

Abstract
Suboptimal ventilator support or regional ventilation heterogeneity in inflamed lungs causes excessive tissue distension, which triggers stretch-induced pathological signaling and may lead to vascular leak and lung dysfunction. Focal adhesions (FAs) are cell-substrate adhesive complexes participating in cellular mechanotransduction and regulation of the Rho GTPase pathway. Stretch-induced Rho regulation remains poorly understood. We used human lung endothelial cells (ECs) exposed to pathological cyclic stretch (CS) at 18% distension to test the hypothesis that FA protein paxillin participates in CS-induced Rho activation by recruiting the Rho-specific guanine nucleotide exchange factor GEF-H1. CS induced phosphorylation of paxillin and activated p42/44-MAP kinase, Rho GTPase, and paxillin/GEF-H1/p42/44-MAPK association. CS caused nearly 2-fold increase in EC permeability, which was attenuated by paxillin knockdown. Expression of the paxillin-Y31/118F phosphorylation mutant decreased the CS-induced paxillin/GEF-H1 association (16.3±4.1%), GEF-H1 activation (28.9±9.2%), and EC permeability (28.7±8.1%) but not CS-induced p42/44-MAPK activation. Inhibition of p42/44-MAPK suppressed CS-induced paxillin/GEF-H1 interactions (15.9±7.9%), GEF-H1 activation (11.7±4.3%), and disruption of EC monolayer. Expression of GEF-H1T678A lacking p42/44-MAPK phosphorylation site attenuated Rho activation (31.2±11.6%). We conclude that MAPK-dependent targeting of GEF-H1 to paxillin is involved in the regulation of CS-induced Rho signaling and EC permeability. This study proposes a novel concept of paxillin-GEF-H1-p42/44-MAPK module as a regulator of pathological mechanotransduction.—Gawlak, G., Tian, Y., O'Donnell, J. J., III, Tian, X., Birukova, A. A., Birukov, K. G. Paxillin mediates stretch-induced Rho signaling and endothelial permeability via assembly of paxillin-p42/44MAPK-GEF-H1 complex.
Keywords: mechanotransduction, focal adhesions, cytoskeleton, guanine nucleotide exchange factors
Excessive mechanical forces in the form of mechanical strain or cyclic stretch (CS) from mechanical ventilation may increase vascular leak and culminate in life-threatening conditions such as brain and pulmonary edema. Lung perturbations caused by mechanical ventilation increase vascular permeability (1) and activate inflammatory cytokine production (2, 3) and apoptosis (4–6). These events mirror the in vivo alveolar flooding, leukocyte infiltration, and hypoxemia associated with high morbidity and mortality rates in patients with acute respiratory distress syndrome (ARDS; refs. 7, 8).
The lung endothelium forms a semiselective barrier between circulating blood and interstitial fluid, which is dynamically regulated by bioactive molecules present in the circulation and mechanical forces imposed by blood flow and lung expansion during respiratory cycles. A working model of paracellular endothelial cell (EC) barrier regulation suggests that paracellular gap formation is regulated by the balance of competing contractile forces imposed by actomyosin cytoskeleton, which generate centripetal tension, and adhesive cell-cell and cell-matrix tethering forces imposed by focal adhesions and adherens junctions, which together regulate cell shape changes and control EC permeability (9, 10).
Biomechanical forces acting on vascular endothelium stimulate a variety of signaling cascades triggered by MAP kinases (p42/44 MAPK, JNK, and p38), nonreceptor tyrosine kinases [p60Src and FA kinase (FAK)], integrin-mediated signaling, ion channels, and Rho family GTPases, which altogether define the endothelial barrier properties (11–13). CS-induced activation of Rho GTPase signaling pathway exacerbates agonist-induced EC barrier disruption, while attenuation of Rho activity reduces lung vascular leak and promotes barrier recovery in the in vitro and in vivo models of acute lung injury (14–18). Although several intriguing hypotheses have been proposed to explain mechanisms of mechanosensing and stretch-induced Rho activation by focal adhesions (FAs; refs. 19, 20), they await further experimental validation.
FAs are formed by the association of substrate adhesion receptors called integrins with intracellular protein complexes containing paxillin, talin, zyxin, vinculin, Src and FAK tyrosine kinases, and other proteins (21). Paxillin is a multidomain adapter FA protein containing 4 LD domains in the N-terminal part and 4 LIM domains in the C-terminal part that functions as a molecular scaffold for protein recruitment to FAs, which is essential for protein networking and efficient signal transmission. Phosphorylation of paxillin Tyr31 and Tyr118 residues by FAK or Src family nonreceptor tyrosine kinases creates docking sites for various paxillin binding partners (21). Specific patterns of FA protein interactions and site-specific phosphorylation have been associated with barrier-protective and barrier-disruptive EC responses to mechanical and chemical stimuli (14, 22–26). In addition, paxillin and FAK may locally regulate agonist-induced Rho GTPase signaling depending on physiological context, but these mechanisms are much less understood.
Guanine nucleotide exchange factor H1 (GEF-H1) has been originally described as a microtubule-associated activator of Rho (27). However, GEF-H1 was also found at the tight junctions of epithelial cells, where it directly interacts with a junctional adaptor protein, cingulin. This interaction regulates GEF-H1 activity toward Rho and represents a molecular mechanism of Rho regulation by cell junctions (28). GEF-H1 can be also activated by mechanical forces generated during hyperosmotic stress (29) or by direct cell mechanical stimulation with magnetic beads (20). We have previously reported the involvement of GEF-H1 in the CS-induced cell remodeling and modulation of endothelial responses to agonists in cell culture and in the animal model of ventilator induced lung injury (30). However, the mechanism of GEF-H1 activation and subcellular redistribution induced by cell mechanical stimulation remains obscure. This study tested the hypothesis that CS-induced Rho activation is mediated by recruitment of GEF-H1 to FA via formation of a functional complex with paxillin, which controls the CS-induced increase in EC permeability.
