

SUMMARY
We have applied a super-resolution fluorescence imaging method, STochastic Optical Reconstruction Microscopy (STORM), to visualize the structure of functional telomeres and telomeres rendered dysfunctional through removal of shelterin proteins. The STORM images showed that functional telomeres frequently exhibit a t-loop configuration. Conditional deletion of individual components of shelterin showed that TRF2 was required for the formation and/or maintenance of t-loops, whereas deletion of TRF1, Rap1, or the POT1 proteins (POT1a and POT1b) had no effect on the frequency of t-loop occurrence. Within the shelterin complex, TRF2 uniquely serves to protect telomeres from two pathways that are initiated on free DNA ends: classical non-homologous end-joining (NHEJ) and ATM-dependent DNA damage signaling. The TRF2-dependent remodeling of telomeres into t-loop structures, which sequester the ends of chromosomes, can explain why NHEJ and the ATM signaling pathway are repressed when TRF2 is present.
INTRODUCTION
The telomere concept arose from cytological data indicating that natural chromosome ends are resistant to a fusion reaction that joins broken chromosomes (McClintock, 1938; McClintock, 1941). DNA ends of linear plasmids, when introduced into cells, recombine with chromosomal DNA (Orr-Weaver et al., 1981) and double strand breaks (DSBs), induced by genotoxic agents, activate a signaling pathways that can halt cell cycle progression (reviewed in (Callegari and Kelly, 2007)). As the natural ends of chromosomes are stable and do not activate the DNA damage response (DDR), a view has emerged that telomeres have an inherent ability to repress inappropriate DSB repair and DNA damage signaling. How telomeres solve this end-protection problem is a question relevant to understanding telomeropathies and the role of telomere dysfunction in human cancer (reviewed in (Artandi and DePinho, 2010; Savage and Bertuch, 2010)).
Mammalian cells solve the end-protection problem through the agency of shelterin, a multi-subunit protein complex bound to the telomeric TTAGGG repeats (reviewed in (Palm and de Lange, 2008; O'Sullivan and Karlseder, 2010)). Shelterin is anchored on the telomeric DNA by two duplex DNA binding factors, TRF1 and TRF2. These two proteins interact with TIN2, which in turn binds the TPP1-POT1 heterodimer. In the mouse, there are two functionally distinct forms of POT1, POT1a and POT1b. Once tethered to telomeres through this TPP1-TIN2 link, the POT1 proteins bind the single-stranded TTAGGG repeats present at all mammalian chromosome ends in the form of a 50-400 nucleotide (nt) 3’ overhang. An additional member of the shelterin complex, Rap1, associates with TRF2.
Simultaneous deletion of TRF1 and TRF2 from mouse embryo fibroblasts (MEFs) has allowed the creation of telomeres devoid of all shelterin proteins (Sfeir and de Lange, 2012). These shelterin-free telomeres are equivalent to the unprotected DNA ends, whose instability provided the first clues to telomere function. Together with prior data, this telomere deconstruction established that the telomeric DNA at the ends of mouse chromosomes is potentially a substrate for four distinct DSB processing reactions: classical Ku70/80- and DNA ligase 4-dependent NHEJ (c-NHEJ), micro-homology-dependent alternative NHEJ (a-NHEJ) mediated by PARP1 and DNA ligase 3, homology-directed repair (HDR), and CtIP-dependent 5’ end resection. In addition, the shelterin-free telomeres activate DSB signaling by the ATM and ATR kinase pathways. Thus, telomeres require protection from six distinct pathways that together define the telomere end-protection problem in mammalian cells.
Among these six pathways, c-NHEJ and ATM kinase signaling are the purview of TRF2 (Karlseder et al., 1999; van Steensel et al., 1998; Celli and de Lange, 2005; Denchi and de Lange, 2007; Smogorzewska et al., 2002). Deletion of TRF2 results in activation of the ATM kinase cascade at telomeres and very frequent c-NHEJ that generates long trains of chromosomes fused at their telomeres. Deletion of other shelterin components does not produce these phenotypes. Removal of POT1a results in activation of the ATR kinase whereas POT1b loss changes post-replicative processing of the telomere terminus, resulting in extended 3’ overhangs (Denchi and de Lange, 2007; Hockemeyer et al., 2008; Hockemeyer et al., 2006; Wu et al., 2012; Guo et al., 2007). Even at telomeres lacking both POT1a and POT1b or their TPP1 tether, ATM kinase signaling is not elicited and telomere fusions are infrequent (Kibe et al., 2010; Tejera et al., 2010; Denchi and de Lange, 2007). Similarly, deletion of TRF1 does not activate ATM signaling or c-NHEJ, although TRF1 removal compromises the replication of the telomeric DNA and activates the ATR kinase (Sfeir et al., 2009; Martinez et al., 2009). Likewise, telomeres lacking Rap1 do not activate the ATM kinase and remain impervious to NHEJ (Sfeir et al., 2010; Martinez et al., 2010). However, the outcome of TIN2 deletion is complex and includes ATM kinase signaling and c-NHEJ, but this is in part due to the loss of TRF2 from telomeres (Takai et al., 2011).
The mechanism by which TRF2 represses ATM kinase signaling and c-NHEJ has not been established. Electron microscopy (EM) of isolated telomeric DNA has revealed that telomeres can exhibit a t-loop configuration (Griffith et al., 1999). T-loops are lariat structures formed through the invasion of the telomeric 3’ overhang into the double-stranded telomeric repeat array. Because t-loops essentially sequester the terminus of the telomere, it has been proposed that this altered architecture could block DDR reactions that require an accessible DNA end for their activation (de Lange, 2009).
Several in vitro studies have implicated a role for TRF2 in t-loop formation. Recombinant TRF2 can remodel artificial telomeric DNA substrates into a looped configuration in vitro, suggesting that TRF2 has the ability to generate t-loops (Griffith et al., 1999; Stansel et al., 2001). TRF2 also promotes supercoiling in telomeric DNA and induces strand invasion in vitro, possibly by unwinding nearby sequences (Amiard et al., 2007; Poulet et al., 2009; Verdun and Karlseder, 2006). Finally, TRF2 can bind and protect Holliday junctions (HJ) in vitro, even when they are not composed of telomeric DNA (Fouche et al., 2006; Poulet et al., 2009; Nora et al., 2010). Thus, it is possible that TRF2 can promote t-loop formation by mediating strand-invasion of the telomere end and stabilizing the resulting the HJ-like structure. However, whether t-loops form in vivo and, if so, whether TRF2 is involved in their formation or maintenance has not been established.
The t-loop model of TRF2-mediated repression of ATM signaling and NHEJ predicts that deletion of TRF2 will diminish or eliminate t-loops at chromosome ends. In vivo detection of t-loops and quantitative assessment of t-loop frequency have, however, been hampered by technical challenges. Detection of t-loops by EM analysis requires the preparation of protein-free genomic DNA, which has been interstrand crosslinked in order to stabilize the strand-invasion of the 3’ overhang. Because telomeric DNA cannot be distinguished from other genomic DNA in EM images, telomeric sequences have to be enriched. This partial purification is achieved by fragmentation of the genomic DNA with restriction enzymes that spare TTAGGG repeats, followed by purification of the long telomeric fragments on a Biogel sizing column (Griffith et al., 1999). Although the resulting fractions are highly enriched for telomeric DNA, they are contaminated with variable amounts of non-telomeric DNA fragments, introducing uncertainty with regard to the frequency of t-loops. More importantly, the large quantity of genomic DNA required for the purification steps has so far prevented systematic genetic analysis of the proteins required for t-loop formation.
