vasorum. However, we did not find any difference in abundance of lipid-laden macrophages in adventitia of two regions of aorta with different predisposition to atherosclerosis. Third, extraluminal atherosclerosis is a consistent feature of the venous bypass grafts (27). We speculate that in the absence of inflammatory stimuli, monocytes enter vessel wall via vasa vasorum, but do not continue migration to the intima, as Nef causes accumulation of cholesterol in these cells while still in adventitia, reducing their mobility.
In apoE−/− mice, treatment with Nef caused reduction in HDL-C levels, a finding consistent with our previous report in HIV-infected humanized mice (28). The most likely mechanism is the effect of Nef on liver ABCA1, the main contributor to the regulation of plasma HDL-C levels (16, 17). Hypo-α-lipoproteinemia, a consistent feature of HIV disease (22, 29), is likely to have a similar mechanism. This effect was not seen in C57BL/6 mice despite reduced levels of ABCA1 in the livers of Nef-treated animals. The reasons why reduction of liver ABCA1 on the background of HFHC diet did not cause hypo-α-lipoproteinemia in this model are not clear, but most likely reflect differences in HDL metabolism between the two strains, which are currently not fully understood. In both models, Nef caused a significant elevation of plasma triglyceride levels. Hypertriglyceridemia is also a common feature of HIV disease (22, 29). The exact mechanism of how Nef may cause hypertriglyceridemia is yet to be elucidated; however, it is most likely also related to the deficiency of liver ABCA1. ABCA1-knockout mice are characterized by hypertriglyceridemia due to enhanced secretion of VLDL and decreased triglyceride clearance and plasma lipolytic activity (30). Consistent with this suggestion, we observed increased concentrations of cholesterol and triglycerides in VLDL fractions of apoE−/− mice treated with Nef. Another possible contributor is the effect of Nef on ABCA1 in adipose tissue triggering its dysfunction (31), a suggestion supported by significant weight gain in Nef-injected mice in a dietary model of atherosclerosis. Nef also caused modest systemic inflammation in genetic model of atherosclerosis. Combination of hypo-α-lipoproteinemia, hypertriglyceridemia, systemic inflammation, and obesity is a feature of metabolic syndrome common in patients with HIV, suggesting that Nef may contribute to this comorbidity of HIV disease.
An unexpected finding was that while Nef caused a reduction of ABCA1 abundance in the liver, it increased the abundance of ABCA1 in the plaque macrophages. In vitro Nef reduced the abundance of ABCA1 in cells infected with HIV (3) or treated with Nef (4). Two explanations of this finding are possible. One is that most of heterologous Nef accumulates in the liver, and this is where its concentration reaches an effective level. Another explanation is that in vivo accumulation of cholesterol in macrophages over prolonged periods of time may lead to a compensatory elevation of ABCA1. The effects of rising cholesterol level may override the effects of Nef in macrophages; liver cells having much more effective pathways controlling cholesterol homeostasis did not accumulate cholesterol. This finding makes it more likely that development of atherosclerosis is caused by the effect of Nef on plasma lipids rather than on the cells in the vessel wall, at least at the late stages of plaque development. On the other hand, we have recently demonstrated that Nef may affect ABCA1 localization and functionality independently of its abundance (32). Finally, Nef may have effects on atherosclerosis and vessel remodeling that are independent of ABCA1.
If confirmed, our findings indicate an intriguing possibility. Within a limitation of being derived from animal models and at conditions relevant to only HIV-induced atherosclerosis, it appears that formation of foam cells does not necessarily depend on local inflammation on the one hand, and does not necessarily cause it on the other. This is consistent with recent findings of Klingenberg et al. (33) demonstrating a dissociation between increases in the size of atherosclerotic plaques and level of inflammation in the plaques. While being speculative, a hypothesis of disconnection between inflammation and accumulation of cholesterol in the atherosclerotic plaque may be a consideration for mechanistic understanding of initiation and progression of atherosclerosis.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council of Australia (GNT1019847, GNT1036352) and the U.S. National Institutes of Health (NIH; HL093818), and in part by the Victorian government's OIS program and the District of Columbia Developmental Center for AIDS Research (DC D-CFAR), an NIH-funded program (1P30AI087714). H.L.C. was supported by The May Stewart Bursary and The Elizabeth Mary Sweet Scholarship (Faculty of Medicine, Dentistry, and Health Sciences, The University of Melbourne). D.S. is a fellow of the National Health and Medical Research Council of Australia.
The authors declare no conflicts of interest.
