

Abstract
Intermediate filaments (IFs) form a dense and dynamic network that is functionally associated with microtubules and actin filaments. We used the GFP-tagged vimentin mutant Y117L to study vimentin-cytoskeletal interactions and transport of vimentin filament precursors. This mutant preserves vimentin interaction with other components of the cytoskeleton, but its assembly is blocked at the unit-length filament (ULF) stage. ULFs are easy to track, and they allow a reliable and quantifiable analysis of movement. Our results show that in cultured human vimentin-negative SW13 cells, 2% of vimentin-ULFs move along microtubules bidirectionally, while the majority are stationary and tightly associated with actin filaments. Rapid motor-dependent transport of ULFs along microtubules is enhanced ≥5-fold by depolymerization of actin cytoskeleton with latrunculin B. The microtubule-dependent transport of vimentin ULFs is further regulated by Rho-kinase (ROCK) and p21-activated kinase (PAK): ROCK inhibits ULF transport, while PAK stimulates it. Both kinases act on microtubule transport independently of their effects on actin cytoskeleton. Our study demonstrates the importance of the actin cytoskeleton to restrict IF transport and reveals a new role for PAK and ROCK in the regulation of IF precursor transport.—Robert, A., Herrmann, H., Davidson, M. W., and Gelfand, V. I. Microtubule-dependent transport of vimentin filament precursors is regulated by actin and by the concerted action of Rho- and p21-activated kinases.
Keywords: cytoskeleton, particle tracking, motor proteins
There is increasing evidence demonstrating that intermediate filaments (IFs) form a dynamic network contributing to many cellular processes in concert with microtubules and actin filaments (1–3). Vimentin, a type III IF protein, is the major IF protein expressed in mesenchymal cells. In the past 2 decades, the importance of vimentin expression for physiological processes like wound healing, metastasis, and angiogenesis has been documented (4–6). Indeed, vimentin contributes to a panoply of cellular processes, such as organelle positioning, cell migration, focal adhesion assembly, and stress response (7–11). Many of these functions reside in the motile properties of the vimentin IF (VIF) network and are likely caused by VIF interactions with microtubules and actin. It is known that microtubules and actin filaments affect the distribution of VIFs (12, 13). Microtubule depolymerization results in the rapid reorganization of the majority of VIFs from a well-extended network to a tight cap in the perinuclear region (14). The actin cytoskeleton is required for the reorganization of vimentin after microtubule depolymerization (13). However, the molecular connections between VIF and the two other cytoskeletal systems remain elusive.
Bidirectional movement of both mature filaments and their nonfilamentous precursors (vimentin particles) has been attributed to the microtubule motors, kinesin and dynein (15–17). Detailed live-cell imaging of VIF precursor transport along microtubules has never been performed. This is essential in order to gain insights into the regulation of IF transport within cells. Typically, the studies of IF transport have used changes in filament distribution as a readout of filament transport. This type of measurement could, however, be seriously misleading, since distribution of filaments may be determined by their binding to other cellular components rather than by their active transport. The difficulty in live-cell imaging of IF transport is attributable to the presence of dense arrays of complex fully polymerized IF networks radiating from the nucleus to the cell surface (18, 19), making it extremely difficult to follow the transport of individual IF elements. To overcome this problem, we have taken advantage of the tyrosine 117 to leucine vimentin mutant, i.e., Y117L-vimentin. In vitro assembly assays have shown that this mutant does laterally associate into full-width filaments but fails to longitudinally anneal and thus fails to form elongated VIFs (20). The expression of this mutant in vimentin-deficient cells leads to the formation of uniform oligomers, and this property facilitates the quantitative analysis of motility. Utilizing this system, we directly tested the roles of microtubules and actin microfilaments, respectively, in VIF precursor transport in live cells.
It is well established that IFs are major phosphoproteins. Vimentin is a target for several kinases, for instance PI3K, Rho-kinase (ROCK), p21-activated kinase (PAK), PKC, PKA, and CaMK (21–23). Hence, several reports indicate a role for vimentin phosphorylation in the regulation of the assembly state and the organization of VIF (24–28). However, the kinases responsible for the regulation of IF transport have never been investigated. In this study, we used live cell imaging to track the transport of vimentin unit-length filaments (ULFs) along microtubules in order to understand how the interaction of IFs with the actin cytoskeleton and phosphorylation by ROCK and PAK affect vimentin transport. We found that the two GTPase-regulated kinases ROCK and PAK have opposite effects on the regulation of ULF transport independent from the effect of these kinases on the actin cytoskeleton.
MATERIALS AND METHODS
DNA constructs, cell culture, transfection, and stable cell lines
The generation of the Y117L-vimentin mutant cDNA has been described previously (29). Using appropriate PCR primers, a cDNA was generated to be cloned with BspEI/HindIII into the pEGFP-C1 vector. To construct the mTagRFP-T-EB3-7 plasmid, mTagRFP-T cDNA was amplified with a 5′ primer encoding an AgeI site and a 3′ primer encoding a NotI site, gel purified, and digested with the appropriate restriction enzymes for insertion into an EGFP-N1 (Clontech-type) vector, yielding mTagRFP-T N1. The human microtubule-associated protein EB3 (NM_012326.2; a gift from Lynne Cassimeris, Lehigh University, Bethlehem, PA, USA), was amplified with a 5′ primer encoding a BglII site and a 3′ primer encoding a BamHI site, gel purified, and digested with the appropriate restriction enzymes. The resulting PCR product was ligated with a similarly digested mEmerald N1 cloning vector, forming mEmerald-EB3-N-7 with a 7-aa linker separating the proteins. The EB3 fusion was first created using an advanced EGFP variant (mEmerald) with several mutations designed to enhance brightness and folding efficiency. This acted as a pilot vector, to verify proper localization and expression of the construct. The resulting plasmid, along with mTagRFP-T N1 cloning vector were double digested with BamHI and NotI, gel purified, and ligated to form mTagRFP-T-EB3-7. The DNA used for transfection was prepared and purified using the Plasmid Maxi kit (Qiagen, Valencia, CA, USA), according to manufacturer's instructions. Proper localization of the mTagRFP-T EB3 was characterized by transfection in HeLa cells (CCL2 line; American Type Culture Collection, Manassas, VA, USA) using Effectene (Qiagen) and ∼1-μg vector.
