

Abstract
Pax genes encode evolutionarily conserved transcription factors that play critical roles in development. Pax3 and Pax7 constitute one of the four Pax subfamilies. Despite partially overlapping expression domains, mouse mutations for Pax3 and Pax7 have very different consequences. To investigate the mechanism of these contrasting phenotypes, we replaced Pax3 by Pax7 by using gene targeting in the mouse. Pax7 can substitute for Pax3 function in dorsal neural tube, neural crest cell, and somite development, but not in the formation of muscles involving long-range migration of muscle progenitor cells. In limbs in which Pax3 is replaced by Pax7, the severity of the muscle phenotype increases as the number of Pax7 replacement alleles is reduced, with the forelimb more affected than the hindlimb. We show that this hypomorphic activity of Pax7 is due to defects in delamination, migration, and proliferation of muscle precursor cells with inefficient activation of c-met in the hypaxial domain of the somite. Despite this, overall muscle patterning is retained. We conclude that functions already prefigured by the single Pax3/7 gene present before vertebrate radiation are fulfilled by Pax7 as well as Pax3, whereas the role of Pax3 in appendicular muscle formation has diverged, reflecting the more recent origin of this mode of myogenesis.
Keywords: Pax3, Pax7, myogenesis, appendicular muscle, evolution, neural tube
Gene duplication is a key mechanism for increasing diversity and complexity during evolution. Multigene families arise as a result of successive duplications. One such family with important functions during development codes for the Pax transcription factors (Tremblay and Gruss 1994). Pax genes are divided into four sub-families based on sequence similarities (Gruss and Walther 1992; Noll 1993; Mansouri et al. 1996a). Among these, the Pax3 and Pax7 genes arose by duplication from a unique ancestral Pax3/7 gene, and similarities in their protein sequence and expression pattern reflect this common origin. All vertebrates examined so far have at least one of each of these genes. Mammals and birds have one Pax3 and one Pax7 gene, whereas in the genome of the teleost fish Danio rerio, one Pax3 gene and at least four Pax7 variants are found, one of which encodes a protein with a sequence close to the mammalian Pax7 (Seo et al. 1998). Alternatively spliced forms of Pax3 and Pax7 have also been reported in mouse and human, although their distribution and function remains poorly understood (Ziman et al. 1997; Barber et al. 1999).
A unique Pax3/7 gene is found in many vertebrates, such as in the ascidians (Wada et al. 1996, 1997), in the chelicerate Tetranychus urticae (Dearden et al. 2002), in the nematode Caenorhabditis elegans (Hobert and Ruvkun 1999) or in Amphioxus (Holland et al. 1999). Because the genome of Amphioxus, a cephalochordate regarded as the closest living invertebrate relative to the vertebrate, contains only one Pax3/7 gene, it is likely that the complexity of this Pax subfamily, like that of other Pax subfamilies, has arisen by gene duplication at the onset of vertebrate evolution (Holland et al. 1999). In Drosophila, however, there are three Pax3/7-related genes (Prd, Gsb, and Gsbn), which probably arose from independent duplications (Baumgartner et al. 1987; Li and Noll 1994; Xue et al. 2001).
The spatiotemporal expression pattern of the ascidian HrPax-37 gene suggests involvement in two distinct developmental processes: specification of dorsal cell fates in ectoderm during neurulation, and regional differentiation of the neural tube at later stages (Wada et al. 1996). Hence, Pax3/7 transcription in the dorsal neural tube is believed to be the primitive Pax3/7 expression domain, and indeed, this is a common feature of all species examined to date. In the mouse, Pax3 and Pax7 are expressed in the dorsal neural tube, where only Pax3 transcription extends into the dorsal-most region. Splotch (Sp) mice, in which a mutation disrupts Pax3 function (Tremblay and Gruss 1994), have defects in dorsal neural tube closure (spina bifida and exencephaly). In addition, analysis of Pax3 and Pax7 double-mutant mice demonstrates that in the absence of both genes, neural cell fate is affected such that some cells located dorsally acquire ventral interneuron identity (Mansouri and Gruss 1998).