MATERIALS AND METHODS
Cell culture and reagents
Human pulmonary artery endothelial cells (HPAECs) and human lung microvascular endothelial cells (HLMVECs; Lonza, Allendale, NJ, USA) were cultured according to the manufacturer's recommendations. Unless specified, biochemical reagents were obtained from Sigma (St. Louis, MO, USA). Reagents for immunofluorescence were purchased from Molecular Probes (Eugene, OR, USA). Antibodies to GEF-H1, p-FAK Y397, p-FAK Y576/577, p-Src Y416, p-MYPT T696, pp-MLC T18/S19, p-p42/44 MAPK T202/Y204, and p-paxillin Y118 were from Cell Signaling (Beverly, MA, USA); paxillin, p-paxillin Y118, p42/44 MAPK, and RhoA were from BD Transduction Laboratories (San Diego, CA, USA). Plasmids encoding paxillin-wild-type and paxillin-Y31/118F were provided by C. Turner (State University of New York, Syracuse, NY, USA). A plasmid encoding GFP-tagged wild-type GEF-H was provided by G. Bokoch (Scripps, La Jolla, CA, USA). A plasmid encoding phosphorylation-deficient GFP-tagged GEF-H1T678A mutant was generous gift from K. Szaszi (St. Michael's Hospital, Toronto, ON, Canada). Cells were used for transient transfections according to protocol described previously (31).
The express micromolecule permeability testing (XPerT) assay has been previously developed by our group. This method is based on high-affinity binding of avidin-conjugated FITC-labeled tracer to the biotinylated ligand in the underlying matrix (32).
Cell culture under CS
CS experiments were performed according to previously described protocol (1). Briefly, a FX-4000T Flexcell Tension Plus system (Flexcell International, McKeesport, PA, USA) equipped with a 25 mm BioFlex Loading station was employed, which provides uniform radial and circumferential strain across a membrane surface along all radii. Each BioFlex membrane is stretched over the post when under vacuum pressure, creating a single-plane uniformly stretched circle. The radial and circumferential strain was experimentally determined by the vendor. Cells were exposed to acute high-magnitude (18% linear elongation, sinusoidal wave, 25 cycles/min) CS to recapitulate the mechanical stresses experienced by the alveolar endothelium at high-tidal volume (HTV) mechanical ventilation (1, 33). At the end of experiment, cell lysates were collected. Coimmunoprecipitation studies and Western blot analysis were performed using confluent HPAEC monolayers as described previously (34). In immunofluorescence experiments, CS-exposed endothelial monolayers were fixed with 3.7% formaldehyde and used for immunofluorescence staining as described previously (34).
RhoA and GEF-H1 activity assays
Active (GTP-bound) RhoA was captured using GST-rhotekin beads as previously reported (35). Briefly, HPAECs grown to confluence on 60 mm dishes were exposed to 18% CS or static conditions for indicated time periods. After mechanical stimulation, cells were lysed with ice-cold Rho assay buffer containing 100 mM NaCl, 50 mM Tris-HCl (pH 7.6), 20 mM NaF, 10 mM MgCl2, 1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS, 1 mM Na3VO4, and protease inhibitors. After centrifugation, the supernatants were incubated at 4°C for 45 min with 20–25 μg of GST-rhotekin beads, followed by extensive washing. Total cell lysates and the rhotekin-captured proteins were analyzed by Western blotting using RhoA antibody. Active GEF-H1 was affinity precipitated from cell lysates as described previously (36) using a RhoA(G17A) mutant, which cannot bind nucleotide and therefore has high affinity for activated GEFs (37). Activated GEF-H1 in RhoA(G17A) pulldowns was detected by immunoblotting and normalized to total GEF-H1 in cell lysates for each sample.
Immunoblotting
After CS stimulation, cells were lysed, and protein extracts were separated by SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membrane, and probed with specific antibodies. Equal protein loading was verified by reprobing membranes with antibody to β-actin or specific protein of interest. The relative intensities of immunoreactive protein bands (relative density units) were analyzed and quantified by scanning densitometry using Image Quant software (Molecular Dynamics, Sunnyvale, CA, USA).
Quantitative RT-PCR
Reverse transcription was performed with 1 μg of total RNA isolated from EC. RT-PCR reactions were performed as described previously (38). The following human GEF-H1 gene specific primers were used: forward, 5′-ACACGCTTCCTCAGCCAGCTATTA-3′; reverse, 5′-AATTGCTGGAAGCGTTTGTCTCGG-3′. For the normalization, commercially available β2-microglobulin (39) primers were used.
Mechanical ventilation protocol
All experimental protocols involving the use of animals were approved by the University of Chicago Institutional Animal Care and Use Committee for the humane treatment of experimental animals. C57BL/6J mice (8–10 wk old, males) with average weight 20–25 g (Jackson Laboratories, Bar Harbor, ME, USA) were anesthetized and subjected to mechanical ventilation at HTV (30 ml/kg) as described previously (30). Homogenized tissue was used for Western blot analysis.
Statistical analysis
The data are expressed as means ± sd. Three to 6 independent experiments were performed for each type of assay performed in this study. Experimental samples were compared with controls by unpaired Student's t test. For multiple-group comparisons, a 1-way ANOVA and post hoc multiple comparisons tests were used. Values of P < 0.05 were considered statistically significant.