SW13 vim+ and SW13 vim− cells were derived from human adenocarcinoma cells (30) and were maintained in Leibovitz's L-15 medium supplemented with 10% fetal bovine serum. The GFP-ULF SW13 stable cell line was created by the cotransfection of SW13 vim− cells with vimentin Y117L pEGFP-C1 and pcDNA3 (carrier of neomycin gene) plasmids using Polyfect transfection reagent (Qiagen), according to the manufacturer's instructions. Positive cells were selected using 0.8 mg/ml G418 for 3 wk, and isolated clones were tested for GFP-ULF expression. Transient transfection of SW13 vim− cells with TagRFP-EB3 was performed using Xfect transfection reagent (Clontech Laboratories, Mountain View, CA, USA), according to the manufacturer's recommendations.
Reagents
The following drugs were used as described in figure legends: Cytochalasin D (MP Biomedicals, Santa Ana, CA, USA), latrunculin B (Lat B; Biomol Research Laboratories, Plymouth Meeting, PA, USA), nocodazole and vinblastine (Sigma, St. Louis, MO, USA), sodium azide (Fisher Scientific, Pittsburgh, PA, USA), Y-27632 and IPA-3 (Tocris Bioscience, Minneapolis, MN, USA), Lysotracker Red dye and rhodamine-conjugated phalloidin (Molecular Probes/Life Technologies, Eugene, OR, USA). Ciliobrevin was kindly gifted by Dr. James K. Chen (Stanford University School of Medicine, Stanford, CA, USA).
Immunostaining and microscopy
For actin staining, cells were fixed for 10 min at room temperature with 3.7% formaldehyde in cytoskeleton (CSK) buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, and 10 mM PIPES, pH 6.8) supplemented with 0.1% Triton X-100. Filamentous actin (F-actin) was labeled with rhodamine-phalloidin (1:5000 in PBS; Molecular Probes) for 15 min at room temperature. For microtubule labeling, cells were fixed with ice-cold methanol for 5 min at −20°C. Immunolabeling of α-tubulin was performed as described previously (31) by using DM1A monoclonal antibody (1:1000; Sigma). Images of fixed cells were captured on a Nikon Eclipse U2000 inverted microscope (Nikon Instruments, Melville, NY, USA) equipped with Plan-Apo ×100 1.4 NA and Plan-Apo TIRF ×60 1.45 NA objectives and a CoolSnap ES CCD camera (Roper Scientific, Planegg/Martinsried, Germany), controlled by MetaMorph 7.7.5.0 software (Molecular Devices, Downingtown, PA, USA). Fluorescence excitation was achieved using a mercury lamp. To image microtubule-dependent transport of ULFs, cells were treated with 5 μM Lat B for 30 min before imaging. For live cell imaging, we used a Nikon Eclipse U2000 inverted stand equipped with a Yokogawa CSU10 spinning disc confocal head (Yokogawa Electric Corporation, Sugar Land, TX, USA), a humidified 5% CO2/thermoregulated chamber and a ×100 1.45 NA lens. Images were acquired using Evolve EMCCD (Photometrics, Tucson, AZ, USA) driven by Nikon Elements software. Time-lapse images were collected 1×/s for 1 min.
Particle tracking and temporal color coding
Time-lapse sequences of ULF motility were analyzed using DiaTrack 3.01 software (SemaSopht, North Epping, NSW, Australia). Each condition of a single experiment consisted of between 1500 and 5000 ULFs from ≥10 cells. ULFs were tracked over 60 frames, and ULF trajectories that persisted for ≥10 frames in a single direction longer than 1 μm were isolated. Trajectories of single ULFs were summed to give the distance traveled per ULF during the 60-s imaging period. The frequency distribution of all trajectories per condition was determined using Prism 5 software (GraphPad Software, La Jolla, CA, USA) and was divided by the total number of ULFs analyzed per condition. These values were averaged across ≥3 independent experiments, and standard deviations were found. Multicolor merge images of time-lapse sequences were made using the Temporal-Color Code plug-in in Fiji image analysis software (http://fiji.sc). Briefly, the 60 frames were color coded and merged for a qualitative representation of ULF trajectories. Rainbows correspond to motile ULFs, and white dots correspond to stationary ULFs.
PAK small interfering RNA (siRNA) transfection
The following siRNAs against human PAK1 and PAK2 were purchased from Qiagen; there were 2 sequences for each isoform: Hs_PAK1_8: 5′-TCCACTGATTGCTGCAGCTAA-3′; Hs_PAK1_9: 5′-TTGAAGAGAACTGCAACTGAA-3′; Hs_PAK2_6: 5′-AAGAAAAGGAACGGCCAGAAA-3′; and Hs_PAK2_9: 5′-CCGGATCATACGAAATCAATT-3′. GFP-ULF SW13 cells were grown to 60% confluence and transfected by the calcium-phosphate method for 16 h on 2 consecutive days with either 10 nM of each siRNA to the same PAK isoform (20 nM total siRNA) or with 10 nM of each purchased siRNA (targeting both isoforms, 40 nM total siRNA). Transfected cells were split on coverslips 24 h after the second transfection. At 72 h after the second transfection with siRNAs, live-cell imaging was performed to look at ULF motility, some cells were fixed and stained/labeled for actin or microtubules, and cell lysates were prepared for Western blot analysis. Knockdown efficiency of PAK1 and PAK2 was evaluated by Western blot analysis using PAK1 polyclonal antibody and PAK2 (C17A10) monoclonal antibody from Cell Signaling Technology (Danvers, MA, USA). Vimentin polyclonal antibody (Covance, Princeton, NJ, USA) was used as a loading control.
RESULTS
Y117L-vimentin forms motile ULFs in vimentin-negative cells
To study VIF precursor transport, we have used the SW13 adenocarcinoma cell line lacking cytoplasmic IFs to stably express a GFP-tagged version of Y117L-vimentin. Coexpression of GFP-tagged Y117L-vimentin and wild-type vimentin results in incorporation of the mutant protein into a morphologically normal IF network (Fig. 1A). However, when the mutant protein is expressed in vimentin-deficient cells, it forms uniform dot-like structures, most likely corresponding to individual ULFs, as reported previously (Fig. 1B and ref 20). To study the dynamics of ULF interaction with other cytoskeletal components, we performed live imaging of the vimentin-null SW13 cells stably expressing GFP-tagged vimentin ULFs. We found that the majority of particles remain mostly stationary, but ∼2% of them are transported along linear tracks traveling over 6 μm during 1 min of imaging (Fig. 1C, D and Supplemental Movie S1).
Figure 1.