To address whether t-loops occur in cells and whether TRF2 and other shelterin proteins play a role in the formation and/or maintenance of t-loops, we turned to a super-resolution fluorescence imaging method, STORM. STORM relies on stochastic switching and high-precision localization of individual photoswitchable fluorescent probes to achieve imaging with sub-diffraction-limit resolution (Rust et al., 2006). Recent advances in STORM have allowed imaging of cellular structures with near molecular-scale resolution (Huang et al., 2010). Being a fluorescence imaging approach, STORM is compatible with methods that fluorescently label and identify specific DNA sequences. In particular, when combined with fluorescence in situ hybridization (FISH) here, STORM allowed us to image the telomeric DNA in situ without the need of purification, to directly visualize the t-loop structure in chromatin, and to systematically assess the role of shelterin components in t-loop formation. Our results unambiguously demonstrate that, among the shelterin proteins, TRF2 is the main component that is required for the formation/maintenance of t-loops.
RESULTS
STORM imaging of mouse telomeres
To visualize telomeres in fixed mouse cells, we employed a peptide nucleic acid (PNA) FISH probe that was complementary to the TTAGGG repeats and labeled with a photoswitchable dye. We used astigmatism-based three-dimensional (3D) STORM imaging to determine the x, y, and z coordinates of individual probes and reconstruct images with a resolution of ~20 nm in xy directions and ~50 nm in the z direction (Huang et al., 2008b). In conventional images, the telomeres of MEFs detected by FISH appeared as diffraction-limited entities (Fig. 1A). In STORM images, individual telomeres were readily separated and appeared as ovoid signal clusters with a mean diameter of ~180 nm, but with little discernable substructure (Fig. 1A-C). This size estimate for the unperturbed telomeric domains is consistent with the results obtained by immuno-gold detection of telomeric DNA in EM images of interphase cells (Luderus et al., 1996; Pierron and Puvion-Dutilleul, 1999).
Figure 1. STORM imaging of telomeres in intact MEFs and mouse splenocytes.
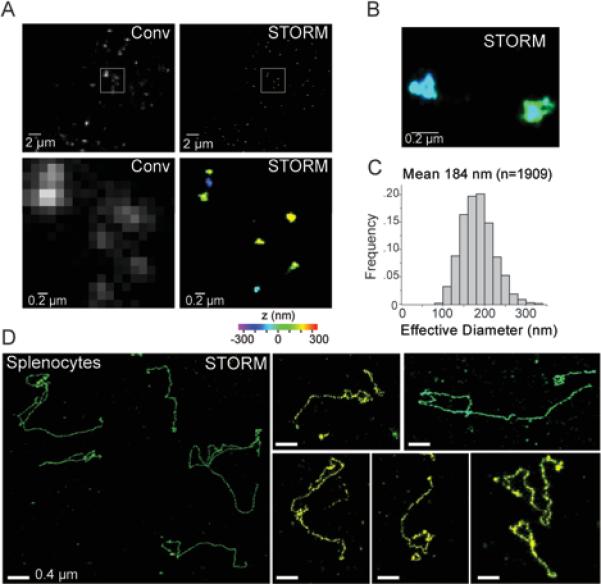
(A) Comparison of conventional (Conv) and 3D-STORM (STORM) images of MEF telomeres detected by FISH. MEFs fixed on coverslips were hybridized with an Alexa 647-labeled [CCCTAA]3 PNA probe. A conventional fluorescence image was taken (left), before the same area was imaged with 3D-STORM (right). The z-coordinates in the 3D STORM images shown here and in subsequent figures are color-coded according to the colored scale bar beneath the STORM image. The lower panels: zoom-in images of the boxed regions in the upper panels.
(B) Enlarged area showing two telomeres imaged by 3D-STORM.
(C) Distribution of the effective diameter of the telomere signals calculated as the diameter of a sphere of equivalent volume.
(D) T-loop like architectures visualized in mouse splenocytes. Left: large field view showing several telomeres. Right: examples of individual telomeres. The images are from a small subset of nuclei that show telomeres in a relaxed configuration after cytocentrifugation.
As mammalian telomeres are known to associate with the nuclear matrix (Luderus et al., 1996; de Lange, 1992), we considered the possibility that disruption of this interaction might relax the telomeric chromatin and allow the visualization of the underlying DNA structure. The telomeres in mouse splenocytes are not bound to the nuclear matrix and telomere-enriched chromatin fragments purified from these cells show lariat structures (Nikitina and Woodcock, 2004). We therefore tested whether splenocytes yield a more open structure of telomeres that can be detected by STORM imaging. Indeed, we occasionally detected extended telomeres in minimally perturbed mouse splenocyte nuclei fixed after cytocentrifugation, which releases chromatin from some of these fragile nuclei. A subset of these ‘relaxed‘ telomeres exhibited a looped configuration (Fig. 1D), suggesting that t-loops indeed exist in vivo. However, as most cells showed telomeres with a compact shape, quantification of the t-loop frequency was difficult. Furthermore, compared to MEFs, mouse splenocytes are suboptimal for dissecting the genetic requirements for t-loop formation.
To better evaluate the architecture of telomeres using STORM, we developed a chromatin spreading protocol of the native chromatin in MEF nuclei, which involved DNA interstrand crosslinking by psoralen/UV treatment. Psoralen crosslinking has been previously shown to stabilize t-loops (Griffith et al., 1999). We then subjected the nuclei to mild detergent lysis before spreading of the chromatin onto glass slides with cytocentrifugation (Fig. 2A). Conventional fluorescence images showed that the bulk DNA detected with YOYO1 was stretched under these conditions, whereas the telomeric DNA detected by the PNA-FISH appeared as discrete dots as well as extended structures (Fig. 2B).
Figure 2. STORM imaging revealing t-loops after chromatin spreading.
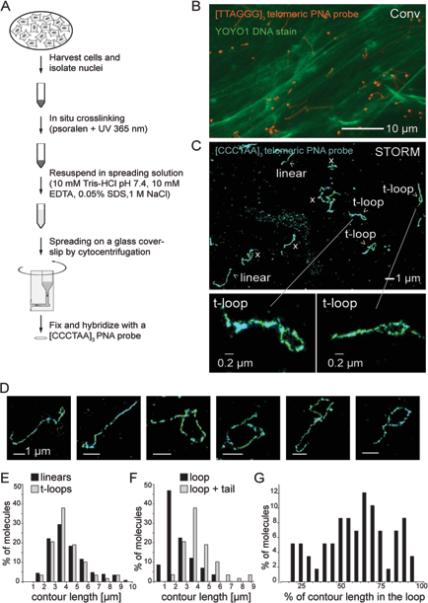
(A) Schematic of the chromatin spreading procedure.
(B) Conventional fluorescence image of a spread sample. A dense layer of decondensed string-like bulk DNA labeled with YOYO-1 (green) and FISH-labeled telomeres (FITC labeled [TTAGGG]3 PNA probe, red) are visible.
(C) 3D-STORM image of t-loops after the chromatin spreading procedure shown in (A) and (B). Linear, t-loop, and ambiguous “x” structures are classified according to the criteria described in the text and Fig. 3. Bottom, two enlarged t-loops.
(D) Examples of t-loops detected as in (C).
(E) Distributions of the total contour lengths of linear telomeric DNAs (n=224) and telomeric DNA exhibiting a t-loop configuration (n=58).
(F) Distribution of the loop portion and the total contour length (loop + tail) of telomeric DNAs in a t-loop configuration (n=58).
(G) Distribution of t-loops based on the relative size of the loop part as a fraction of the total contour length (n=58).