RESULTS
CS (18%) activates focal adhesion signaling and Rho pathway of endothelial permeability
Direct measurement of permeability was performed using a novel assay recently developed by our group (32). Cell monolayers were grown on Flex plates coated with biotinylated collagen. Application of 18% CS induced rapid permeability increase detected after 15 min of CS exposure by increased mobilization of FITC-labeled avidin on the biotinylated collagen-coated BioFlex plate bottom monitored by increased fluorescence of culture plate elastic bottom (Fig. 1A). Application of 18% CS to EC monolayers induced rapid phosphorylation of Src at Y416 reflecting its activation. 18% CS also induced FAK phosphorylation at Y397 and Y576, p42/44 MAP kinase at T202/Y204, and paxillin at Tyr118 (Fig. 1B). Activation of FA signaling observed in CS-stimulated EC culture was also observed in vivo. Increased levels of phosphorylated paxillin, FAK, p42/44 MAPK, and myosin light chains (MLCs) were observed in the lung samples from mice exposed to HTV mechanical ventilation (30 ml/kg; 4 h; Fig. 1C). It is important to note that HTV induced activation of FAK and p42/44 MAPK signaling kinases and increased phosphorylation of paxillin, and MLCs detected in total lung tissue lysates (Fig. 1C) can be brought about by both ECs and non-ECs. However, the analysis of pulmonary EC culture exposed to 18% CS in vitro (Fig. 1B) demonstrates similar patterns of signaling protein activation, suggesting that these signaling mechanisms are activated in mechanically stimulated lung endothelium.
Figure 1.

Effect of acute CS on vascular permeability and activation of FA signalosome. HPAECs were subjected to 18% CS for 15 min. A) Permeability measurements were performed using FITC-avidin fluorescence XPerT assay. AU, arbitrary units. *P < 0.05 vs. static conditions; n = 6. B) Western blot analysis of CS-induced phosphorylation of Src, FAK, paxillin, and p42/44 MAPK. Probing for total protein was used as normalization control. Results are representative of 3–5 independent experiments. C) Mice were subjected to mechanical ventilation, followed by analysis of p-FAK, p-paxillin, p-p42/44 MAPK, and p-MLC in lung homogenates; n = 4 independent experiments. Bar graphs depict the quantitative densitometry analysis of Western blot data. *P < 0.05 vs. control.
Paxillin mediates stretch-induced activation of GEF-H1, Rho signaling, cytoskeletal remodeling, and EC permeability
Short-term CS used in this study did not affect expression levels of GEF-H1 and RhoA GTPase in EC culture (Fig. 2A, B) but activated of RhoA GTPase and Rho/Rho-kinase-dependent phosphorylation of MLC phosphatase (MYPT) and regulatory MLC (Fig. 2C, D). These signaling events are known to trigger Rho-dependent cytoskeletal remodeling and disruption of EC monolayer integrity (9). Indeed, 18% CS induced rapid EC monolayer remodeling characterized by formation of actin stress fibers and paracellular gaps (Fig. 2E), and increased accumulation of phosphorylated MLC on stress fibers (Fig. 2F), which reflects assembly and activation of actomyosin contractile complex (35). This CS-induced cytoskeletal activation was abolished by siRNA-induced knockdown of paxillin. The effects of paxillin knockdown on CS-induced EC permeability were tested using permeability assay described above. Paxillin knockdown significantly attenuated CS-induced permeability increase (Fig. 3A).
Figure 2.

Effect of acute CS on activation of Rho signaling. HPAECs were subjected to 18% CS for 15 min. A) GEF-H1 expression was analyzed by qRT-PCR. B) GEF-H1 and RhoA expression was analyzed by qRT-PCR Western blotting. C) CS-induced Rho activation was assessed by RhoGTP pulldown assay. D) Western blot analysis of CS-induced phosphorylation of MYPT and MLC. Probing for total protein was used as normalization control, n = 4 independent experiments. Bar graphs depict the quantitative densitometry analysis of Western blot data. *P < 0.05 vs. control. E, F) Human pulmonary ECs treated with paxillin-specific or nonspecific siRNA (nsRNA) were subjected to 18% CS for 15 min. E) F-actin remodeling in control and stretched EC monolayers was analyzed by immunofluorescence staining for F-actin. F) Intracellular localization of phosphorylated MLC was determined by immunofluorescence staining with phospho-MLC antibody.
Figure 3.

Role of paxillin in CS-induced EC permeability. Human pulmonary ECs were transfected with paxillin-specific or nonspecific siRNA. A) Permeability measurements of EC monolayers exposed to 18% CS, 15 min, were performed using FITC-avidin fluorescence XPerT assay. *P < 0.05 vs. nonspecific RNA; n = 6. B) GEF-H1 activation was evaluated by GEF pulldown assay as described in Materials and Methods. Western blot detection of GEF-H1 in corresponding total lysates was used as normalization control. RDU, relative density units; WCL, whole cell lysate. C) Rho activation was evaluated by Rho GTP pulldown assay. D) MLC phosphorylation was analyzed by Western blotting with corresponding antibody. siRNA-induced paxillin protein depletion was confirmed by Western blot. Probing for β-actin was used as normalization control. *P < 0.05 vs. nsRNA; n = 4.
Previous studies by our and other groups suggested a role of GEF-H1 in the mechanism of RhoA activation by mechanical forces (20, 30, 40). Direct activation of GEF-H1 was evaluated in a pulldown assay with a RhoA(G17A) mutant that cannot bind nucleotide and therefore has high affinity for activated GEFs (37). CS (18%) caused GEF-H1 activation that was attenuated by paxillin depletion (Fig. 3B). The inhibitory effect of paxillin depletion on 18% CS-induced EC barrier dysfunction and GEF-H1 activation was also accompanied by attenuation of CS-induced Rho activation and MLC phosphorylation (Fig. 3C, D).