Movement of vimentin ULFs in SW13 cells. A, B) GFP-tagged Y117L mutant vimentin was expressed in SW13 vim+ cells containing endogenous vimentin (A) or in vimentin-deficient SW13 vim− (B). C) Temporal color-code representation of GFP-ULF movement in a cell shown in B. Images were captured every second for 1 min, then color coded and merged for a qualitative representation of ULF trajectories. Time color-code bar is at left. Rainbows correspond to motile ULFs, and white dots correspond to stationary ULFs. Enlargement of the boxed area (at right) shows an example of a linear trajectory (rainbow picture) and the corresponding image sequence. See also Supplemental Movie S1. Scale bar = 10 μm. D) Trajectories of ULFs were analyzed using Diatrack software in ≥10 cells/experiment. Graph shows the mean frequency distribution of trajectory length for trajectories that are ≥3 μm long.
Vimentin-ULFs interact with the two other cytoskeletal systems
The linear tracks followed by the motile ULFs suggested that they are transported by a microtubule-dependent mechanism. To test this, we visualized microtubules in GFP-ULF-expressing cells by transfecting them with a construct encoding a microtubule-binding protein EB3 tagged with a red fluorescent protein mTagRFP-t (TagRFP-EB3). EB3 normally accumulates at the tips of growing microtubules. However, we found that at higher levels of expression, EB3 decorates microtubules along their whole length. Unlike other methods of microtubule network visualization that we have tried, TagRFP-EB3 labeling did not interfere with the distribution of VIFs or ULFs and did not cause any changes in the morphology of the microtubule network. Coimaging of GFP-ULF and TagRFP-EB3 revealed that around 40% of ULFs were found in proximity to microtubules (Fig. 2A, left panel). Moreover, analysis of time-lapse image sequences demonstrated that ULFs can rapidly move along microtubule (see enlargement of image sequence, Fig. 2A, and Supplemental Movie S2) suggesting that microtubules participate in ULF transport.
Figure 2.

ULFs interact with microtubules and F-actin. A) GFP-ULF-expressing cells were transfected with TagRFP-EB3 that decorates microtubules. Microtubule network images were captured at the beginning and end of the time-lapse sequence of GFP-ULF. Cells were treated with vinblastine (5 nM) to minimize microtubule dynamics during the imaging period, and the image sequence of GFP-ULF was superimposed with the static microtubule image. Left panel: first frame of the image sequence, showing alignment of GFP-ULF with microtubules. Right panel: enlargement of the boxed area shows movement of ULFs along a microtubule (white arrows). See also Supplemental Movie S2. B) GFP-ULF interaction with actin. GFP-ULF-expressing cells were fixed after the following treatment: control (untreated), nocodazole (10 μM for 1 h), and Lat B (5 μM for 1 h). F-actin was stained using rhodamine-phalloidin. Enlargements show colocalization of ULF with actin cables in untreated and nocodazole-treated cells, or with residual actin aggregates into Lat B-treated cells. Scale bars = 10 μm.
To evaluate whether ULFs interact with the actin cytoskeleton, GFP-ULF-expressing cells were fixed and stained with rhodamine-phalloidin. This revealed a striking alignment of ≥75% of ULFs along actin cables (Fig. 2B, top panels). This association is enhanced to 90% of ULFs after depolymerization of microtubules with nocodazole (Fig. 2B, middle panels). The affinity of ULFs to F-actin is likely very high, because even after depolymerization of the majority of actin filaments with Lat B, a fraction of ULFs stays tightly associated with residual phalloidin-positive actin aggregates (Fig. 2B, bottom panels). Similar results were obtained following the disruption of actin filaments with cytochalasin D (not shown), suggesting high affinity of ULFs to F-actin.
Anchorage to F-actin prevents microtubule-dependent transport of ULFs
Our data show that ULFs interact with the two other cytoskeletal systems, microtubules, and actin filaments. To evaluate the relative contributions of these structures to ULF transport, we depolymerized either microtubules or actin filaments and analyzed the effect of depolymerization on ULF transport. Microtubules were depolymerized by incubating cells at 4°C for 1 h, followed by transfer to 37°C in the medium containing 10 μM nocodazole. Immunostaining with a tubulin antibody confirmed that this treatment was sufficient to depolymerize most of the microtubules (not shown). The results revealed that the combination of cold and nocodazole treatment completely eliminated long ULF trajectories (>6 μm) that are observed in control cells, thus demonstrating that at least trajectories longer than 6 μm for the 1-min imaging period represent ULFs that are transported in a microtubule-dependent manner (Fig. 3 and Supplemental Movie S3).
Figure 3.

F-actin suppresses microtubule-dependent transport of ULFs. GFP-ULF expressing cells were incubated for 30 min at 4°C to depolymerize microtubules, then transferred to 37°C for 1 h in medium containing: nocodazole (10 μM to prevent microtubule polymerization), Lat B (5 μM to depolymerize F-actin), or nocodazole and Lat B together (to prevent microtubule and F-actin polymerization). Control cells were incubated at 30 min at 4°C and then warmed at 37°C 60 min before imaging. A) Temporal color-code representation of ULF motility (see Materials and Methods and Fig. 1). Time color-code bar is shown in the control panel. Note that Lat B treatment increased ULF transport, as revealed by the number of rainbow tracks corresponding to motile ULFs. See also Supplemental Movie S3. Scale bar = 10 μm. B, C) Quantitative analysis of motility. Graphs show the frequency distribution of ULF trajectory length (B) and the percentage of trajectories longer than 6 μm (C).
Next, we used Lat B to depolymerize actin filaments, and we analyzed ULF movement in corresponding Lat B-treated cells (Fig. 2B, bottom panels). Both visual analysis and quantitative comparison demonstrate that F-actin depolymerization dramatically (∼5-fold) increased the fraction of ULFs moving at a distance > 6 μm during 60 s recording (Fig. 3A, B and Supplemental Movie S3). Like transport in control cells, long-distance transport in Lat B-treated cells was dependent on microtubules, since it was inhibited by nocodazole (Fig. 3B, C). These results indicate that anchorage to F-actin supresses microtubule-dependent transport of ULFs.