See Fig. S1 for additional t-loop images.
STORM imaging of the spread chromatin revealed telomeric signal tracts in conspicuous looped configurations (Fig. 2C, D; Fig. S1A,B). With the exception of a few large, well-spread loops, most t-loops could not be discerned by conventional fluorescence imaging (Fig. S1A). The t-loops showed variable loop sizes and variable loop-to-tail ratios (Fig. 2E-G), suggesting that there is no preferential position for the strand-invasion point along the double-stranded telomeric DNA. The size distribution of the linear structures was similar to that of the lariat shaped structures, consistent with both linear and lariat structures representing mouse telomeres.
The molecules detected by STORM were not removed upon treatment of the nuclei with RNaseA and RNaseH (9.1±4 FISH tracts > I.0 μm per field before and 12.9±4 after RNaseA/RNaseH treatment; based on images of 38 fields of view per condition, ~40 μm × 40 μm per field of view). Since northern blotting showed that treatment by RNaseA and RNaseH removed greater than 90% of total RNA as well as the telomeric TERRA transcripts (data not shown) (Azzalin et al., 2007; Feuerhahn et al., 2010), the persistence of the FISH signals after the treatment confirms that they represent telomeres rather than RNA.
Based on EM analysis, mouse telomeric restriction fragments are 20±6 kb long, whereas estimates from genomic blotting suggests a size range of 10-50 kb (Griffith et al., 1999; Kipling and Cooke, 1990). Both methods overestimate the length of the telomeric repeat array since the telomeric fragments analyzed contain subtelomeric segments of unknown size. The telomeric lariat structures detected by STORM showed a total contour length ranging from 2-9 μm with an average of 4.0±1.4 μm (Fig. 2E). Assuming ~0.3 μm per kb of double-stranded DNA, these measurements would indicate that mouse telomeres range from 7 to 30 kb with a mean of 13 kb. The STORM images likely underestimate of the length of the telomeric repeat array because some fine wavy patterns in the DNA path may be blurred by the finite resolution of the STORM images and because some of the DNAs might be broken during the spreading procedure or might not be fully spread out.
T-loops are lost upon telomere fusion
If the detected t-loops reflect a native architecture of telomeres, their occurrence should diminish when the telomeres have undergone covalent fusions so that the telomeric DNA is no longer terminal. Telomere fusions generated by c-NHEJ are frequent after Cre-mediated deletion of TRF2 from SV40LT-immortalized TRF2F/−Cre-ERT1 MEFs (Fig. 3A-C). The majority of the telomeres became fused as was evident from the long trains of joined chromosomes in metaphase spreads (Fig. 3B) and from the larger molecular weights of the telomeric restriction fragments (Fig. 3C). The telomere fusion led to a drastic change in the telomeric structure detected by STORM (Fig. 3D). T-loop structures were no longer prominent and nearly all telomeres were present as linear, highly elongated signal tracts. The more elongated shape of the telomeric signal tracts after telomere fusion is likely because the stretching forces exerted on internal telomeric sequences flanked by two chomosomes during cytocentrifugation-induced chromatin spreading were greater than those exerted on terminal telomere of a single chromosome.
Figure 3. The frequency of t-loop occurrence is diminished when telomeres fuse.
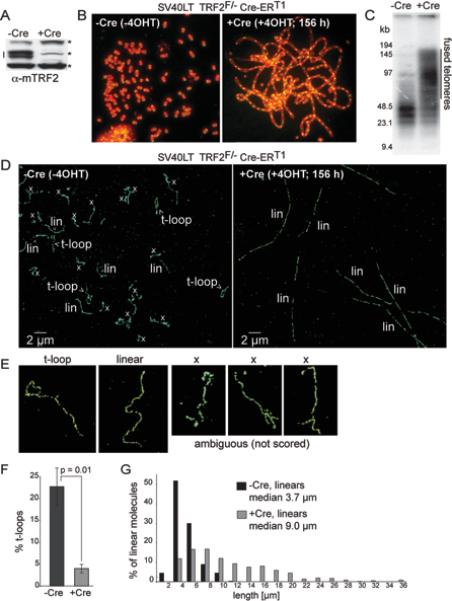
(A) Immunoblot for TRF2 (doublet indicated by the line) in SV40LT TRF2F/−Cre-ERT1 cells treated with 0.5 mM 4-OH tamoxifen (4OHT) and harvested after 156 h (+Cre). *, non-specific bands (loading control).
(B) Metaphase spreads showing telomere fusion before and after TRF2 removal from cells treated as in (A). Green: telomeric FISH with a FITC-conjuaged PNA [CCCTAA]3 probe. Red: DNA stained with DAPI.
(C) Genomic blot for telomeric DNA demonstrating telomere fusions (indicated) after deletion of TRF2. The gel image shows AluI/MboI-digested DNA hybridized with a TTAGGG repeat probe. AluI/MboI restriction enzymes digest non-telomeric genomic DNA and spare the telomeric TTAGGG repeats.
(D) Representative STORM images before and after TRF2 deletion with Cre. T-loops, linear telomere structures (lin), ambiguous molecules (x) are indicated.
(E) Examples of molecules scored as t-loops, linear telomeres, and ambiguous ‘x’ structures.
(F) Percentage of the scored molecules (ambiguous ‘x’ structures excluded) that are in a t-loop configuration. Cells were treated as described in (A) and imaged as in (D). Graphs show mean and standard deviation (SD) values from 3 independent experiments (n≥200 molecules per experiment). P value from unpaired two-tailed Student's t-test.
(G) Length distribution of linear telomeric DNAs detected by STORM imaging in TRF2F/−Cre-ERT1 cells before (-Cre; n=224) and 156 h after (+Cre; n=357) Cre treatment.
See Fig. S2 for t-loop counts with and without exclusion of ‘x’ structures and the results obtained at 72 h after Cre.
To determine the change in the frequency of t-loop appearance after deletion of TRF2, the STORM images were quantified. For unambiguous identification of t-loops, telomeric signal tracks < 1 μm in length were excluded. Furthermore, we only considered molecules that were well spread without any obviously condensed, branched, or knob-like structures throughout their length and that were not entangled with other telomeres (see Fig. 3E for examples of excluded molecules). We also excluded molecules interrupted by gaps longer than 0.5 μm that prevent accurate tracing of the DNA path. Based on these criteria, approximately 60-80% of the FISH signal tracts (marked ‘x’ in the images) were not scored because they cannot be unambiguously classified. Among the 20-40% of the signal tracts that were scored, telomeres were classified as t-loops when containing a single terminal loop with an aperture of at least 0.01 μm2 or as linear structures if they lacked a loop (Fig. 3D, E). Using these criteria, approximately one fourth of the scored telomeres (22.7±4.1%) were in the t-loop configuration before the deletion of TRF2 (Fig. 3F). Because of the stringent scoring criteria, we likely underestimated the frequency of t-loops. For instance, t-loops with a loop smaller than 0.01 μm2 or not readily discernable due to incomplete spreading were not scored, and any breakage of the telomeric molecules during spreading would tend to reduce the frequency of t-loops preferentially.
When TRF2F/−Cre-ERT1 MEFs were induced to express Cre recombinase, which caused TRF2 deletion, and examined after 156 h, the t-loop frequency decreased dramatically and only 4.0±1.0% of the scored telomeres were in the t-loop configuration (Fig. 3F). Many of the linear telomeric tracts ranged from 10 to 30 μm, which were substantially longer than the lengths of the telomere tracts in TRF2-expressing cells and consistent with being products of telomere fusion (Fig. 3G). The 6-fold reduction in t-loop frequency is statistically significant (p=0.01, 3 independent experiments) and consistent with the t-loops being dependent on the functional and terminal state of the telomeric DNA. Similarly, when the cells were examined at 72 h after induction of Cre, the frequency of t-loops dropped by 6 fold, from 36% to 6% (Fig. S2A, B).