CS (18%) induces functional interactions between paxillin, p42/44 MAPK, and GEF-H1
The role of the FA complex in the mechanism of GEF-H1 regulation by CS remains poorly understood. Application of 18% CS induced rapid GEF-H1 association with paxillin and p42/44 MAPK detected in HPAECs and HLMVECs using coimmunoprecipitation assays (Fig. 4A). CS-induced formation of a paxillin-GEF-H1-p42/44 MAPK complex was further enhanced by EC stimulation with the Rho-activating agonist thrombin. This activation of p44-MAPK was essential for paxillin-GEF-H1 interaction, since an upstream inhibitor of p42/44-MAPK activation, UO126, abolished CS-induced formation of this paxillin-GEF-H1-p42/44-MAPK complex (Fig. 4B). In accord with the role of p42/44 MAPK in CS-induced formation of the Rho activating signaling complex at FA, pretreatment with UO126 attenuated 18% CS-induced rapid gap formation in pulmonary EC (Fig. 4C).
Figure 4.

Effect of 18%CS on paxillin-GEF-H1 interactions. Pulmonary macrovascular or microvscular ECs were subjected to 18% CS for 15 min, followed by immunoprecipitation (IP) of paxillin. Detection of coimmunporecipitated GEF-H1 and p44/42 MAPK was performed by Western blot. A) Cells were treated with thrombin (0.2 U/ml) in the last 5 min of CS exposure. B) Cells were pretreated with U0126 (5 μM) for 30 min before CS stimulation. Reprobing with paxillin antibody was used as normalization control. Nonimmune IgG was used as negative control in immunoprecipitation assays. Bar graphs depict the quantitative analysis of Western blot densitometry data. *P < 0.05 vs. static conditions, **P < 0.05 vs. vehicle; n = 5. C) Cytoskeletal remodeling in control and stretched EC monolayers with our without U0126 preconditioning was analyzed by immunofluorescence staining for F-actin. Arrows indicated paracellular gaps. Results are representative of 3 independent experiments.
CS-induced activation of GEF-H1 is mediated by p42/44 MAPK
The role of p44/42 MAPK in CS-induced GEF-H1 activation was directly evaluated in pulldown assay with RhoA(G17A) mutant, as described above. CS (18%) increased GEF-H1 activity, while EC pretreatment with UO126 abolished this effect (Fig. 5A). Stimulation of GEF-H1 nucleotide exchange activity by p44/42 MAPK involves GEF-H1 phosphorylation at T678 (41). Using immunoprecipitation under denaturing conditions, we tested threonine phosphorylation of GEF-H1 using phosphothreonine antibody and observed increased phosphothreonine immunoreactivity in CS-stimulated ECs, which was abolished by pretreatment with upstream p44/42 MAPK inhibitor UO126 (Fig. 5B). The effect of site-specific GEF-H1 phosphorylation at T678 mediated by p44/42 MAPK was further tested in EC overexpressing GEF-H1 phosphorylation-deficient mutant at this site, GEF-H1-T678A. In contrast to cells transfected with the wild-type GEF-H1, overexpression of GEF-H1-T678A attenuated CS-induced Rho activation (Fig. 5C) and phosphorylation of downstream Rho effector MLC (Fig. 5D). Of note, expression of GEF-H1 mutant did not affect the phosphorylation levels of p42/44 MAPK. These results suggest a GEF-H1-independent mechanism of p42/44 MAPK activation by CS.
Figure 5.

Role of p44/42 MAPK mechanism in CS-induced GEF-H1 activation. A, B) Pulmonary ECs were pretreated with U0126 (5 μM, 30 min) before CS stimulation. A) GEF-H1 activation was evaluated by GEF pulldown assay. Western blot detection of GEF-H1 in corresponding total lysates was used as normalization control. B) GEF-H1 was immunoprecipitated under denaturing conditions, followed by probing with phosphothreonine-specific antibody and reprobing with GEF-H1 antibody. C, D) Cells were transfected with GEF-H1 wild-type (GEF-H1-FL) or phosphorylation-deficient mutant (GEF-H1T678A), followed by 18% CS, 15 min. C) Rho activation was evaluated by RhoGTP pulldown assay, D) MLC and p42/44 MAPK phosphorylation was analyzed by Western blotting with corresponding antibody. GEF-H1 transfection efficiency was verified by probing with GFP tag antibody. Bar graphs depict the quantitative analysis of Western blot densitometry data. *P < 0.05; n = 5.
Role of paxillin tyrosine phosphorylation in CS-induced recruitment of GEF-H1 and increased MLC phosphorylation
The association of paxillin and GEF-H1 following CS was attenuated by cell pretreatment with PP2, a Src family inhibitor which inhibits CS-induced paxillin tyrosine phosphorylation (43, 44; and data not shown (Fig. 6A). Paxillin signaling and interaction with binding partners is critically dependent on phosphorylation status of its tyrosine residues Y31 and Y118 (21). Expression of phosphorylation-deficient Pxn-Y31/118F in human pulmonary EC abolished CS-induced association of GEF-H1 with this paxillin mutant, which was observed in Pxn-Y31/118F pulldown assays using GFP-tag antibody (Fig. 6B).
Figure 6.

Role of paxillin phosphorylation in CS-induced paxillin-GEF-H1 association. A) Pulmonary ECs were pretreated with PP2 (2 μM) for 30 min before CS followed by immunoprecipitation with paxillin antibody and Western blot detection of GEF-H1 in immunoprecipitates. Equal loading was confirmed by reprobing with paxillin antibody. B) Paxillin wild-type (Pxn-WT) or phosphorylation-deficient mutant (Pxn-Y31/118F) bearing EGFP-tags were overexpressed in pulmonary ECs followed by 18% CS for 15 min. Paxillin-bound GEF-H1 was determined in GFP immunoprecipitates. Bar graphs depict the quantitative analysis of Western blot densitometry data. *P < 0.05; n = 3.