ULF transport is independent of microtubule dynamics and requires ATP
There is evidence suggesting that microtubule dynamics can contribute to IF organization (32). To determine whether the microtubule-dependent transport of ULFs requires the maintenance of the dynamic properties of microtubules, cells were treated with a low dose of vinblastine (10 nM). This substoichiometric concentration of vinblastine is sufficient to block microtubule dynamics (by capping the microtubule plus-ends) but does not cause their depolymerization (33). To validate the inhibition of microtubule dynamics, cells were transfected with the TagRFP-EB3 construct. In control cells expressing TagRFP-EB3 at a low level, TagRFP-EB3 accumulates at the plus-ends of growing microtubules forming comets. The velocity of EB3 comets is a good measure of plus-end dynamics. Live-cell imaging of TagRFP-EB3 showed that incubation with 10 nM vinblastine for 5 min is sufficient to stop EB3 comets (Fig. 4A). Immunostaining of vinblastine-treated cells shows that this concentration of the drug does not cause depolymerization or redistribution of microtubules for the duration of the experiment (30 min). Movement of GFP-ULFs was monitored in the presence of Lat B to maximize the proportion of microtubule-dependent movement of ULFs. Figure 4B shows that 10 nM vinblastine had no effect on ULF movement and, therefore, that the dynamic properties of microtubules are not essential for ULF transport.
Figure 4.
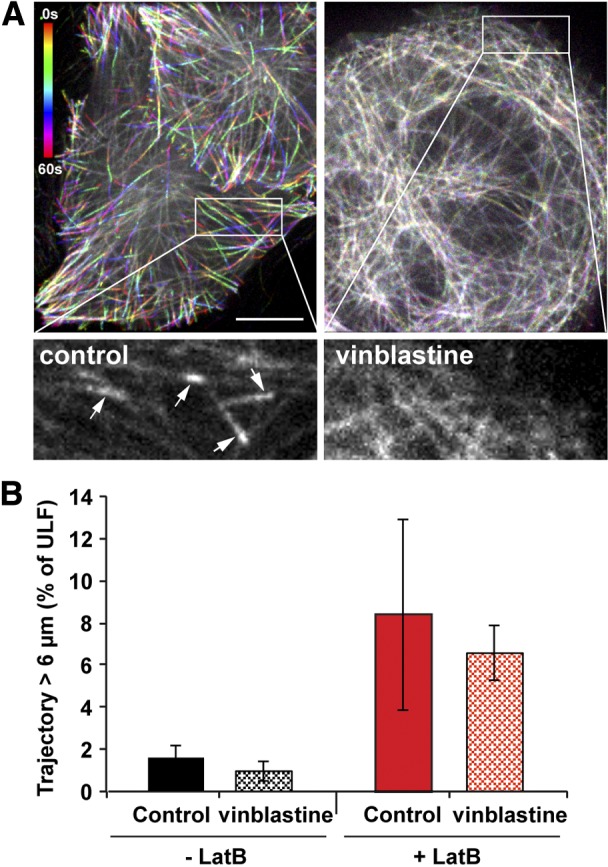
Microtubule dynamics is not required for ULF transport. GFP-ULF-expressing cells were transfected with TagRFP-EB3. Left panel; temporal color coding from the 60-frame projection of EB3 (1 frame/s) revealed the EB3 comet evolution at the tip of growing microtubules as an indicator of microtubule dynamics. Right panel; 10 nM vinblastine for 5 min is sufficient to stop microtubule dynamics, since EB3 comets at the microtubule tips are absent. Time color-code bar is shown in the control panel. Scale bar = 10 μm. Insets: enlargement of the first frame corresponding to the boxed area. Arrows point to the EB3 comets, which are present in the control cells. B) Vinblastine (10 nM) was added for 5 to 30 min to GFP-ULF cells treated or not with 5 μM Lat B. Graph shows the percentage of ULF tracks longer than 6 μm. Inhibition of microtubule dynamics does not substantially change the movement.
To determine whether the movement of ULF along microtubules is ATP dependent, we depleted ATP in cells by treatment with sodium azide and monitored the movement of ULFs. The sodium azide treatment was performed in the absence of glucose to prevent ATP production by glycolysis. Analysis of ULF trajectories revealed that the transport of ULFs is dramatically blocked after 15 min of treatment with sodium azide (Fig. 5A, B). This effect was delayed by the addition of glucose to the medium (Fig. 5B). The effects of ATP depletion were reversible, as ULFs partially recovered their motility after sodium azide washout (Fig. 5B). Finally, to confirm the integrity of microtubule tracks after sodium azide treatment, cells were fixed and stained for tubulin. Coimaging of GFP-ULF with stained tubulin revealed a striking colocalization of ULFs with microtubules in ATP-depleted cells (Fig. 5C), most likely caused by microtubule motors being locked to microtubules in a rigor complex.
Figure 5.

ATP depletion inhibits ULF transport. ATP in cells was depleted by incubation with 20 mM sodium azide (NaN3) in Ca2+, Mg2+-containing PBS. Lat B (5 μM) was added to stimulate microtubule-dependent transport. Controls include NaN3 washout and incubation with NaN3 in the presence of 50 mM glucose to allow ATP production by glycolysis. A) Temporal color-code representation of ULF motility before and after NaN3 treatment. B) Graph shows the percentage of ULF tracks longer than 6 μm, which is completely inhibited by NaN3 treatment. C) GFP-ULF binding to microtubules in Lat B- and NaN3-treated cells. GFP-ULF-expressing cells were fixed and stained by an antibody against tubulin. Scale bar = 10 μm.
Inhibition of dynein with ciliobrevin inhibits ULF transport
The results of the vinblastine and ATP depletion experiments described above are consistent with the fact that VIFs are transported by microtubule motors (kinesin and dynein; refs. 15–17). More than 50 kinesin anterograde motors have been identified in mammalian cells (34), while only one dynein retrograde motor is present (35). It has been reported that inhibition of the motor responsible for the retrograde transport of one cargo also inhibits the anterograde transport of the same cargo and vice versa (36). Therefore, we used ciliobrevin, a cytoplasmic dynein inhibitor (37) to inhibit the only candidate for driving the retrograde transport of ULFs along microtubules. GFP-ULF-expressing cells were treated with Lat B to enhance microtubule-dependent transport. Then, cells were treated for 1 h with 50 μM ciliobrevin before imaging. Strikingly, a complete inhibition of ULF transport was observed after ciliobrevin treatment (Fig. 6). The same inhibition was also observed in the absence of Lat B (not shown). To test the viability of the ciliobrevin-treated cells, they were washed twice with PBS and incubated for 1 h with complete medium in the presence of Lat B. The quantitative analysis of ULF trajectories indicated that ULF transport resumed after ciliobrevin washout, thus providing evidence that the ciliobrevin treatment was not toxic to the cells (Fig. 6B). These results demonstrate that dynein participates in ULF retrograde transport.
Figure 6.