The exclusion of the ambiguous ‘x’ structures did not affect the conclusion that TRF2 deletion reduces the frequency of t-loops. Even when all ‘x’ structures were included in the statistics and counted as a separate category from the t-loop and linear structures, deletion of TRF2 still resulted in a substantial and statistically significant reduction in the percentage of t-loops (Fig. S2C, D). It is noteworthy that the percentage of ambiguous ‘x’ molecules dropped from ~80% to ~60% upon deletion of TRF2 (Fig. S2A). A possible reason for this drop is that the ‘x’ fraction of TRF2-proficient cells included t-loops that cannot be unambiguously scored.
TRF2 is required for t-loop formation/maintenance
Having established an assay to detect the presence of t-loops in MEFs, we set out to determine whether TRF2 is important for t-loop formation and/or maintenance. As deletion of TRF2 results in fusion of telomeres, the disappearance of the t-loops under these conditions does not inform on the role of TRF2 in their formation/maintenance. We therefore analyzed the effect of TRF2 removal in ATM-deficient setting where telomere fusions are rare. ATM signaling is required for the c-NHEJ of telomeres, in part because the ATM target 53BP1 prevents resection at deprotected telomeres and increases their mobility (Lottersberger et al., 2013; Zimmermann et al., 2013; Denchi and de Lange, 2007; Dimitrova et al., 2008; Chapman et al., 2013). As a result, ATM-deficient cells show a ~100-fold reduction in telomere fusions after TRF2 deletion as compared to ATM-proficient cells (Denchi and de Lange, 2007) (Fig. 4A-C). In addition, the use of TRF2F/−ATM−/− cells is advantageous because there is no DNA damage signal emanating from the TRF2-depleted telomeres (Denchi and de Lange, 2007), thus removing potentially confounding effects of chromatin alterations associated with the DNA damage response.
Figure 4. TRF2 is required for the formation and/or maintenance of t-loops.
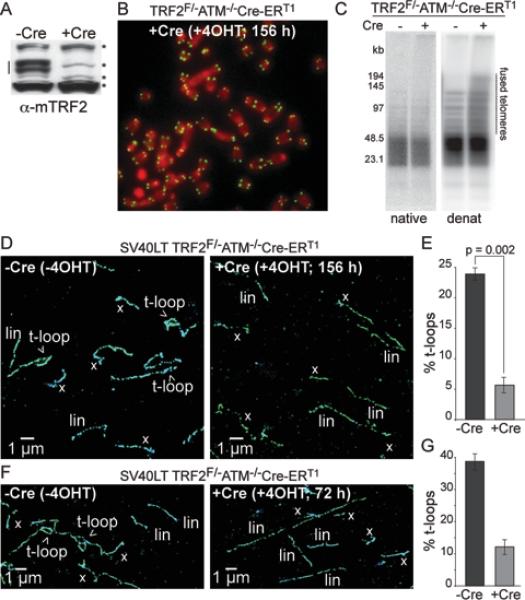
(A) Immunoblot for TRF2 (line) in SV40LT TRF2F/−ATM−/−Cre-ERT1 cells before (-Cre) and after (+Cre) 4OHT treatment (156 h). *, non-specific bands (loading control).
(B) Representative metaphase spread of cells as in (A).
(C) In-gel assay for the telomeric 3’ overhang (native) and total telomeric DNA analysis (denat). Note minimal telomere fusions and telomeric overhang loss (<30%) after TRF2 deletion from ATM−/− cells.
(D, F) Representative STORM images of telomeres in cells as in (A) or at 72 h after induction of Cre with 4-OHT (F).
(E) Percentage of the scored molecules that are in a t-loop configuration. Cells as in (A) were imaged as in (D). Graphs show mean and SD values from 3 independent experiments (n≥200 molecules per experiment). P value was derived from unpaired two-tailed Student's t-test.
See Fig. S3 for details of the t-loop counting with and without exclusion of ‘x’ structures.
(G) T-loop fraction of the total scored telomeric molecules detected by STORM imaging as in (F). -Cre: n=286; +Cre (72 h): n=287. Graphs show means and SEMs.
STORM imaging of SV40LT-immortalized TRF2F/−ATM−/−Cre-ERT1 MEFs not expressing Cre showed the expected presence of both t-loops and linear telomeres (Fig. 4D). The frequency of t-loops in these control samples was 23.9±1.0% (Fig. 4D, E), which is quantitatively similar to the frequency observed in the ATM-proficient cells, and the t-loops have similar size distributions (Fig. S3A, B). Thus, the ATM kinase did not have an appreciable effect on these structural features of telomeres. At 156 h after induction of Cre, the deletion of TRF2 from the ATM-deficient cells resulted in a drastic reduction of the t-loop frequency to 5.8±1.3% (Fig. 4D, E; p=0.002, 3 independent experiments). A similar reduction was obtained when the telomeres were examined at 72 h after induction of Cre (Fig. 4F, G). As was the case for the ATM-proficient cells, inclusion of the ambiguous ‘x’ structures in the scoring did not affect the conclusion that the deletion of TRF2 resulted in substantial reduction in the t-loop frequency (Fig. S3C, D). Taken together, the results indicate that TRF2 is essential for the formation and/or maintenance of the t-loop structures.
We also considered alternative interpretations for the decrease in t-loop frequency upon TRF2 deletion but found them to be unlikely due to the following observations. In principle, a reduction in t-loop frequency after TRF2 deletion could be caused by a loss of the single-stranded 3’ overhang, which is required for the strand-invasion event. However, the 3’ overhang was minimally affected when TRF2 was deleted from ATM-deficient cells (Fig. 4C). The lowered frequency of t-loops could also be explained if TRF2 deletion reduced the frequency of psoralen/UV-induced crosslinks in the telomeric DNA. We therefore designed a method to determine the frequency of interstrand crosslinks in bulk and telomeric DNA (Fig. 5). To first monitor the presence of crosslinks in bulk DNA, we determined what fraction of the duplex DNA rapidly reannealed after heat denaturation, as would be expected for DNA with interstrand crosslinks (Fig. 5A). We performed this test on mono-, di-, tri-, and tetra-nucleosomal DNA fragments obtained by MNase digestion of the crosslinked nuclei. Consistent with the preferential crosslinking in the nucleosomal linker sequence, the mono-, di-, tri- and tetranucleosomal bands showed increasing crosslinking efficiencies (Fig. 5A). Importantly, TRF2 deletion from either ATM-proficient or ATM-deficient cells did not show a significant change in crosslinking efficiency in bulk DNA (Fig. 5B, upper and middle panel). In order to determine the crosslinking efficiency in telomeric DNA, a [CCCTAA]4 probe was hybridized to heat-denatured dinucleosomal DNA in the agarose gel without further denaturation. As only the noncrosslinked fraction hybridizes under these conditions, the crosslinking frequency can be deduced from the detected signal. Again, deletion of TRF2 did not substantially change the crosslinking frequency of the telomeric DNA (Fig. 5B, lower panel). Hence, the decrease in the t-loop frequency upon TRF2 deletion could not be caused by changes in either the 3’ overhang length or the crosslinking efficiency, but likely indicate a direct role of TRF2 in t-loop formation and/or maintenance.
Figure 5. Deletion of TRF2 does not affect crosslink efficiency in telomeric DNA.