Decreased GEF-H1 targeting to paxillin in cells expressing Pxn-Y31/118F mutant attenuated CS-induced activation of GEF-H1 (Fig. 7A) and phosphorylation of Rho/Rho-kinase downstream targets MYPT and MLC (Fig. 7B). Although overexpression of Pxn-Y31/118F attenuated CS-induced MLC phosphorylation, it did not affect CS-induced activation of p44/42 MAPK (Fig. 7B), suggesting a paxillin-independent mechanism of p44/42 MAPK activation by 18% CS. Measurements of EC permeability using the XPerT assay showed that expression of Pxn-Y31/118F attenuated EC permeability induced by 18% CS (Fig. 7C). Altogether, these results demonstrate a role of site-specific paxillin tyrosine phosphorylation in CS-induced targeting of GEF-H1 and activation of Rho signaling.
Figure 7.

Role of paxillin phosphorylation at Tyr31 an Tyr118 in CS-induced EC permeability. Human pulmonary ECs were transfected with wild-type (Pxn-FL) or phosphorylation-deficient mutant (Pxn-Y31/118F). A) GEF-H1 activation was evaluated by GEF pulldown assay. Western blot detection of GEF-H1 in corresponding total lysates was used as normalization control. B) Phosphorylation of MYPT, MLC, and p44/42 MAPK was evaluated by Western blotting with corresponding phospho-antibody. Probing for β-actin was used as normalization control. Bar graphs depict the quantitative analysis of Western blot densitometry data. *P < 0.05 vs. paxillin wild type; n = 3. C) Permeability measurements of EC monolayers exposed to 18% CS, 15 min, were performed using FITC-avidin fluorescence XPerT assay. *P < 0.05 vs. paxillin wild type; n = 5.
DISCUSSION
Mechanical ventilation, even in the range of barrier protective amplitudes, remains a factor contributing to increased lung vascular leak and preexisting lung injury. The pathological role of Rho signaling in mechanically challenged cells and animal models of ventilator-induced lung injury (VILI) is well recognized (14, 15, 30, 42–44), and Rho inhibition has been considered as potential approach to alleviate lung injury. However, precise mechanisms of Rho activity regulation by excessive mechanical forces remain to be elucidated.
Mechanical strain induces activation of nonreceptor tyrosine kinases p60Src and FAK (45, 46) and integrin-mediated signaling (13, 47–51) and activates phosphorylation of FA protein, paxillin (52). These events are important for CS-induced cytoskeletal response. In addition, mechanical forces stmiulate MAP kinases, including p42/44 MAPK (also known as Erk-1,2), Erk-5, JNK, and p38-MAPK (1, 53, 54). This study shows that 18% CS-induced EC permeability was associated with paxillin-p42/44 MAPK-GEF-H1 complex formation. In turn, pharmacologic inhibition of p42/44 MAPK activity by UO126 abolished CS-induced association of paxillin, p42/44 MAPK, and GEF-H1, suggesting that p42/44 MAPK is critical for this process. Therefore, this study focused on evaluation of the p42/44 MAPK role, although potential association of other MAP kinases with this complex is an interesting possibility that deserves further investigation.
EC exposure to 18% CS activates Src kinase and induces FAK autophosphorylation at Y397 and Src-dependent FAK phosphorylation at Y576 and Y577. FAK phosphorylation at these sites is required for full activation of FAK (55). Both FAK and Src mediate paxillin phosphorylation at Y31 and Y118, which creates binding sites for SH2 domain-containing signaling proteins (21, 56). Src-dependent phosphorylation of paxillin at Y31 and Y118 was essential for CS-induced paxillin interaction with GEF-H1 and p42/44 MAPK (Fig. 7A, B) and stimulation of GEF-H1 activity (Fig. 8A, B). CS-induced targeting of GEF-H1 to the paxillin complex was also critical for CS-induced activation of Rho signaling, MLC phosphorylation, and increases in endothelial permeability.
Figure 8.
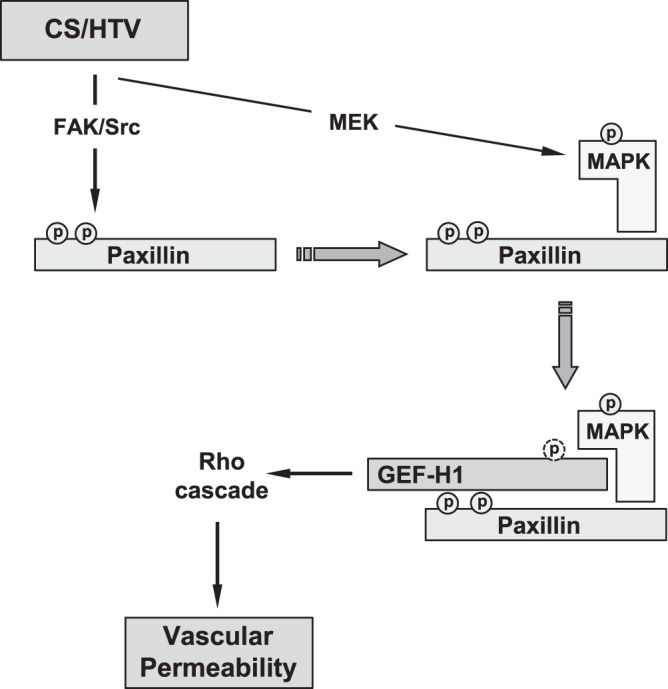
Proposed model of stretch-induced Rho signaling regulation at focal adhesions. HTV-relevant cyclic stretch activates Src/FAK-dependent phosphorylation of paxillin at Tyr31 and Tyr118 and MEK-dependent phosphorylation/activation of p44/42 MAPK and MAPK association with paxillin. Paxillin–p44/42 MAPK then recruits and activates GEF-H1 via p44-MAPK-mediated Thr678 phosphorylation. CS-induced GEF-H1–paxillin association requires paxillin LD4 domain. As a result, stretch-induced formation of paxillin/p44/42-MAPK/GEF-H1 signalosome stimulates local Rho signaling, leading to activation of MLC phosphorylation, actomyosin contraction, and increased endothelial permeability.