Pax3 and Pax7 are also expressed in neural crest, which is a characteristic of the vertebrate embryo (Le Douarin and Kalcheim 1999). Neural crest cells emerge from the dorsal neural tube and migrate throughout the developing embryo, where they give rise to a range of cell types, including neurons and glial cells of the peripheral nervous system and melanocytes. In Pax3-mutant embryos, neural crest cells fail to migrate correctly, resulting in either a severe reduction or a complete absence of neural crest cell derivatives (Auerbach 1954; Franz 1989; Tremblay et al. 1995). The severity of these defects increases along the rostrocaudal axis, so that no neural crest derivatives are present in the caudal portion of the Splotch-mutant embryo (Auerbach 1954). In contrast, Pax7-deficient mice show anterior (cephalic) neural crest cell defects (Mansouri et al. 1996b). Amphi-Pax3/7 neural expression suggests that a population of cells comparable to the premigratory neural crest cells of vertebrates may have been present in the common ancestor of all extant chordates (Holland et al. 1999).
The function of the Pax3/7 genes in mesoderm has been acquired more recently during evolution. Amphioxus, similar to a vertebrate, has segmented axial muscles and expresses Amphi-Pax3/7 in this muscle lineage, suggesting that Pax3/7 function in mesoderm is present in cephalochordates (Holland et al. 1999). In the mouse, Pax3 is expressed in presomitic mesoderm (Schubert et al. 2001) and then throughout the somite, before becoming restricted to the dermomyotome as somites mature (Goulding et al. 1991). At later stages, Pax3 expression becomes concentrated in the epaxial (adjacent to the axis) and hypaxial extremities of the dermomyotome (see Tajbakhsh and Buckingham 2000). Pax3-mutant embryos have somite defects, including abnormalities in segmentation (Schubert et al. 2001) as well as loss of the epaxial and, most notably, hypaxial dermomyotome (Tajbakhsh and Buckingham 2000). In consequence, the myotome fails to form correctly. This leads to trunk muscle defects (Tremblay et al. 1998). Pax3 also has a critical function in the formation of muscles that depend on the migration of muscle progenitor cells from the hypaxial dermomyotome. Most notably, in the absence of functional Pax3, the limb muscles are absent in Splotch-mutant embryos (Auerbach 1954; Franz et al. 1993; Bober et al. 1994; Goulding et al. 1994). Pax3 is required for the activation of c-met (Epstein et al. 1996; Yang et al. 1996; Relaix et al. 2003), which encodes a tyrosine kinase receptor, Met, essential for the delamination/migration of muscle progenitor cells (Bladt et al. 1995). Pax3 also has an essential function in regulating the gene hierarchy which leads to the activation of MyoD and the formation of skeletal muscle (Tajbakhsh et al. 1997).
In contrast, no embryonic muscle defect has been described in Pax7-mutant mice (Mansouri et al. 1996b), although Pax7 is expressed in part of the dermomyotome (Jostes et al. 1990; Tajbakhsh et al. 1997). However Pax7 plays a critical role postnatally when it is required for the presence of satellite cells, the precursor cells responsible for the growth and regeneration of skeletal muscle (Seale et al. 2000). Pax3 is also expressed in satellite cells (Conboy and Rando 2002; Buckingham et al. 2003); however, due to the perinatal death of Pax3 mutants, its role in adult muscle remains to be elucidated.
Although Pax3 and Pax7 are apparently involved in different stages of myogenesis, evidence for shared functions comes from the observation that translocations in humans of both PAX3 and PAX7 with FKHR to give PAX3-FKHR and PAX7-FKHR fusion proteins lead to the formation of alveolar rhabdomyosarcomas (ARMSs), a pediatric tumor of skeletal muscle (Galili et al. 1993; Davis et al. 1994; Barr 2001).
A key question in understanding the relative functions of Pax3 and Pax7 is whether these are determined by biochemical differences between the proteins or by differences in the spatiotemporal expression of the two genes. To address this, we replaced Pax3 by Pax7 by using gene targeting in the mouse. Analysis of mutant embryos with one or two alleles of Pax7 replacing Pax3 shows that Pax7 can replace Pax3 in the dorsal neural tube, in neural crest cells, and in somite development. However, Pax3 function in the long-range migration of muscle progenitor cells is only partially rescued. Different combinations of the Pax7 replacement alleles act as hypomorphic alleles of Pax3 in the formation of limb muscles, and reveal a multistep requirement for Pax3 activity in appendicular muscle development. These results provide new insights into the evolution of the Pax3/7 functions.