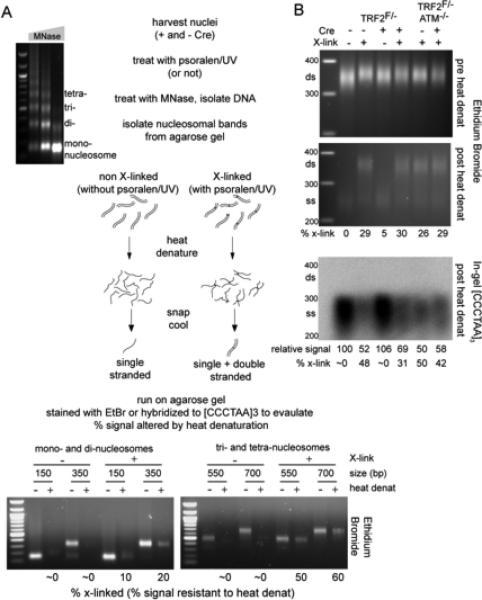
(A) Schematic of the experimental procedure. MEF nuclei either treated with psoralen/UV or not were digested with increasing amounts of MNase (agarose gel on left). DNA from the mono-, di, tri-, and tetra-nucleosomal MNase products was isolated and half of each sample was heat-denatured. Re-annealing of interstrand crosslinked DNAs regenerates double-stranded (ds) DNA while non-crosslinked DNAs remains single-stranded (ss). Bottom: The indicated samples from wt MEFs were separated on agarose gels and bands were visualized with Ethidium Bromide (EtBr), which preferentially stains dsDNA. Signals migrating at the position of dsDNA fragments were quantified with ImageJ and the percentage of signal remaining for each heat-denatured sample relative to its non-denatured control reflects the % crosslinking.
(B) TRF2 deletion does not alter on the psoralen/UV crosslinking efficiency in bulk and telomeric DNA substantially. Top, middle: dinucleosomal MNase products were isolated and processed as in (A). EtBr signals for the region between 300 and 400 bp (marked by the line) were quantified with ImageJ and the heat resistant signal intensity (a measure for the crosslinking fraction) was calculated from comparison of the signals in the top and middle gels. Bottom: The gel containing the heat-denatured samples was dried and hybridized with a 32P-labeled [AACCCT]4 probe. Note that only the non-crosslinked DNAs will hybridize. Signal intensities were quantified with ImageJ and normalized to the EtBr value of the non-denatured samples in the top gel. The telomeric DNA signal intensity value for the TRF2F/− sample not treated with Cre, and not treated with psoralen/UV was set to 100% and the values for the other samples were expressed relative to this value. The inferred % crosslinking of the dinucleosomal telomeric DNA band is given below the image.
The crosslinking frequency in telomeric DNA appears higher than that in bulk DNA (Fig. 5B), as may be expected since psoralen preferentially crosslinks at the A-T steps, which occur every six bp in telomeric DNA. Despite this, the crosslinking efficiency is still substantially below 100% for the ~350 bp dinucleosomal fragment. Given that the 3’ overhang is only 50-400 nt long, it is possible that the crosslinking efficiency in the duplex DNA formed by strand invasion of the 3’ overhang that secures the t-loop is also less than 100%, which likely accounts for another source of underestimation of the t-loop frequency. Stronger crosslinking conditions could not be used because they resulted in reduced FISH signals and compromised STORM images.
TRF1 and Rap1 are not required for t-loop formation/maintenance
The data presented above indicate that TRF2 is required to establish and/or maintain the t-loop configuration. It has been proposed that TRF1 could also contribute to t-loop formation based on the biochemical evidence that TRF1 has the ability to loop and pair duplex telomeric DNA in vitro (Bianchi et al., 1997; Bianchi et al., 1999; Griffith et al., 1998; Griffith et al., 1999). Furthermore, recent biochemical data suggested that the TRF2 interacting partner Rap1 increases the affinity of TRF2 for telomeric DNA ends and promotes t-loop formation (Arat and Griffith, 2012). It was therefore of interest to determine whether deletion of either TRF1 or Rap1 affects the occurrence of t-loops.
STORM imaging of telomeres was performed on MEFs from which TRF1 or Rap1 was deleted with Cre recombinase. The deletion of TRF1 and Rap1 was confirmed by immunoblotting (Fig. 6A, E) and these treatments did not change the telomeric 3’ overhang signal substantially (Fig. 6B, F). Interestingly, the removal of TRF1 or Rap1 did not affect the frequency of t-loop occurrence (Fig. 6C,D,G,H; Fig. S4A). Although the frequency of t-loops appeared to be slightly lower in the cells lacking TRF1, this small change is unlikely to be significant given the variability in these experiments.
Figure 6. TRF1 and Rap1 are not required for t-loop formation/maintenance.
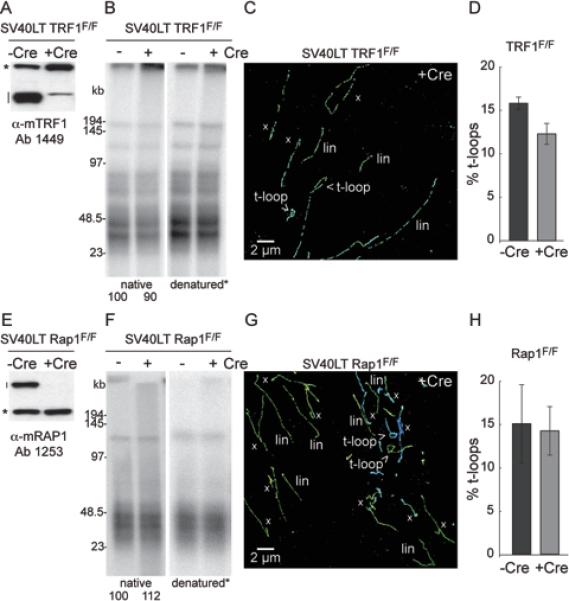
(A) Immunblot for loss of TRF1 (line) in the indicated cells treated Cre. *, non-specific band (loading control).
(B) In-gel assay for the telomeric 3’ overhang (native) and total telomeric DNA (denatured) before and after deletion of TRF1. Numbers below the overhang gel indicate the relative signal intensity of the ssDNA overhang normalized to the total telomeric DNA.
(C) Example of telomere imaging by STORM of cells lacking TRF1 (144 h post 4OHT).
(D) T-loop frequency before and after deletion of TRF1. Data are presented as mean +/− SEM from 2 independent experiments.
(E-H) As for (A-D) but with experiments performed on conditional Rap1 knockout cells.
See Fig. S4 for t-loop counts of both experiments.
The POT1 proteins are not involved in t-loop formation/maintenance
Based on their biochemical features, it is expected that both POT1a and POT1b bind to the single-stranded TTAGGG repeats in the D-loop formed upon strand-invasion of the 3’ overhang (Palm et al., 2009). As the binding of POT1a and/or POT1b to the D-loop might exert a stabilizing effect on the t-loop structure, it was also of interest to determine the requirement for the POT1 proteins in the formation/maintenance of t-loops. Surprisingly, deletion of either POT1b alone or POT1a and POT1b together did not show a sizable change in t-loop frequency (Fig. 7A-D; Fig. S4A), even though molecular data showed that the deletion had taken place and the cells showed the expected phenotype of an increase in the telomeric 3’ overhang signal (Fig. S4B, C).
Figure 7. TRF2 is the main shelterin protein required for t-loop formation and/or maintenance.