Our study demonstrates the p42/44 MAPK-dependent mechanism of CS-induced activation of GEF-H1 leading to stimulation of Rho signaling and EC barrier disruption. This pathway employs a recently described mechanism of GEF-H1 activation by MAPK-mediated phosphorylation at T678 (41). In agreement with this mechanism, expression of GEF-H1T678A mutant lacking p42/44MAPK phosphorylation site attenuated CS-induced Rho activation (Fig. 6C) and inhibited Rho-dependent phosphorylation of MYPT and MLC (Fig. 6D). Unexpectedly, our experiments showed that p44/42 MAPK activity was also required for assembly of a paxillin-GEF-H1-p44/42 MAPK complex, suggesting a novel role for p44/42 MAPK in the local regulation of GEF-H1 function at FA. These data and experiments with paxillin Y31/118 phosphorylation-deficient mutant demonstrate that GEF-H1 recruitment to paxillin complex requires FAK/Src-dependent phosphorylation of paxillin at Y31 and Y118 and p44/42 MAPK activity. Does the interaction with paxillin promote GEF-H1 activation at FA or does it serve as a GEF-H1 deactivation mechanism? Our data show that CS-induced formation of paxillin-GEF-H1 complex is linked to CS-induced activation of GEF-H1 and Rho signaling.
Based on our results and published studies, we propose a model of stretch-induced regulation of Rho signaling at focal adhesions (Fig. 8). Pathological CS associated with suboptimal lung mechanical ventilation activates Src and FAK leading to paxillin phosphorylation at Y31 and Y118, which creates binding sites for GEF-H1-p44/42 MAPK complex. In turn, CS-induced activation of p42/44 MAPK independent on paxillin phosphorylation provides additional signal to formation of paxillin-p44/42-MAPK-GEF-H1 complex at focal adhesions. CS-induced p44/42-MAPK activation is necessary for GEF-H1 binding to paxillin. CS-activated p44/42-MAPK also phosphorylates GEF-H1 at T678 and triggers GEF-H1 nucleotide exchange activity toward RhoA. As a result, stretch-induced formation of paxillin-p44/42-MAPK-GEF-H1 signalosome stimulates local Rho signaling leading to activation of MLC phosphorylation, actomyosin contraction and increased endothelial permeability. This mechanism contributes to vascular barrier dysfunction in organs and systems experiencing excessive mechanical forces. Investigations into the mechanism of mechanoactivated Rho signaling in ALI may have a substantial effect on the drug design strategies toward generation of “mechanoselective” inhibitors of Rho pathway. Thus, the results of this study suggest GEF-H1 as potential drug target for pharmacological interventions to prevent vascular barrier dysfunction induced by pathological mechanical environment.
Acknowledgments
The authors thank C. Turner (State University of New York, Syracuse, NY, USA) for providing paxillin constructs; G. Bokoch (Scripps, La Jolla, CA, USA) and K. Szaszi (St. Michael's Hospital, Toronto, ON, Canada) for providing GEF-H1 constructs and RhoA(G17A) mutant.
This work was supported by Public Health Service grants HL-76259 and HL1-07920 from the National Heart, Lung, and Blood Institute, U.S. National Institutes of Health.
Footnotes
- ARDS
- acute respiratory distress syndrome
- CS
- cyclic stretch
- EC
- endothelial cell
- FA
- focal adhesion
- FAK
- focal adhesion kinase
- GEF
- guanine nucleotide exchange factor
- HLMVEC
- human lung microvascular endothelial cell
- HPAEC
- human pulmonary artery endothelial cell
- HTV
- high tidal volume
- MLC
- myosin light chain
- MYPT
- myosin light-chain phosphatase
- XPerT
- express micromolecule permeability testing
REFERENCES
- 1. Birukov K. G., Jacobson J. R., Flores A. A., Ye S. Q., Birukova A. A., Verin A. D., Garcia J. G. (2003) Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L785–L797 [DOI] [PubMed] [Google Scholar]
- 2. Jafari B., Ouyang B., Li L. F., Hales C. A., Quinn D. A. (2004) Intracellular glutathione in stretch-induced cytokine release from alveolar type-2 like cells. Respirology 9, 43–53 [DOI] [PubMed] [Google Scholar]
- 3. Yang G., Im H. J., Wang J. H. (2005) Repetitive mechanical stretching modulates IL-1beta induced COX-2, MMP-1 expression, and PGE2 production in human patellar tendon fibroblasts. Gene 363, 166–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanchez-Esteban J., Wang Y., Cicchiello L. A., Rubin L. P. (2002) Cyclic mechanical stretch inhibits cell proliferation and induces apoptosis in fetal rat lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L448–L456 [DOI] [PubMed] [Google Scholar]
- 5. Pimentel D. R., Amin J. K., Xiao L., Miller T., Viereck J., Oliver-Krasinski J., Baliga R., Wang J., Siwik D. A., Singh K., Pagano P., Colucci W. S., Sawyer D. B. (2001) Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ. Res. 89, 453–460 [DOI] [PubMed] [Google Scholar]
- 6. Hammerschmidt S., Kuhn H., Grasenack T., Gessner C., Wirtz H. (2004) Apoptosis and necrosis induced by cyclic mechanical stretching in alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 30, 396–402 [DOI] [PubMed] [Google Scholar]
- 7. Matthay M. A., Zimmerman G. A., Esmon C., Bhattacharya J., Coller B., Doerschuk C. M., Floros J., Gimbrone M. A., Jr., Hoffman E., Hubmayr R. D., Leppert M., Matalon S., Munford R., Parsons P., Slutsky A. S., Tracey K. J., Ward P., Gail D. B., Harabin A. L. (2003) Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am. J. Respir. Crit. Care. Med. 167, 1027–1035 [DOI] [PubMed] [Google Scholar]