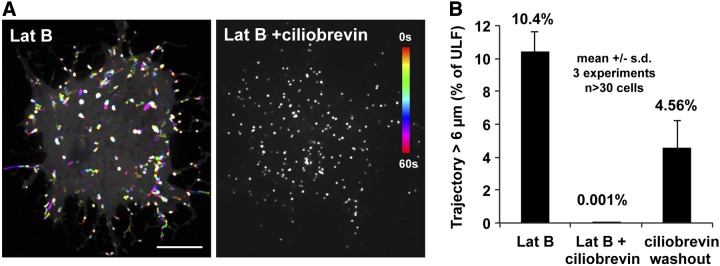
Inhibition of dynein with ciliobrevin blocks ULF transport. GFP-ULF cells were incubated with serum-free medium supplemented with 5 μM Lat B for 15 min; then 50 μM ciliobrevin was added for 1 h. A) Temporal color coding of GFP-ULF position shows that ciliobrevin treatment inhibits ULF. Scale bar = 10 μm. B) Graph shows the percentage of ULF tracks longer than 6 μm, which are completely inhibited by the ciliobrevin treatment. For the ciliobrevin washout condition, ciliobrevin-treated cells were washed twice with PBS and incubated for 1 h in completed medium in the presence of Lat B to reverse ciliobrevin action.
ROCK is a negative regulator of ULF transport
There is increasing evidence that site-specific phosphorylation of IF proteins can affect their assembly, structural organization, and distribution. Interestingly, many vimentin phosphorylation sites are targets of kinases involved in regulating cell cytoskeleton remodeling and cell motility (23). For example, VIF organization is regulated by RhoA kinase (ROCKα), a well-known promoter of stress fiber and focal adhesion complex formation (24). It is very likely that signaling pathways that affect vimentin phosphorylation could also affect the affinity of vimentin for microtubules or F-actin, as well as ULF transport.
To test the involvement of ROCK, we used the specific ROCK inhibitor Y-27632 to determine whether ROCK inhibition alters ULF association with F-actin. Staining with rhodamine-phalloidin performed 1 h after treatment with 10 μM Y-27632 showed the expected disassembly of stress fibers, demonstrating the efficient inhibition of the major target of ROCK, i.e., myosin-II (38, 39). However, a dense meshwork of disorganized actin filaments remained after this treatment, and association of ULFs with actin filament was still observed (Fig. 7A). This observation suggested that binding of ULFs to actin is independent of the ROCK/myosin II pathway. Then we determined whether Y-27632 treatment affected ULF transport using time-lapse imaging. The results revealed a slight increase in ULF transport in cells treated with this inhibitor compared to controls; however, this difference was not statistically significant (not shown). It is likely that the overall increase of the actin network density caused by Y-27632 treatment could interfere with a direct effect of the ROCK inhibition on ULF transport. Therefore, we tested the effect of ROCK inhibition on ULF transport in the absence of actin filaments by treating the cells with Y-27632 and Lat B simultaneously. When ROCK was inhibited in the absence of actin filaments (Y-27632+Lat B), the fraction of ULFs moving along microtubules was significantly increased compared to the control cells (Lat B; Fig. 7B, C). These observations show that ROCK regulates ULF motility along microtubules without modifying their interaction with actin filaments.
Figure 7.

ROCK is a negative regulator of ULF transport along microtubules. A) F-actin staining of GFP-ULF cells fixed 1 h after treatment with 10 μM Y-27632. Note that the Y-27632 treatment inhibited actin bundling into stress fibers but did not affect ULF interaction with F-actin. B) Lat B-treated GFP-ULF cells were treated with 10 μM Y-27632 for 1 h before and during live-cell imaging of GFP-ULF motility. Temporal color coding of ULF position revealed the presence of very long trajectories in Y-27632-treated cells as compared to control. Scale bars = 10 μm. C) Graph of the frequency distribution of ULF trajectory length shows that Y-27632 treatment increased the frequency of motile ULFs and the length of trajectories.
PAK stimulates ULF transport along microtubules
Many processes regulated by ROCK activity are antagonized by the activity of another GTPase-regulated kinase, PAK. For example, while ROCK proteins promote and maintain actomyosin contraction and stress fiber assembly, activated PAK1 phosphorylates the myosin light chain kinase (MLCK), resulting in decreased MLCK activity and reduced actomyosin assembly (40). Because our data established that ROCK is a negative regulator of vimentin transport, we hypothesized that PAK could be the positive counterpart. PAK protein kinases are subdivided into 2 groups, PAK 1–3 (group I) and PAK 4–6 (group II). The IPA-3 compound is a potent inhibitor of the group I but not group II PAKs (41). We found that IPA-3 dramatically inhibited ULF movement in Lat B-treated cells (Fig. 8A, B) pointing to a direct role of group I PAK in the stimulation of VIF precursor transport along microtubules. It has been reported that inhibition of PAK by IPA-3 can affect the microtubule network (42), but staining with the tubulin antibody showed that microtubules were not affected by 10 μM IPA-3 that we used (Supplemental Fig. S1B). Therefore, IPA-3 inhibitory effect is attributable to down-regulation of transport rather than microtubule depolymerization.
Figure 8.

PAK2 is a positive regulator of ULF transport along microtubules. A, B) GFP-ULF cells were incubated for 1 h with 5 μM Lat B in the absence or presence of 10 μM IPA-3 before imaging of GFP-ULF motility. A) Temporal color coding of GFP-ULF position shows that IPA-3 treatment inhibits ULF transport along microtubules. Scale bar = 10 μm. B) Graph of the frequency distribution of ULF trajectory length shows that IPA-3 treatment decreased the frequency of motile ULF and the length of trajectories. C) GFP-ULF cells were transfected twice with scrambled (all star), PAK1, PAK2, or PAK1 and PAK2 siRNAs. Cell lysates were prepared 96 h after the first transfection. Western blot analysis reveal ≥80 and 90% knockdown efficiency for PAK1 and PAK2, respectively. Level of expression of GFP-ULF revealed by the vimentin antibody was used as a loading control. D) Imaging of GFP-ULF motility was performed 96 h after transfection with the indicated siRNAs. Graph shows the frequency distribution of ULF trajectory length > 6 μm. Note that PAK2 siRNA treatment decreased the frequency of motile ULFs and the length of trajectories, while PAK1 siRNA alone had no effect. E) Lysosomes were labeled using Lysotracker Red (Molecular Probes) in control or 10 μM IPA-3-treated GFP-ULF-expressing cells. Cell imaging was performed in the presence of 5 μM Lat B to reproduce the same conditions where PAK inhibition inhibited GFP-ULF transport. Graph of the frequency distribution of lysosome trajectory length shows that IPA-3 treatment had no effect on lysosome transport.