Footnotes
- ABCA1
- ATP binding cassette transporter A1
- FPLC
- fast protein liquid chromatography
- HDL
- high-density lipoprotein
- HDL-C
- high-density lipoprotein cholesterol
- HFD
- high-fat diet
- HFHCD
- high-fat/high-cholesterol diet
- IEL
- internal elastic lamina
- LCAT
- lecithin cholesterol acyltransferase
- ROS
- reactive oxygen species
- TC
- total cholesterol
- TG
- triglyceride
- VLDL
- very low density lipoprotein
REFERENCES
- 1. Sudano I., Spieker L. E., Noll G., Corti R., Weber R., Luscher T. F. (2006) Cardiovascular disease in HIV infection. Am. Heart J. 151, 1147–1155 [DOI] [PubMed] [Google Scholar]
- 2. Glesby M. J. (2005) Coronary heart disease in HIV-infected patients. Curr. HIV/AIDS Rep. 2, 68–73 [DOI] [PubMed] [Google Scholar]
- 3. Mujawar Z., Rose H., Morrow M. P., Pushkarsky T., Dubrovsky L., Mukhamedova N., Fu Y., Dart A., Orenstein J. M., Bobryshev Y. V., Bukrinsky M., Sviridov D. (2006) Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol. 4, e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asztalos B. F., Mujawar Z., Morrow M. P., Grant A., Pushkarsky T., Wanke C., Shannon R., Geyer M., Kirchhoff F., Sviridov D., Fitzgerald M. L., Bukrinsky M., Mansfield K. G. (2010) Circulating Nef induces dyslipidemia in simian immunodeficiency virus-infected macaques by suppressing cholesterol efflux. J. Infect. Dis. 202, 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Getz G. S., Reardon C. A. (2012) Animal models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breuer S., Gerlach H., Kolaric B., Urbanke C., Opitz N., Geyer M. (2006) Biochemical indication for myristoylation-dependent conformational changes in HIV-1 Nef. Biochemistry 45, 2339–2349 [DOI] [PubMed] [Google Scholar]
- 7. Fujii Y., Otake K., Tashiro M., Adachi A. (1996) Soluble Nef antigen of HIV-1 is cytotoxic for human CD4+ T cells. FEBS Lett. 393, 93–96 [DOI] [PubMed] [Google Scholar]
- 8. Huang M. B., Jin L. L., James C. O., Khan M., Powell M. D., Bond V. C. (2004) Characterization of Nef-CXCR4 interactions important for apoptosis induction. J. Virol. 78, 11084–11096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warnick G. R., Benderson J., Albers J. J. (1982) Dextran sulfate-Mg2+ precipitation procedure for quantitation of high-density-lipoprotein cholesterol. Clin. Chem. 28, 1379–1388 [PubMed] [Google Scholar]
- 10. Ditiatkovski M., D'Souza W., Kesani R., Chin-Dusting J., de Haan J. B., Remaley A., Sviridov D. (2013) An apolipoprotein A-I mimetic peptide designed with a reductionist approach stimulates reverse cholesterol transport and reduces atherosclerosis in mice. PLoS ONE 8, e68802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. D'Souza W., Stonik J. A., Murphy A., Demosky S. J., Sethi A. A., Moore X. L., Chin-Dusting J., Remaley A. T., Sviridov D. (2010) Structure/function relationships of apolipoprotein A-I mimetic peptides: implications for antiatherogenic activities of high-density lipoprotein. Circ. Res. 107, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hanna Z., Priceputu E., Kay D. G., Poudrier J., Chrobak P., Jolicoeur P. (2004) In vivo mutational analysis of the N-terminal region of HIV-1 Nef reveals critical motifs for the development of an AIDS-like disease in CD4C/HIV transgenic mice. Virology 327, 273–286 [DOI] [PubMed] [Google Scholar]
- 13. Quehenberger O. (2005) Thematic review series: the immune system and atherogenesis. molecular mechanisms regulating monocyte recruitment in atherosclerosis. J. Lipid Res. 46, 1582–1590 [DOI] [PubMed] [Google Scholar]
- 14. Bentzon J. F., Pasterkamp G., Falk E. (2003) Expansive remodeling is a response of the plaque-related vessel wall in aortic roots of apoE-deficient mice: an experiment of nature. Arterioscler. Thromb. Vasc. Biol. 23, 257–262 [DOI] [PubMed] [Google Scholar]
- 15. Asztalos B. F., Schaefer E. J. (2003) High-density lipoprotein subpopulations in pathologic conditions. Am. J. Cardiol. 91, 12E–17E [DOI] [PubMed] [Google Scholar]
- 16. Basso F., Freeman L., Knapper C. L., Remaley A., Stonik J., Neufeld E. B., Tansey T., Amar M. J. A., Fruchart-Najib J., Duverger N., Santamarina-Fojo S., Brewer H. B., Jr. (2003) Role of the hepatic ABCA1 transporter in modulating intrahepatic cholesterol and plasma HDL cholesterol concentrations. J. Lipid Res. 44, 296–302 [DOI] [PubMed] [Google Scholar]