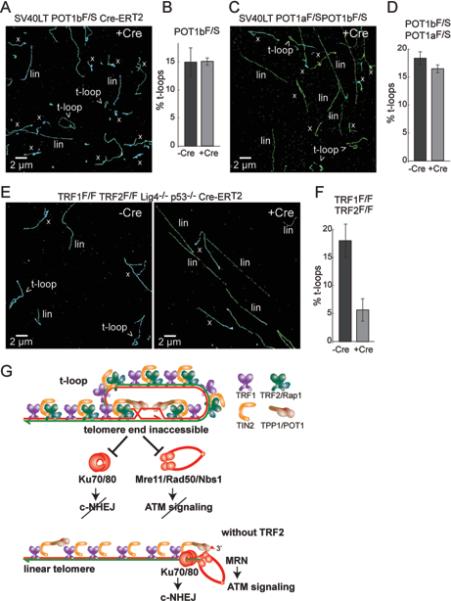
(A) Representative STORM image of telomeres after deletion of POT1b from the indicated cells (144 h post 4OHT)
(B) T-loop frequencies before and after deletion of POT1b. Data are presented as mean +/− SEM from 2 independent experiments.
(C, D) As for (A, B) but involving co-deletion of POT1a and POT1b from the indicated cells with Cre (144 h time point).
(E) Representative STORM images of telomeres before and after removal of the shelterin complex through co-deletion of TRF1 and TRF2 (156 h post 4OHT).
(F) T-loop frequencies before and after deletion of TRF1 and TRF2. Data are presented as mean +/− SEM from 2 independent experiments.
See Fig. S4 for details of the t-loop counts in B, D, and F and the pertinent analysis of the protein and DNA in the cells.
(G) Model explaining how TRF2-mediated t-loop formation/maintenance protects telomeres from MRN-initiated ATM signaling and Ku70/80-initiated NHEJ. When TRF2 is absent, telomeres are converted into the linear structure thereby allowing access to Ku70/80 and MRN. Ku70/80 and MRN are excluded from the telomere terminus when telomeres are in the t-loop configuration. While Ku70/80 and the MRN complex are also found in association with the shelterin complex, these Ku70/80 and MRN are not depicted here. The functions of these shelterin-associated Ku70/80 and MRN are not known.
These data would argue that, among the shelterin components, TRF2 is the only factor critical for t-loop formation/maintenance. If this conclusion is correct, the removal of the whole shelterin complex should yield the same phenotype as removal of TRF2 alone. We tested this notion using TRF1/TRF2 double-knockout cells, in which Cre treatment generates shelterin-free telomeres (Sfeir and de Lange, 2012). Deletion of shelterin in the context of DNA ligase 4 (Lig4) proficient cells yielded the expected drop (from 18% to 3.2%) in t-loop frequency due to the massive fusion of telomeres (Fig. S4A). Thus, we deleted TRF1 and TRF2 from Lig4-deficient cells, where telomere fusion are minimal (Fig. S4C). Importantly, the t-loop frequency was also reduced in the TRF1/TRF2/Lig4-deficient cells, and the extent of reduction was no greater than that observed after removal of TRF2 alone from Lig4-deficient cells (Fig. 7E,F; Fig. S4A). These results are consistent with t-loop formation being largely dependent on TRF2 but not other components of shelterin. We did not separately test the role of TPP1 since its deletion is functionally equivalent to loss of POT1a and POT1b (Kibe et al., 2010). We also did not separately test TIN2 because its deletion results in a complex phenotype reflecting the loss of both POT1a/b and some TRF2, which would make the data difficult to interpret (Takai et al., 2011).
DISCUSSION
In this work, we demonstrated the visualization of t-loop structures at telomeres using super-resolution fluorescence imaging, either directly in the nuclei of minimally processed mouse splenocytes or in spread chromatin from MEFs. Our results provide both direct evidence for the presence of t-loops at telomeres and new insights into how the t-loop structure forms. The imaging assay developed for t-loop visualization does not require any telomeric DNA purification and thus allows the investigation of t-loops under many different genetic settings. By imaging the telomere structure in wild type and eight different mutant MEFs, we observed a specific dependence of t-loop persistence on a single shelterin subunit, TRF2, which provides crucial insight into how TRF2 protects telomeres ends from being recognized as DSBs by the ATM kinase and prevent their processing by c-NHEJ.
The structure and frequency of t-loops
The STORM images of the t-loops indicate that the strand invasion point is highly variable, taking place all along the duplex telomeric DNA region, resulting in t-loops ranging from short loops with long tails to very large loops with minimal tails. The simplest interpretation of the t-loop structure observed here is that insertion point of the telomere terminus and the size of the loop are not predetermined. This finding is consistent with the insertion of the terminus being the critical aspect of the t-loop structure rather than the creation of a specific segment of the telomeric DNA that is in the loop.
The frequency of t-loops detected in cells with fully functional telomeres varied from 10-40% (average 24%). This number likely represents an underestimate of the actual t-loop frequency in vivo because our scoring criteria for t-loops were stringent, leading to exclusion of telomeres with structures that closely resembled a lariat, and because any breakage or incomplete crosslinking of the DNA would preferentially reduce the t-loop fraction. Thus, our estimates of the t-loop frequency do not exclude the possibility that the majority of telomeres are in a t-loop configuration, but further work will be required to determine the prevalence of t-loops in vivo.
The role of t-loops in repressing NHEJ and ATM signaling
TRF2 is the main factor responsible for preserving the t-loop structure at telomeres. Among the five shelterin components that we analyzed, only the deletion of TRF2 lowered the t-loop frequency by 4.5±0.9 fold (based on 11 experiments). In contrast, deletion of the other shelterin components did not affect the frequency of t-loops (1.1±0.1 fold average change in 8 experiments). TRF2 is also largely responsible for the repression of ATM signaling and c-NHEJ (Celli and de Lange, 2005; Denchi and de Lange, 2007), whereas the deletion of TRF1, Rap1, and POT1a/b does not elicit ATM signaling or telomere fusion (Sfeir et al., 2010; Martinez et al., 2010; Sfeir et al., 2009; Martinez et al., 2009; Denchi and de Lange, 2007; Hockemeyer et al., 2006). Therefore, our data indicate a strong correlation between the loss of protection from the ATM and c-NHEJ pathways and the disappearance of the t-loops. The simplest interpretation is that the t-loop structures provide the main mechanism by which telomeres protect themselves from ATM signaling and c-NHEJ.
The ability of TRF2 to promote a t-loop configuration at the telomeres can explain how telomeres avoid being processed by c-NHEJ, because this pathway is initiated by loading the ring-shaped Ku70/80 heterodimer on DNA ends (reviewed in (Lieber, 2010)). Since Ku70/80 binds to exposed DNA ends, the strand-invasion of the telomeric 3’ overhang will prevent loading of the Ku70/80 heterodimer onto the telomere terminus (Fig. 7G). Similarly, the activation of the ATM kinase pathway starts with the association of the MRN complex (composed of Mre11, Rad50, and Nbs1) with DNA ends (reviewed in (Stracker and Petrini, 2011)), which is also inhibited by the t-loop structure (Fig. 7G). In addition to its role in t-loop formation/maintenance, TRF2 was recently shown to have a second mechanism repressing aspects of the DDR that lead to NHEJ (Okamoto et al., 2013), which may serve to protect the linear telomeres from NHEJ when t-loops are absent. It is generally assumed that t-loops are resolved during DNA replication, thereby making the telomeres vulnerable to NHEJ. On the other hand, the resolution of the t-loop structure during DNA replication may provide a window of opportunity for telomerase to access the 3’ terminus of the telomere. Whether t-loops actually prevent the telomere end from being accessed by the telomerase and to what extent telomerase action at the telomere is regulated by the t-loop structure remains to be determined.