To distinguish between the two ubiquitously expressed group I PAK isoforms, PAK1 and PAK2, we used siRNAs specific to these isoforms. Western blot with specific antibodies (Fig. 8C) shows that these treatments reduced PAK1 protein by >80% and PAK2 protein by 90%. Transfection of a rhodamine-labeled scrambled siRNA (all stars) had no effect on PAK isoforms' protein levels and revealed that >95% of the cell population was effectively transfected (not shown). Rhodamine-phalloidin staining shows that both siRNAs stimulated stress fiber formation, thus confirming a significant decrease of PAK isoform expression by the siRNA treatments (Supplemental Fig. S1A). Analysis of ULF motility in siRNA-treated cells in the presence of Lat B demonstrated that depletion of PAK2 but not PAK1 decreased ULF motility. However, simultaneous depletion of both isoforms mimics the effect of IPA-3 treatment by causing a major reduction of ULF motility (Fig. 8D). These results indicated that PAK2 is a major regulator of ULF transport but did not exclude additional contribution of PAK1. To verify that the effect of PAK2 inhibition was specific to ULF motility and not attributed to the total down-regulation of microtubule-dependent transport, we analyzed transport of an unrelated cargo, lysosomes, labeled by Lysotracker Red (Molecular Probes). The transport of lysosomes was not affected by either the IPA-3 (Fig. 8E) or PAK siRNA (not shown) treatments showing that the effect of PAK inhibition is specific to ULF transport.
DISCUSSION
IFs form a dynamic network that constantly adapts its distribution and connections with the two other cytoskeletal systems, microtubules and actin (43, 44). Vimentin in the cell is present in 3 principal forms, which correlate with the 3 main levels of filament assembly observed in vitro: filament precursors called particles, short filaments or squiggles, and mature long filaments (45, 46). Bidirectional movement of all three forms of vimentin has been previously observed (15, 47), but the high density of IF network usually prevents a quantitative analysis of transport and its regulation.
In this study, we decided to use the Y117L-mutant of vimentin. Assembly of this mutant is arrested at the ULF stage, on average 8 laterally associated tetramers of vimentin that represent the smallest stable filamentous form of vimentin in vitro (20, 48). When the GFP-tagged version of this mutant was expressed in vimentin-deficient cells, it formed uniform fluorescent particles that were capable of moving along linear tracks. These particles could easily be tracked in the cytoplasm of cultured cells with technologies typically used for tracking transport of membrane organelles. By coexpressing GFP-ULF with a microtubule binding protein TagRFP-EB3, we showed that GFP-ULFs are transported along microtubules. Quantitative analysis revealed that the fraction of ULFs moving over a long distance is very small (around 2%), suggesting the existence of a mechanism that restricts ULF movement.
Our data demonstrate that ULFs, like mature VIFs (49–51), interact with the actin cytoskeleton. Most notably, our functional data demonstrate that interaction with actin restricts ULF transport along microtubules. The nature of the ULF association with actin is not known; it could be either direct binding (51) or association via an actin-binding protein, such as plectin (50), filamin A (49), or fimbrin (52). However, we know that the ULF association with actin is so robust that it withstands treatment of cells with CytoD or Lat B; ULFs retain their association with the aggregates of F-actin resistant to this treatment. Moreover, we have evidence that binding of ULFs to actin is independent of the activity of myosin-II, as actin-ULF binding was resistant to the inhibition of ROCK, a major activator of myosin II.
Vimentin harbors 25 serine and 11 threonine residues in its N-terminal non-α-helical domain, many of which are targets for several kinases (21, 22). Intriguingly, most of these kinases, for example, PI3K, ROCK, PAK, PKC, PKA, and CaMK, are classically involved in the regulation of cytoskeleton remodeling (23). Accordingly, it is very likely that signaling pathways that affect vimentin phosphorylation could also affect vimentin filament precursor transport and affinity to microtubules or F-actin. Therefore, we decided to screen kinase inhibitors for their effect on the regulation of ULF transport. We found that two GTPase-regulated kinases, ROCK and PAK, have opposite effects on ULF transport along microtubules. ROCK inhibition stimulates transport, while PAK2 inhibition drastically blocks it. Both kinases have been demonstrated to phosphorylate vimentin at different sites in vitro (26, 53, 54), which some were confirmed in vivo (25, 27, 28, 54). Actin and myosin II are well-described downstream effectors. However, changes in ULF transport are not explained by an effect of kinases on actin and associated proteins because effects on ULF transport by ROCK and PAK inhibition were observed after depolymerization of F-actin. Alternatively, ROCK and PAK could phosphorylate ULFs directly, modifying their affinity to microtubule motors. Another possibility is that the effect of the kinases on ULF transport could be explained not by the phosphorylation of vimentin but by the phosphorylation of motors or other components of the transport machinery, affecting the ability of these components to transport ULFs. To date, there is evidence for both plus-end (kinesin) and minus-end (dynein) microtubule motor-dependent movement of VIF along microtubules (15–17). Kinesin and dynein motors are multiprotein complexes, and the adapters required for vimentin binding has not been investigated yet. Our data confirm the involvement of dynein in ULF retrograde transport (Fig. 6). To understand how phosphorylation by PAK and ROCK could affect binding of ULFs to dynein, we will need to identify the dynein subunits and/or adapters that are responsible for the dynein-vimentin interaction.
In this study, we made use of a vimentin mutant to model VIF precursors since the mutant forms a full-width filament of one unit-length that is unable to elongate because of a single amino acid substitution impeding longitudinal annealing. However, we can hypothesize that the mode of regulation described here for VIF precursors could also apply to the transport of mature filaments. As previously mentioned, it is known that VIF can connect to actin filaments (49–51). It is known that the actin cytoskeleton is required for the retraction of the VIF network after microtubule depolymerization (13). However, the role of F-actin in the regulation of VIF transport, as well as the importance of VIF precursor transport for the integrity of the VIF network remains to be determined. On the basis of our results, we can suppose that the binding of vimentin to F-actin will restrict VIF transport to stabilize the network. Moreover, we can hypothesize that the anchoring of VIF precursors to F-actin may prevent their incorporation into longer filaments. This is a likely scenario because formation of actin-rich structures is known to correlate with VIF disassembly. For example, there is evidence that polymerized VIFs inhibit podosome formation (55). Also, disassembly of VIFs into vimentin particles was observed during lamellipodia formation in migrating cells (56). In both examples, as in many others, disassembly of VIF correlated with the hyperphosphorylation of vimentin. We found here that in addition to known effects on assembly, vimentin phosphorylation regulates filament precursor transport. Additional studies are needed to clarify how the regulation of VIF precursor transport described here contributes to the dynamics of the VIF network and affect other important vimentin functions.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Science of the U. S. National Institutes of Health under awards P01GM09697 and R01 GM52111. A.R. was supported by a fellowship from the American Heart Association (13POST16210010).