- 17. Singaraja R. R., Stahmer B., Brundert M., Merkel M., Heeren J., Bissada N., Kang M., Timmins J. M., Ramakrishnan R., Parks J. S., Hayden M. R., Rinninger F. (2006) Hepatic ATP-binding cassette transporter A1 is a key molecule in high-density lipoprotein cholesteryl ester metabolism in mice. Arterioscler. Thromb. Vasc. Biol. 26, 1821–1827 [DOI] [PubMed] [Google Scholar]
- 18. Duffy P., Wang X., Lin P. H., Yao Q., Chen C. (2009) HIV Nef protein causes endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J. Surg. Res. 156, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. El-Sadr W. M., Lundgren J. D., Neaton J. D., Gordin F., Abrams D., Arduino R. C., Babiker A., Burman W., Clumeck N., Cohen C. J., Cohn D., Cooper D., Darbyshire J., Emery S., Fatkenheuer G., Gazzard B., Grund B., Hoy J., Klingman K., Losso M., Markowitz N., Neuhaus J., Phillips A., Rappoport C. (2006) CD4+ count-guided interruption of antiretroviral treatment. N. Engl. J. Med. 355, 2283–2296 [DOI] [PubMed] [Google Scholar]
- 20. Libby P., Ridker P. M., Maseri A. (2002) Inflammation and atherosclerosis. Circulation 105, 1135–1143 [DOI] [PubMed] [Google Scholar]
- 21. Libby P., Ridker P. M., Hansson G. K., and the Leducq Transatlantic Network on Atherothrombosis (2009) Inflammation in atherosclerosis: from pathophysiology to practice. J. Am. Coll. Cardiol. 54, 2129–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rose H., Hoy J., Woolley I., Tchoua U., Bukrinsky M., Dart A., Sviridov D. (2008) HIV infection and high density lipoprotein metabolism. Atherosclerosis 199, 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kay D. G., Yue P., Hanna Z., Jothy S., Tremblay E., Jolicoeur P. (2002) Cardiac disease in transgenic mice expressing human immunodeficiency virus-1 nef in cells of the immune system. Am. J. Pathol. 161, 321–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galkina E., Ley K. (2009) Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 27, 165–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamashita A., Shoji K., Tsuruda T., Furukoji E., Takahashi M., Nishihira K., Tamura S., Asada Y. (2008) Medial and adventitial macrophages are associated with expansive atherosclerotic remodeling in rabbit femoral artery. Histol. Histopathol. 23, 127–136 [DOI] [PubMed] [Google Scholar]
- 26. Jongstra-Bilen J., Haidari M., Zhu S. N., Chen M., Guha D., Cybulsky M. I. (2006) Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med. 203, 2073–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pourabdollah M., Hasssantash S. A., Bikdeli B., Sadeghian M., Afshar H., Kalantarian S., Sabeti S., Ahmadi H., Marzban M., Mohammadi F. (2013) Extraluminal atherosclerosis: an under-recognised finding in human aortocoronary venous bypass grafts. Heart Lung Circ. 22, 19–24 [DOI] [PubMed] [Google Scholar]
- 28. Dubrovsky L., Duyne R. V., Senina S., Guendel I., Pushkarsky T., Sviridov D., Kashanchi F., Bukrinsky M. (2012) Liver X receptor agonist inhibits HIV-1 replication and prevents HIV-induced reduction of plasma HDL in humanized mouse model of HIV infection. Biochem. Biophys. Res. Commun. 419, 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rose H., Woolley I., Hoy J., Dart A., Bryant B., Mijch A., Sviridov D. (2006) HIV infection and high-density lipoprotein: the effect of the disease vs the effect of treatment. Metabolism 55, 90–95 [DOI] [PubMed] [Google Scholar]
- 30. Chung S., Timmins J. M., Duong M., Degirolamo C., Rong S., Sawyer J. K., Singaraja R. R., Hayden M. R., Maeda N., Rudel L. L., Shelness G. S., Parks J. S. (2010) Targeted deletion of hepatocyte ABCA1 leads to very low density lipoprotein triglyceride overproduction and low density lipoprotein hypercatabolism. J. Biol. Chem. 285, 12197–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu B. L., Zhao S. P., Hu J. R. (2010) Cholesterol imbalance in adipocytes: a possible mechanism of adipocytes dysfunction in obesity. Obes. Rev. 11, 560–567 [DOI] [PubMed] [Google Scholar]
- 32. Cui H. L., Grant A., Mukhamedova N., Pushkarsky T., Jennelle L., Dubrovsky L., Gaus K., Fitzgerald M. L., Sviridov D., Bukrinsky M. (2012) HIV-1 Nef mobilizes lipid rafts in macrophages through a pathway that competes with ABCA1-dependent cholesterol efflux. J. Lipid Res. 53, 696–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klingenberg R., Gerdes N., Badeau R. M., Gistera A., Strodthoff D., Ketelhuth D. F., Lundberg A. M., Rudling M., Nilsson S. K., Olivecrona G., Zoller S., Lohmann C., Luscher T. F., Jauhiainen M., Sparwasser T., Hansson G. K. (2013) Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Invest. 123, 1323–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]