Results
Pax3 and Pax7 expression in normal and mutant embryos
The expression domains of Pax3 and Pax7 in the mouse embryo were compared, using mice in which Pax3 (Relaix et al. 2003) as well as Pax7 (Mansouri et al. 1996b) genes have been targeted with nLacZ and LacZ reporter genes, respectively. X-Gal staining (Fig. 1A-H) of normal and mutant embryos was complemented by in situ hybridization of transcripts from the endogenous Pax genes (Fig. 1I-Q). In thoracic somites, Pax3 is expressed throughout the dermomyotome, with the strongest expression observed in the hypaxial epithelium (Fig. 1A,K; Relaix et al. 2003). In Pax3-mutant embryos, the somites are reduced mainly in the hypaxial, but also in the epaxial domains, and somite segmentation is perturbed (Fig. 1B; Relaix et al. 2003). Pax7 is also expressed in the dermomyotome (Fig. 1C,L); however, this expression is mainly restricted to the central region and is excluded from the epaxial and hypaxial extremities, which strongly express Pax3 (Fig. 1, cf. L and K). In contrast to the situation in chicken embryos (Marcelle et al. 1995), Pax7 transcripts are not detected in muscle progenitor cells migrating to the limb, which express Pax3 as shown at embryonic day 10.5 (E10.5; Fig. 1, cf. O and N). In the embryonic limb, Pax7 transcripts are first detected at E11.5 in the proximal limb (Fig. 1P) and then by E12.5 in more distal limb muscles as well (Fig. 1Q). In Pax7 mutants, limb muscles are present (Fig. 1G,H) and Pax7-expressing cells show a normal distribution in the somite (Fig. 1C,D). Trunk muscles develop normally (Fig. 1G,H). Pax7, unlike Pax3, is expressed in the branchial arches (Fig. 1I,J) and later in facial muscles (Fig. 1G,H), some of which derive from muscle precursor cells in the arches. In Pax7-mutant mice, facial muscles do not appear to be affected (Fig. 1H), and indeed, further regulators intervene specifically in the formation of muscles derived from the arches (Lu et al. 2002).
Figure 1.

Despite expression in partially overlapping domains, Pax7 does not rescue defects in Pax3-mutant embryos. (A,B) Pax3 expression at E10.5 revealed by X-Gal staining of Pax3nLacZ/+ (A) and Pax3nLacZ/nLacZ (B) embryos. (A) Normal Pax3 expression is observed in somites (arrowhead), muscle progenitor cells migrating to the limb (black arrow), hypoglossal chord (red arrow), frontonasal processes, and dorsal neural tube. (B) Pax3-mutant embryos have somite defects with abnormal segmentation and epaxial and hypaxial truncation (arrowhead), lack of migrating precursor cells to the limbs (black arrow) and hypoglossal chord (red arrow), and failure of dorsal neural tube closure (exencephaly and spina bifida). (C,D) Pax7 expression at E10.5 revealed by X-Gal staining of Pax7LacZ/+ (C) and Pax7LacZ/LacZ (D) embryos. (C) Pax7 expression is detected in somites (arrowhead), frontonasal processes, and the neural tube, but not in the muscle progenitor cells migrating to the limb (black arrow) or in the hypoglossal chord (red arrow). (D) Pax7-deficient embryos do not present any obvious defects at sites of Pax7 expression. (E,F) Expression of Pax3 at E13.5 revealed by X-Gal staining in Pax3nLacZ/+ (E) and Pax3nLacZ/nLacZ (F) embryos. (E) Pax3 expression is observed at this stage in the dorsal neural tube, limb, cervical, and body muscles. (F) Pax3-mutant embryos have defects in neural tube closure (here striking spina bifida), lack of limb muscles, and reduced and disorganized cervical and trunk muscles. (G,H) Expression of Pax7 at E13.5 revealed by X-Gal staining in Pax7LacZ/+ (G) and Pax7LacZ/LacZ (H) embryos. In contrast to Pax3-deficient embryos, lack of Pax7 does not lead to any obvious defect despite expression of Pax7 at this stage in the muscle masses of the limbs as well as the trunk. Head muscles do not express Pax3 (E, arrowhead), but express Pax7, clearly seen in mouse embryos with two Pax7LacZ alleles (H, arrowhead) despite the absence of Pax7. (I,J) Pax7 expression at E10.5 in Pax3nLacZ/+ (I) and Pax3nLacZ/nLacZ (J) embryos revealed by in situ hybridization. (I) In Pax3nLacZ/+ embryos, Pax7 is expressed in somites and in the branchial arches (arrowheads), in cells that will later form facial muscles, but not in limb muscle progenitor cells (arrow). Pax7 transcripts are also present in frontonasal processes and in the dorsal neural tube. (J) In Pax3nLacZ/nLacZ-mutant embryos, Pax7 expression is reduced in the somite, reflecting the loss of cells, but is not affected in the branchial arches, where Pax3 is not expressed (see A). (K) Pax3 expression in the thoracic region at E10.5 in Pax3nLacZ/+ embryos revealed by in situ hybridization. Pax3 is transcribed in the dorsal neural tube (blue arrowhead) and throughout the dermomyotome, with stronger expression in the epaxial and hypaxial domains (black arrowheads). (L,M) Pax7 expression in the thoracic region of Pax3nLacZ/+ (L) and Pax3nLacZ/nLacZ (M) embryos at E10.5 revealed by in situ hybridization. (L) Pax7 is transcribed in the central dermomyotome, but not in the epaxial and hypaxial-most regions in heterozygote embryos (black arrowheads). In the dorsal neural tube, Pax7 transcripts are absent from the dorsal-most region (blue arrowheads). (M) In Pax3-mutant embryos, Pax7 expression in the somite is severely reduced due to loss of cells (black arrowhead), whereas neural tube expression seems unaffected (blue arrowheads). (N) Pax3 expression in muscle progenitor cells in the forelimb of E10.5 embryos revealed by in situ hybridization. (O-Q) Pax7 expression in the forelimb of E10.5 (O), E11.5 (P), and E12.5 (Q) embryos monitored by in situ hybridization. Pax7 transcripts are first detected at E11.5 in proximal muscle masses (P) and then detected in the more distal limb muscles at E12.5 (Q). (R-T) Expression of Pax3 (R), Pax7 (S), and a Pax3 transcriptional reporter transgene (T; P34TKZ; Relaix et al. 2003) at E11.5 revealed by X-Gal staining. The black arrowhead shows expression of Pax7 and P34TKZ, but not Pax3, in head muscles, suggesting that Pax7 acts, similarly to Pax3, as a transcriptional activator in vivo.
Pax3 and Pax7 are also expressed in the dorsal neural tube, where, unlike Pax3 (Fig. 1L), Pax7 is excluded from the dorsal most domain (Fig. 1K). Embryos in which both alleles of Pax3 have been targeted with nLacZ have defects in neural tube closure (Fig. 1F). As in the case of the spontaneous Sp mutation (Auerbach 1954; Epstein et al. 1993), spina bifida is observed with a frequency approaching 100%, whereas exencephaly is more frequent (65%) than in Sp-mutant embryos (∼40%). Pax7-mutant embryos do not show neural tube defects (Fig. 1D).
Loss of function in critical developmental genes can lead to up-regulation of closely related factors as a compensation mechanism, as in the myogenic lineage, in which MyoD-mutant mice up-regulate Myf5 (Rudnicki et al. 1992). In Pax3-deficient somites, Pax7 is not up-regulated, and its expression is further reduced due to cell death in the absence of Pax3 (Fig. 1J,M). In the neural tube also, we detect no significant change in Pax7 expression (Fig. 1M; Supplementary Fig. S1). This is in contrast to a previous report for Sp embryos, in which up-regulation of Pax7 was described (Borycki et al. 1999). This may reflect differences in genetic background or differences between the targeted nLacZ allele and the Sp alleles from which a truncated form of Pax3 may still be generated, potentially affecting Pax7 levels. Differences in the frequency of exencephaly may reflect this. We conclude that there is no change in the spatiotemporal expression pattern of Pax7 in the absence of Pax3, and we do not detect major differences in expression levels, suggesting that the two genes are independently regulated.
To determine if Pax3 and Pax7 have similar transcriptional activities, we compared the expression of Pax3nLacZ/+ and Pax7LacZ/+ with that of a Pax3 transcriptional reporter mouse line, P34TKZ (Fig. 1R-T; Relaix et al. 2003). At E11.5, nLacZ transcripts are found at all sites of Pax3 and Pax7 expression (Fig. 1, cf. R and S and T). Notably, P34TKZ expresses nLacZ in head muscles, in which Pax7 but not Pax3 transcripts are detected (Fig. 1R-T, arrowhead). These results suggest that Pax7, similarly to Pax3, acts as a transcriptional activator in vivo.