- 8. Uhlig S. (2002) Ventilation-induced lung injury and mechanotransduction: stretching it too far? Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L892–L896 [DOI] [PubMed] [Google Scholar]
- 9. Dudek S. M., Garcia J. G. (2001) Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 91, 1487–1500 [DOI] [PubMed] [Google Scholar]
- 10. Mehta D., Malik A. B. (2006) Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367 [DOI] [PubMed] [Google Scholar]
- 11. Geiger B., Bershadsky A. (2001) Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13, 584–592 [DOI] [PubMed] [Google Scholar]
- 12. Orr A. W., Helmke B. P., Blackman B. R., Schwartz M. A. (2006) Mechanisms of mechanotransduction. Dev. Cell 10, 11–20 [DOI] [PubMed] [Google Scholar]
- 13. Schwartz M. A., Desimone D. W. (2008) Cell adhesion receptors in mechanotransduction. Curr. Opin. Cell Biol. 20, 551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Birukova A. A., Chatchavalvanich S., Rios A., Kawkitinarong K., Garcia J. G., Birukov K. G. (2006) Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am. J. Pathol. 168, 1749–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nonas S., Birukova A. A., Fu P., Xing J., Chatchavalvanich S., Bochkov V. N., Leitinger N., Garcia J. G., Birukov K. G. (2008) Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit. Care 12, R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jenkins R. G., Su X., Su G., Scotton C. J., Camerer E., Laurent G. J., Davis G. E., Chambers R. C., Matthay M. A., Sheppard D. (2006) Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J. Clin. Invest. 116, 1606–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Singleton P. A., Moreno-Vinasco L., Sammani S., Wanderling S. L., Moss J., Garcia J. G. (2007) Attenuation of vascular permeability by methylnaltrexone: role of mOP-R and S1P3 transactivation. Am. J. Respir. Cell Mol. Biol. 37, 222–231 [DOI] [PubMed] [Google Scholar]
- 18. Xu M. Y., Porte J., Knox A. J., Weinreb P. H., Maher T. M., Violette S. M., McAnulty R. J., Sheppard D., Jenkins G. (2009) Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am. J. Pathol. 174, 1264–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bershadsky A. D., Balaban N. Q., Geiger B. (2003) Adhesion-dependent cell mechanosensitivity. Annu. Rev. Cell Dev. Biol. 19, 677–695 [DOI] [PubMed] [Google Scholar]
- 20. Guilluy C., Swaminathan V., Garcia-Mata R., O'Brien E. T., Superfine R., Burridge K. (2011) The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 13, 722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown M. C., Turner C. E. (2004) Paxillin: adapting to change. Physiol. Rev. 84, 1315–1339 [DOI] [PubMed] [Google Scholar]
- 22. Birukov K. G., Birukova A. A., Dudek S. M., Verin A. D., Crow M. T., Zhan X., DePaola N., Garcia J. G. (2002) Shear stress-mediated cytoskeletal remodeling and cortactin translocation in pulmonary endothelial cells. Am. J. Respir. Cell Mol. Biol. 26, 453–464 [DOI] [PubMed] [Google Scholar]
- 23. Shikata Y., Birukov K. G., Birukova A. A., Verin A. D., Garcia J. G. (2003) Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J. 17, 2240–2249 [DOI] [PubMed] [Google Scholar]
- 24. Shikata Y., Rios A., Kawkitinarong K., DePaola N., Garcia J. G., Birukov K. G. (2005) Differential effects of shear stress and cyclic stretch on focal adhesion remodeling, site-specific FAK phosphorylation, and small GTPases in human lung endothelial cells. Exp. Cell Res. 304, 40–49 [DOI] [PubMed] [Google Scholar]
- 25. Mehta D., Tiruppathi C., Sandoval R., Minshall R. D., Holinstat M., Malik A. B. (2002) Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J. Physiol. 539, 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holinstat M., Knezevic N., Broman M., Samarel A. M., Malik A. B., Mehta D. (2006) Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J. Biol. Chem. 281, 2296–2305 [DOI] [PubMed] [Google Scholar]
- 27. Ren Y., Li R., Zheng Y., Busch H. (1998) Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 273, 34954–34960 [DOI] [PubMed] [Google Scholar]
- 28. Aijaz S., D'Atri F., Citi S., Balda M. S., Matter K. (2005) Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev. Cell. 8, 777–786 [DOI] [PubMed] [Google Scholar]
- 29. Ly D. L., Waheed F., Lodyga M., Speight P., Masszi A., Nakano H., Hersom M., Pedersen S. F., Szaszi K., Kapus A. (2013) Hyperosmotic stress regulates the distribution and stability of myocardin-related transcription factor, a key modulator of the cytoskeleton. Am. J. Physiol. Cell Physiol. 304, C115–C127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Birukova A. A., Fu P., Xing J., Yakubov B., Cokic I., Birukov K. G. (2010) Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L837–L848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Birukova A. A., Zebda N., Cokic I., Fu P., Wu T., Dubrovskyi O., Birukov K. G. (2011) p190RhoGAP mediates protective effects of oxidized phospholipids in the models of ventilator-induced lung injury. Exp. Cell Res. 317, 859–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dubrovskyi O., Birukova A. A., Birukov K. G. (2013) Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab. Invest. 93, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tschumperlin D. J., Margulies S. S. (1999) Alveolar epithelial surface area-volume relationship in isolated rat lungs. J. Appl. Physiol. 86, 2026–2033 [DOI] [PubMed] [Google Scholar]