The authors thank the Goldman Laboratory (Fineberg School of Medicine, Northwestern University) for sharing materials that contributed to this work and Caroline Hookway and Robert Goldman for critical reading of the manuscript.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- F-actin
- filamentous actin
- IF
- intermediate filament
- Lat B
- latrunculin B
- PAK
- p21-activated kinase
- ROCK
- Rho-kinase
- siRNA
- small interfering RNA
- ULF
- unit-length filament
- VIF
- vimentin intermediate filament
REFERENCES
- 1. Minin A. A., Moldaver M. V. (2008) Intermediate vimentin filaments and their role in intracellular organelle distribution. Biochem. Biokhim. 73, 1453–1466 [DOI] [PubMed] [Google Scholar]
- 2. Bayless K. J., Johnson G. A. (2011) Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J. Vasc. Res. 48, 369–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chung B. M., Rotty J. D., Coulombe P. A. (2013) Networking galore: intermediate filaments and cell migration. Curr. Opin. Cell Biol. 25, 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rogel M. R., Soni P. N., Troken J. R., Sitikov A., Trejo H. E., Ridge K. M. (2011) Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 25, 3873–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Satelli A., Li S. (2011) Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 68, 3033–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dave J. M., Kang H., Abbey C. A., Maxwell S. A., Bayless K. J. (2014) Proteomic profiling of endothelial invasion revealed receptor for activated C kinase 1 (RACK1) complexed with vimentin to regulate focal adhesion kinase (FAK). J. Biol. Chem. 288, 30720–30733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nekrasova O. E., Mendez M. G., Chernoivanenko I. S., Tyurin-Kuzmin P. A., Kuczmarski E. R., Gelfand V. I., Goldman R. D., Minin A. A. (2011) Vimentin intermediate filaments modulate the motility of mitochondria. Mol. Biol. Cell 22, 2282–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Potokar M., Stenovec M., Gabrijel M., Li L., Kreft M., Grilc S., Pekny M., Zorec R. (2010) Intermediate filaments attenuate stimulation-dependent mobility of endosomes/lysosomes in astrocytes. Glia 58, 1208–1219 [DOI] [PubMed] [Google Scholar]
- 9. Eckes B., Dogic D., Colucci-Guyon E., Wang N., Maniotis A., Ingber D., Merckling A., Langa F., Aumailley M., Delouvee A., Koteliansky V., Babinet C., Krieg T. (1998) Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 111, 1897–1907 [DOI] [PubMed] [Google Scholar]
- 10. Burgstaller G., Gregor M., Winter L., Wiche G. (2010) Keeping the vimentin network under control: cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol. Biol. Cell 21, 3362–3375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mendez M. G., Kojima S., Goldman R. D. (2010) Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 24, 1838–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Franke W. W., Schmid E., Grund C., Geiger B. (1982) Intermediate filament proteins in nonfilamentous structures: transient disintegration and inclusion of subunit proteins in granular aggregates. Cell 30, 103–113 [DOI] [PubMed] [Google Scholar]
- 13. Hollenbeck P. J., Bershadsky A. D., Pletjushkina O. Y., Tint I. S., Vasiliev J. M. (1989) Intermediate filament collapse is an ATP-dependent and actin-dependent process. J. Cell Sci. 92, 621–631 [DOI] [PubMed] [Google Scholar]
- 14. Goldman R. D. (1971) The role of three cytoplasmic fibers in BHK-21 cell motility. I. Microtubules and the effects of colchicine. J. Cell Biol. 51, 752–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prahlad V., Yoon M., Moir R. D., Vale R. D., Goldman R. D. (1998) Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J. Cell Biol. 143, 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gyoeva F. K., Gelfand V. I. (1991) Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature 353, 445–448 [DOI] [PubMed] [Google Scholar]
- 17. Helfand B. T., Mikami A., Vallee R. B., Goldman R. D. (2002) A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J. Cell Biol. 157, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Starger J. M., Brown W. E., Goldman A. E., Goldman R. D. (1978) Biochemical and immunological analysis of rapidly purified 10-nm filaments from baby hamster kidney (BHK-21) cells. J. Cell Biol. 78, 93–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun T. T., Green H. (1978) Immunofluorescent staining of keratin fibers in cultured cells. Cell 14, 469–476 [DOI] [PubMed] [Google Scholar]
- 20. Meier M., Padilla G. P., Herrmann H., Wedig T., Hergt M., Patel T. R., Stetefeld J., Aebi U., Burkhard P. (2009) Vimentin coil 1A-A molecular switch involved in the initiation of filament elongation. J. Mol. Biol. 390, 245–261 [DOI] [PubMed] [Google Scholar]
- 21. Eriksson J. E., He T., Trejo-Skalli A. V., Harmala-Brasken A. S., Hellman J., Chou Y. H., Goldman R. D. (2004) Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117, 919–932 [DOI] [PubMed] [Google Scholar]
- 22. Izawa I., Inagaki M. (2006) Regulatory mechanisms and functions of intermediate filaments: a study using site- and phosphorylation state-specific antibodies. Cancer Sci. 97, 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sihag R. K., Inagaki M., Yamaguchi T., Shea T. B., Pant H. C. (2007) Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp. Cell Res. 313, 2098–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sin W. C., Chen X. Q., Leung T., Lim L. (1998) RhoA-binding kinase alpha translocation is facilitated by the collapse of the vimentin intermediate filament network. Mol. Cell. Biol. 18, 6325–6339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chan W., Kozma R., Yasui Y., Inagaki M., Leung T., Manser E., Lim L. (2002) Vimentin intermediate filament reorganization by Cdc42: involvement of PAK and p70 S6 kinase. Eur. J. Cell Biol. 81, 692–701 [DOI] [PubMed] [Google Scholar]
- 26. Goto H., Tanabe K., Manser E., Lim L., Yasui Y., Inagaki M. (2002) Phosphorylation and reorganization of vimentin by p21-activated kinase (PAK). Genes Cells 7, 91–97 [DOI] [PubMed] [Google Scholar]