T-loops and ATR signaling
The ATR pathway is unlikely to be dissuaded by the t-loop structure. When POT1a and POT1b are deleted from telomeres, t-loops persist, yet the ATR signaling pathway is activated throughout the cell cycle at the majority of telomeres (Denchi and de Lange, 2007; Gong and de Lange, 2010). We imagine that in absence of the POT1 proteins, the single-stranded DNA in the displaced strand at the base of the t-loop can bind RPA and recruit ATRIP/ATR. In addition to RPA binding, ATR activation also requires a TopBP1-dependent step, which requires the presence of Rad17, the 9-1-1 complex, and RHINO at the transition point between double-stranded (ds) and single-stranded (ss) DNA ((Cotta-Ramusino et al., 2011) reviewed in (Nam and Cortez, 2011)). However, in the t-loop setting, the ds/ss transition is not present on the strand that binds RPA (the D loop) but exists nearby at the 5’ end of the telomere. Perhaps Rad17/9-1-1 and RHINO loaded at the 5’ end of the telomere places TopBP1 in sufficient proximity to activate the ATR kinase bound to RPA on the D loop.
TRF2-mediated t-loops as a telomere end-protection mechanism
Our data suggest that TRF2 provides an architectural solution to specific aspects of the telomere end-protection problem. By remodeling telomeres into t-loops, TRF2 effectively sequesters the telomere terminus away from the threats of c-NHEJ and ATM kinase signaling. Furthermore, the formation of t-loops could potentially help limit 5’ end resection and a-NHEJ, which are blocked by TRF2 as well as other shelterin proteins (Lottersberger et al., 2013; Zimmermann et al., 2013; Rai et al., 2010; Sfeir and de Lange, 2012).
How TRF2 creates t-loops in vivo is of obvious interests. The ability of TRF2 to change DNA topology and bind HJ-like DNA structures are likely to be relevant and merit further exploration (Fouche et al., 2006; Amiard et al., 2007; Poulet et al., 2009). TRF2 separation-of-function mutants will be helpful in this regard. In addition, it will be of interest to determine whether TRF2 is aided by other factors, such as BRCA2 and Rad51, which mediate strand-invasion of 3’ extensions in HDR. Given that telomeres use many DNA repair factors to achieve their protected state (reviewed in (Diotti and Loayza, 2011)), the involvement of bona fide HDR proteins in telomere protection is not excluded. The STORM imaging method reported here will be a valuable tool to address these questions.
EXPERIMENTAL PROCEDURES
Detailed experimental procedures are provided in the Supplemental Material available on line.
MEFs
SV40LT TRF2F/−Cre-ERT1, SV40LT TRF2F/−ATM−/−Cre-ERT1, TRF1F/FTRF2F/FLig4−/−p53−/−Cre-ERT2, TRF1F/FTRF2F/FLig4+/−p53−/−Cre-ERT2 and SV40LT POT1aF/SPOT1bF/S, SV40LT POT1bF/F Cre-ERT2 SV40LT TRF1F/FCre-ERT2, SV40LT Rap1F/FKu80+/− MEFs were described previously (Denchi and de Lange, 2007; Wu et al., 2012; Sfeir et al., 2010; Hockemeyer et al., 2006; Sfeir and de Lange, 2012). Cre expression was induced by treatment with 0.5 mM 4-OH tamoxifen or infection with pWZL-hygro-Cre as described (Wu et al., 2012).
Psoralen crosslinking and chromatin spreading
1-2 × 107 nuclei were isolated as described (Pipkin and Lichtenheld, 2006), resuspended in 3 ml NWB (10 mM Tris-HCl pH 7.4, 15 mM NaCl, 60 mM KCl, 5 mM EDTA, 300 mM sucrose), and incubated for 5 min with 100 μg/ml Trioxsalen (Sigma). The incubation was carried out in a 6 cm dish, on ice, in the dark, while stirring. Nuclei were exposed to 365 nm UV light at 2-3 cm from the light source (model UVL-56, UVP) for 30 min, while stirring on ice. After crosslinking, nuclei were collected, washed once with ice-cold NWB, and resuspended at 2-5 × 106 nuclei/ml. For spreading, nuclei were diluted 1:10 in spreading buffer (10 mM Tris-HCl 7.4, 10 mM EDTA, 0.05% SDS, 1 M NaCl, pre-warmed at 37°C) and 100 μl of the suspension was immediately deposited on a coverslip using a Shandon Cytospin 3 at 600 rpm for 1 min. Samples were fixed in methanol at −20°C for 10 min followed by 1 min in acetone at −20°C. The coverslips were washed in PBS and dehydrated through a 70%, 95%, 100% ethanol series before performing FISH.
STORM imaging and analysis
For STORM imaging, coverslips containing FISH-labeled sample were sealed in a well containing ~100 μl of imaging buffer (see online Experimental Procedure section) and imaged as described previously (Huang et al., 2008a) with the following modifications. Prior to STORM imaging, large areas of the coverslip were imaged at the conventional resolution using a motorized stage scan to identify areas of interest in which telomeres were abundant. Next, an imaging sequence was set up to allow sequential conventional and STORM imaging of dozens of 43 × 43 μm fields of view per sample.
Image analysis was performed as described previously (Bates et al., 2007; Huang et al., 2008b). All instrument control, data acquisition, and data analysis was performed using custom written software.
Analysis of telomeric DNA and telomeric proteins
Telomere analysis by telomeric FISH on metaphase spreads, analysis of telomeric DNA on CHEF gels, analysis of the telomeric overhang, and immunoblotting for telomeric proteins was done using standard procedures according to previously published protocols. Detailed description of the protocols is given in the on line materials.
Supplementary Material
HIGHLIGHTS.
STORM imaging of telomeres reveals t-loops in relaxed mouse chromatin
Deletion of the shelterin component TRF2 converts t-loops into linear telomeres
Other components of shelterin are not required to form/maintain t-loops
TRF2-dependent t-loop formation is proposed to block c-NHEJ and ATM kinase signaling
ACKNOWLEDGMENTS
We are grateful to members of the de Lange and Zhuang labs for comments on this manuscript. Y.D. is an Ellison Medical Foundation/AFAR Fellow of the Life Sciences Research Foundation. J.Y.W. was supported as a National Science Foundation Graduate Research fellow. This work was supported by grants from the NIH to TdL (AG016642, GM049046) and to XZ (GM068518 and GM096450). TdL is an American Cancer Society Research Professor. XZ is a Howard Hughes Medical Institute Investigator.