- 34. Birukova A. A., Malyukova I., Poroyko V., Birukov K. G. (2007) Paxillin-β-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L199–L211 [DOI] [PubMed] [Google Scholar]
- 35. Birukova A. A., Smurova K., Birukov K. G., Kaibuchi K., Garcia J. G. N., Verin A. D. (2004) Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc. Res. 67, 64–77 [DOI] [PubMed] [Google Scholar]
- 36. Kakiashvili E., Speight P., Waheed F., Seth R., Lodyga M., Tanimura S., Kohno M., Rotstein O. D., Kapus A., Szaszi K. (2009) GEF-H1 mediates tumor necrosis factor-alpha-induced Rho activation and myosin phosphorylation: role in the regulation of tubular paracellular permeability. J. Biol. Chem. 284, 11454–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garcia-Mata R., Wennerberg K., Arthur W. T., Noren N. K., Ellerbroek S. M., Burridge K. (2006) Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 406, 425–437 [DOI] [PubMed] [Google Scholar]
- 38. Birukova A. A., Xing J., Fu P., Yakubov B., Dubrovskyi O., Fortune J. A., Klibanov A. M., Birukov K. G. (2010) Atrial natriuretic peptide attenuates LPS-induced lung vascular leak: role of PAK1. Am. J. Physiol. Lung Cell. Mol. Physiol. 299, L652–L663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmittgen T. D., Zakrajsek B. A. (2000) Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J. Biochem. Biophys. Methods 46, 69–81 [DOI] [PubMed] [Google Scholar]
- 40. Heck J. N., Ponik S. M., Garcia-Mendoza M. G., Pehlke C. A., Inman D. R., Eliceiri K. W., Keely P. J. (2012) Microtubules regulate GEF-H1 in response to extracellular matrix stiffness. Mol. Biol. Cell 23, 2583–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Waheed F., Dan Q., Amoozadeh Y., Zhang Y., Tanimura S., Speight P., Kapus A., Szaszi K. (2013) Central role of the exchange factor GEF-H1 in TNF-alpha-induced sequential activation of Rac, ADAM17/TACE, and RhoA in tubular epithelial cells. Mol. Biol. Cell 24, 1068–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Desai L. P., Sinclair S. E., Chapman K. E., Hassid A., Waters C. M. (2007) High tidal volume mechanical ventilation with hyperoxia alters alveolar type II cell adhesion. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L769–L778 [DOI] [PubMed] [Google Scholar]
- 43. Dipaolo B. C., Margulies S. S. (2012) Rho kinase signaling pathways during stretch in primary alveolar epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L992–L1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Su X., Robriquet L., Folkesson H. G., Matthay M. A. (2006) Protective effect of endogenous beta-adrenergic tone on lung fluid balance in acute bacterial pneumonia in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L769–L776 [DOI] [PubMed] [Google Scholar]
- 45. Naruse K., Yamada T., Sai X. R., Hamaguchi M., Sokabe M. (1998) Pp125FAK is required for stretch dependent morphological response of endothelial cells. Oncogene 17, 455–463 [DOI] [PubMed] [Google Scholar]
- 46. Liu M., Qin Y., Liu J., Tanswell A. K., Post M. (1996) Mechanical strain induces pp60src activation and translocation to cytoskeleton in fetal rat lung cells. J. Biol. Chem. 271, 7066–7071 [PubMed] [Google Scholar]
- 47. Chen K. D., Li Y. S., Kim M., Li S., Yuan S., Chien S., Shyy J. Y. (1999) Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J. Biol. Chem. 274, 18393–18400 [DOI] [PubMed] [Google Scholar]
- 48. Ikeda M., Kito H., Sumpio B. E. (1999) Phosphatidylinositol-3 kinase dependent MAP kinase activation via p21ras in endothelial cells exposed to cyclic strain. Biochem. Biophys. Res. Commun. 257, 668–671 [DOI] [PubMed] [Google Scholar]
- 49. Ingram A. J., James L., Ly H., Thai K., Scholey J. W. (2000) Stretch activation of jun N-terminal kinase/stress-activated protein kinase in mesangial cells. Kidney Int. 58, 1431–1439 [DOI] [PubMed] [Google Scholar]
- 50. Kano Y., Katoh K., Fujiwara K. (2000) Lateral zone of cell-cell adhesion as the major fluid shear stress- related signal transduction site. Circ. Res. 86, 425–433 [DOI] [PubMed] [Google Scholar]
- 51. Naruse K., Sai X., Yokoyama N., Sokabe M. (1998) Uni-axial cyclic stretch induces c-src activation and translocation in human endothelial cells via SA channel activation. FEBS Lett. 441, 111–115 [DOI] [PubMed] [Google Scholar]
- 52. Smith P. G., Garcia R., Kogerman L. (1998) Mechanical strain increases protein tyrosine phosphorylation in airway smooth muscle cells. Exp. Cell Res. 239, 353–360 [DOI] [PubMed] [Google Scholar]
- 53. Abdulnour R. E., Peng X., Finigan J. H., Han E. J., Hasan E. J., Birukov K. G., Reddy S. P., Watkins J. E., 3rd, Kayyali U. S., Garcia J. G., Tuder R. M., Hassoun P. M. (2006) Mechanical stress activates xanthine oxidoreductase through MAP kinase-dependent pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L345–L353 [DOI] [PubMed] [Google Scholar]
- 54. Kito H., Chen E. L., Wang X., Ikeda M., Azuma N., Nakajima N., Gahtan V., Sumpio B. E. (2000) Role of mitogen-activated protein kinases in pulmonary endothelial cells exposed to cyclic strain. J. Appl. Physiol. 89, 2391–2400 [DOI] [PubMed] [Google Scholar]
- 55. Parsons J. T., Martin K. H., Slack J. K., Taylor J. M., Weed S. A. (2000) Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene 19, 5606–5613 [DOI] [PubMed] [Google Scholar]
- 56. Schaller M. D. (2001) Paxillin: a focal adhesion-associated adaptor protein. Oncogene 20, 6459–6472 [DOI] [PubMed] [Google Scholar]