- 27. Li Q. F., Spinelli A. M., Wang R., Anfinogenova Y., Singer H. A., Tang D. D. (2006) Critical role of vimentin phosphorylation at Ser-56 by p21-activated kinase in vimentin cytoskeleton signaling. J. Biol. Chem. 281, 34716–34724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu T., Guevara O. E., Warburton R. R., Hill N. S., Gaestel M., Kayyali U. S. (2010) Regulation of vimentin intermediate filaments in endothelial cells by hypoxia. Am. J. Physiol. Cell Physiol. 299, C363–C373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Georgakopoulou S., Moller D., Sachs N., Herrmann H., Aebi U. (2009) Near-UV circular dichroism reveals structural transitions of vimentin subunits during intermediate filament assembly. J. Mol. Biol. 386, 544–553 [DOI] [PubMed] [Google Scholar]
- 30. Sarria A. J., Nordeen S. K., Evans R. M. (1990) Regulated expression of vimentin cDNA in cells in the presence and absence of a preexisting vimentin filament network. J. Cell Biol. 111, 553–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu W., Fox P., Lakonishok M., Davidson M. W., Gelfand V. I. (2013) Initial neurite outgrowth in Drosophila neurons is driven by kinesin-powered microtubule sliding. Curr. Biol. 23, 1018–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sakamoto Y., Boeda B., Etienne-Manneville S. (2013) APC binds intermediate filaments and is required for their reorganization during cell migration. J. Cell Biol. 200, 249–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dhamodharan R., Jordan M. A., Thrower D., Wilson L., Wadsworth P. (1995) Vinblastine suppresses dynamics of individual microtubules in living interphase cells. Mol. Biol. Cell 6, 1215–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hirokawa N., Noda Y., Tanaka Y., Niwa S. (2009) Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696 [DOI] [PubMed] [Google Scholar]
- 35. Kardon J. R., Vale R. D. (2009) Regulators of the cytoplasmic dynein motor. Nat. Rev. Mol. Cell Biol. 10, 854–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ally S., Larson A. G., Barlan K., Rice S. E., Gelfand V. I. (2009) Opposite-polarity motors activate one another to trigger cargo transport in live cells. J. Cell Biol. 187, 1071–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Firestone A. J., Weinger J. S., Maldonado M., Barlan K., Langston L. D., O'Donnell M., Gelfand V. I., Kapoor T. M., Chen J. K. (2012) Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484, 125–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., Kaibuchi K. (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 39. Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., Iwamatsu A., Kaibuchi K. (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273, 245–248 [DOI] [PubMed] [Google Scholar]
- 40. Wirth A., Schroeter M., Kock-Hauser C., Manser E., Chalovich J. M., De Lanerolle P., Pfitzer G. (2003) Inhibition of contraction and myosin light-chain phosphorylation in guinea-pig smooth muscle by p21-activated kinase 1. J. Physiol. 549, 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deacon S. W., Beeser A., Fukui J. A., Rennefahrt U. E., Myers C., Chernoff J., Peterson J. R. (2008) An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 15, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takahashi K., Suzuki K. (2009) Membrane transport of WAVE2 and lamellipodia formation require Pak1 that mediates phosphorylation and recruitment of stathmin/Op18 to Pak1-WAVE2-kinesin complex. Cell. Signal. 21, 695–703 [DOI] [PubMed] [Google Scholar]
- 43. Chang L., Goldman R. D. (2004) Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell Biol. 5, 601–613 [DOI] [PubMed] [Google Scholar]
- 44. Windoffer R., Beil M., Magin T. M., Leube R. E. (2011) Cytoskeleton in motion: the dynamics of keratin intermediate filaments in epithelia. J. Cell Biol. 194, 669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Strelkov S. V., Herrmann H., Aebi U. (2003) Molecular architecture of intermediate filaments. BioEssays 25, 243–251 [DOI] [PubMed] [Google Scholar]
- 46. Goldman R. D., Cleland M. M., Murthy S. N., Mahammad S., Kuczmarski E. R. (2012) Inroads into the structure and function of intermediate filament networks. J. Struct. Biol. 177, 14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoon M., Moir R. D., Prahlad V., Goldman R. D. (1998) Motile properties of vimentin intermediate filament networks in living cells. J. Cell Biol. 143, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Herrmann H., Haner M., Brettel M., Ku N. O., Aebi U. (1999) Characterization of distinct early assembly units of different intermediate filament proteins. J. Mol. Biol. 286, 1403–1420 [DOI] [PubMed] [Google Scholar]
- 49. Kim H., McCulloch C. A. (2011) Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett. 585, 18–22 [DOI] [PubMed] [Google Scholar]
- 50. Svitkina T. M., Verkhovsky A. B., Borisy G. G. (1996) Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J. Cell Biol. 135, 991–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Esue O., Carson A. A., Tseng Y., Wirtz D. (2006) A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J. Biol. Chem. 281, 30393–30399 [DOI] [PubMed] [Google Scholar]
- 52. Correia I., Chu D., Chou Y. H., Goldman R. D., Matsudaira P. (1999) Integrating the actin and vimentin cytoskeletons. adhesion-dependent formation of fimbrin-vimentin complexes in macrophages. J. Cell Biol. 146, 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Inagaki M., Nishi Y., Nishizawa K., Matsuyama M., Sato C. (1987) Site-specific phosphorylation induces disassembly of vimentin filaments in vitro. Nature 328, 649–652 [DOI] [PubMed] [Google Scholar]
- 54. Goto H., Kosako H., Tanabe K., Yanagida M., Sakurai M., Amano M., Kaibuchi K., Inagaki M. (1998) Phosphorylation of vimentin by Rho-associated kinase at a unique amino-terminal site that is specifically phosphorylated during cytokinesis. J. Biol. Chem. 273, 11728–11736 [DOI] [PubMed] [Google Scholar]
- 55. Pan Y. R., Cho K. H., Lee H. H., Chang Z. F., Chen H. C. (2013) Protein tyrosine phosphatase SHP2 suppresses podosome rosette formation in Src-transformed fibroblasts. J. Cell Sci. 126, 657–666 [DOI] [PubMed] [Google Scholar]
- 56. Helfand B. T., Mendez M. G., Murthy S. N., Shumaker D. K., Grin B., Mahammad S., Aebi U., Wedig T., Wu Y. I., Hahn K. M., Inagaki M., Herrmann H., Goldman R. D. (2011) Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 22, 1274–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.