REFERENCES
- Amiard S, Doudeau M, Pinte S, Poulet A, Lenain C, Faivre-Moskalenko C, Angelov D, Hug N, Vindigni A, Bouvet P, Paoletti J, Gilson E, Giraud-Panis MJ. A topological mechanism for TRF2-enhanced strand invasion. Nat. Struct. Mol. Biol. 2007;14:147–154. doi: 10.1038/nsmb1192. [DOI] [PubMed] [Google Scholar]
- Arat NO, Griffith JD. Human Rap1 interacts directly with telomeric DNA and regulates TRF2 localization at the telomere. J. Biol. Chem. 2012;287:41583–41594. doi: 10.1074/jbc.M112.415984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi SE, DePinho RA. Telomeres and telomerase in cancer. 2010;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. 2007;318:798–801. doi: 10.1126/science.1147182. [DOI] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A, Smith S, Chong L, Elias P, de Lange T. TRF1 is a dimer and bends telomeric DNA. EMBO J. 1997;16:1785–1794. doi: 10.1093/emboj/16.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A, Stansel RM, Fairall L, Griffith JD, Rhodes D, de Lange T. TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 1999;18:5735–5744. doi: 10.1093/emboj/18.20.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callegari AJ, Kelly TJ. Shedding light on the DNA damage checkpoint. Cell Cycle. 2007;6:660–666. doi: 10.4161/cc.6.6.3984. [DOI] [PubMed] [Google Scholar]
- Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotta-Ramusino C, McDonald ER, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. 2011;332:1313–1317. doi: 10.1126/science.1203430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. Human telomeres are attached to the nuclear matrix. EMBO J. 1992;11:717–724. doi: 10.1002/j.1460-2075.1992.tb05104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. How telomeres solve the end-protection problem. 2009;326:948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diotti R, Loayza D. Shelterin complex and associated factors at human telomeres. Nucleus. 2011;2:119–135. doi: 10.4161/nucl.2.2.15135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerhahn S, Iglesias N, Panza A, Porro A, Lingner J. TERRA biogenesis, turnover and implications for function. FEBS Lett. 2010;584:3812–3818. doi: 10.1016/j.febslet.2010.07.032. [DOI] [PubMed] [Google Scholar]
- Fouche N, Cesare AJ, Willcox S, Ozgur S, Compton SA, Griffith JD. The basic domain of TRF2 directs binding to DNA junctions irrespective of the presence of TTAGGG repeats. J. Biol. Chem. 2006;281:37486–37495. doi: 10.1074/jbc.M608778200. [DOI] [PubMed] [Google Scholar]
- Gong Y, de Lange T. A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol. Cell. 2010;40:377–387. doi: 10.1016/j.molcel.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith J, Bianchi A, de Lange T. TRF1 promotes parallel pairing of telomeric tracts in vitro. J. Mol. Biol. 1998;278:79–88. doi: 10.1006/jmbi.1998.1686. [DOI] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. Mammalian telomeres end in a large duplex loop. 1999;97:503–14. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- Guo X, Deng Y, Lin Y, Cosme-Blanco W, Chan S, He H, Yuan G, Brown EJ, Chang S. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007;26:4709–4719. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Daniels JP, Takai H, de Lange T. Recent expansion of the telomeric complex in rodents: Two distinct POT1 proteins protect mouse telomeres. 2006;126:63–77. doi: 10.1016/j.cell.2006.04.044. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Palm W, Wang RC, Couto SS, de Lange T. Engineered telomere degradation models dyskeratosis congenita. Genes Dev. 2008;22:1773–1785. doi: 10.1101/gad.1679208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Jones SA, Brandenburg B, Zhuang X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat Methods. 2008a;5:1047–1052. doi: 10.1038/nmeth.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. 2008b;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- Kibe T, Osawa GA, Keegan CE, de Lange T. Telomere Protection by TPP1 Is Mediated by POT1a and POT1b. Mol. Cell. Biol. 2010;30:1059–1066. doi: 10.1128/MCB.01498-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipling D, Cooke HJ. Hypervariable ultra-long telomeres in mice. 1990;347:400–402. doi: 10.1038/347400a0. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc. Natl. Acad. Sci. U S A. 2013;110:2146–2151. doi: 10.1073/pnas.1222617110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luderus ME, van Steensel B, Chong L, Sibon OC, Cremers FF, de Lange T. Structure, subnuclear distribution, and nuclear matrix association of the mammalian telomeric complex. J. Cell Biol. 1996;135:867–881. doi: 10.1083/jcb.135.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Carlos AR, Gomez-Lopez G, Tejera AM, Schoeftner S, Dominguez O, Pisano DG, Tarsounas M, Blasco MA. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat. Cell Biol. 2010;12:768–780. doi: 10.1038/ncb2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. The fusion of broken ends of sister half-chromatids following chromatid breakage at meiotic anaphase. In: Moore JA, editor. The collected papers of Barbara McClintock. Garland Publishing, Inc.; New York and London: 1938. pp. 1–48. [Google Scholar]
- McClintock B. The stability of broken ends of chromosomes in Zea mays. 1941;26:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem. J. 2011;436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitina T, Woodcock CL. Closed chromatin loops at the ends of chromosomes. J. Cell Biol. 2004;166:161–165. doi: 10.1083/jcb.200403118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora GJ, Buncher NA, Opresko PL. Telomeric protein TRF2 protects Holliday junctions with telomeric arms from displacement by the Werner syndrome helicase. Nucleic Acids Res. 2010;38:3984–3998. doi: 10.1093/nar/gkq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell. Biol. 2010;11:171–181. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Bartocci C, Ouzounov I, Diedrich JK, Yates JR, Denchi EL. A two-step mechanism for TRF2-mediated chromosome-end protection. 2013;494:502–505. doi: 10.1038/nature11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW, Rothstein RJ. Yeast transformation: a model system for the study of recombination. Proc Natl Acad Sci USA. 1981;78:6354–6358. doi: 10.1073/pnas.78.10.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Palm W, Hockemeyer D, Kibe T, de Lange T. Functional dissection of human and mouse POT1 proteins. Mol. Cell. Biol. 2009;29:471–482. doi: 10.1128/MCB.01352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierron G, Puvion-Dutilleul F. An anchorage nuclear structure for telomeric DNA repeats in HeLa cells. Chromosome Res. 1999;7:581–592. doi: 10.1023/a:1009253917125. [DOI] [PubMed] [Google Scholar]
- Pipkin ME, Lichtenheld MG. A reliable method to display authentic DNase I hypersensitive sites at long-ranges in single-copy genes from large genomes. Nucleic Acids Res. 2006;34:e34. doi: 10.1093/nar/gkl006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet A, Buisson R, Faivre-Moskalenko C, Koelblen M, Amiard S, Montel F, Cuesta-Lopez S, Bornet O, Guerlesquin F, Godet T, Moukhtar J, Argoul F, Declais AC, Lilley DM, Ip SC, West SC, Gilson E, Giraud-Panis MJ. TRF2 promotes, remodels and protects telomeric Holliday junctions. EMBO J. 2009;28:641–651. doi: 10.1038/emboj.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, Chang S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010;29:2598–2610. doi: 10.1038/emboj.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12:753–764. doi: 10.1097/GIM.0b013e3181f415b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. 2012;336:593–597. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. 2010;327:1657–1661. doi: 10.1126/science.1185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. DNA Ligase IV-Dependent NHEJ of Deprotected Mammalian Telomeres in G1 and G2. Curr. Biol. 2002;12:1635. doi: 10.1016/s0960-9822(02)01179-x. [DOI] [PubMed] [Google Scholar]
- Stansel RM, de Lange T, Griffith JD. T-loop assembly in vitro involves binding of TRF2 near the 3’ telomeric overhang. EMBO J. 2001;20:5532–5540. doi: 10.1093/emboj/20.19.5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell. Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol. Cell. 2011;44:647–659. doi: 10.1016/j.molcel.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejera AM, Stagno d'Alcontres M, Thanasoula M, Marion RM, Martinez P, Liao C, Flores JM, Tarsounas M, Blasco MA. TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev. Cell. 2010;18:775–789. doi: 10.1016/j.devcel.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. 2006;127:709–720. doi: 10.1016/j.cell.2006.09.034. [DOI] [PubMed] [Google Scholar]
- Wu P, Takai H, de Lange T. Telomeric 3’ Overhangs Derive from Resection by Exo1 and Apollo and Fill-In by POT1b-Associated CST. 2012;150:39–52. doi: 10.1016/j.cell.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.