Strategy for the replacement of Pax3 by Pax7
To investigate the functional difference between these factors, we replaced Pax3 by Pax7, using gene targeting in the mouse. The targeting strategy is outlined in Figure 2A and B. We first generated the Pax3DsRed(Pax7-ILZ) allele (Fig. 2A,B). The construct replaces the coding region in the first Pax3 exon and deletes the splice donor site to ensure that no functional Pax3 protein is made from the targeted locus (similarly to the previously reported Pax3(PAX3-FKHR-IRESnLacZ) allele, see Relaix et al. 2003). The Pax3Pax7-IRESnLacZ allele (abbreviated Pax3Pax7-ILZ) was then generated by crossing the Pax3DsRed(Pax7-ILZ) allele with a transgenic line driving ubiquitous expression of the Cre recombinase (Lallemand et al. 1998). This allele contains a mouse Pax7 coding sequence (Seale et al. 2000) followed by an IRESnLacZ reporter gene and is used in the studies presented here. The Pax3Pax7-ILZ/+ adult mice are viable and fertile and do not present the pigmentation phenotype of Pax3 heterozygous mice (Auerbach 1954), suggesting that Pax7 can complement Pax3 activity in migration of the neural crest cells, which give rise to melanocytes. The Pax3DsRed(Pax7-ILZ)/+ embryos presented the pigmentation phenotype of Pax3 (Sp) heterozygous mice, and when crossed to generate homozygous embryos, mutant embryos presented Pax3-mutant defects (such as failure of neural tube closure, deficient neural crest cell migration, lack of dorsal root ganglia and limb muscles), suggesting that no functional Pax3 is generated from this allele.
Figure 2.

Strategy and specificity of Pax7 targeting into the Pax3 locus. (A) Schematic diagram of the Pax3 locus and targeting construct. The construct contains 2.4 kb and 4 kb, respectively, of 5′ and 3′ Pax3 genomic sequence. A floxed DsRed reporter gene followed by a FRT site and then a puromycin-pA (Puro) selection marker replaces the coding sequence in exon 1 of Pax3, followed by a dicistronic cassette containing 1.8 kb of the murine Pax7 cDNA comprising the whole coding region, followed by an IRESnLacZ cassette surrounded by FRT sites and a pA signal. A counter-selection cassette encoding the A subunit of Diphtheria Toxin (DTA) was inserted at the 5′ end of the vector. The probes and restriction enzymes are indicated, with the size of the resulting wild type and recombined restriction fragments. (B) Schematic diagram of the Pax3DsRed(Pax7-ILZ) and Pax3Pax7-ILZ alleles. After homologous recombination in embryonic stem (ES) cells, Pax7-IRESnLacZ expression from the Pax3DsRed(Pax7-IRESnLacZ) allele is blocked by the floxed DsRed-FRT-puromycin-pA cassette and is therefore conditional to removal by crossing with a Cre mouse (Lallemand et al. 1998). This generates the Pax3Pax7-IRESnLacZ allele (abbreviated Pax3Pax7-ILZ) with the Pax7-FRT-IRESn-LacZ-FRT-pA preceded by a single LoxP site. Pax3Pax7-ILZ/+ mice are viable and fertile. The probes and restriction enzymes are indicated, with the size of the resulting wild type and recombined restriction fragments. (C-K′) Immunohistochemistry on transverse sections of neural tube (C-H′) and thoracic somites (I-K′) from E10.5 Pax3nLacZ/+ (C,C′,F,F′,I,I′), Pax3Pax7-ILZ/+ (D,D′,G,G′,J,J′), and Pax3Pax7-ILZ/Sp (E,E′, H,H′,K,K′) embryos using antibodies recognizing β-Gal (C-K), Pax3 (C′-K′), and Pax7 (F′-H′). Note Pax3-specific staining in the dorsal-most neural tube (C′, arrowhead) and dorsal root ganglia (C′, arrow), whereas Pax7 is excluded from these domains (F′, arrowhead and arrow). In Pax3Pax7-ILZ/+ (G′) and Pax3Pax7-ILZ/Sp (H′) embryos, Pax7 is now expressed in these structures (dorsal neural tube, arrowhead; dorsal root ganglia, arrow). Whereas Pax3 is expressed in Pax3nLacZ/+ (C′,I′) and Pax3Pax7-ILZ/+ (D′,J′), no expression is detected in Pax3Pax7-ILZ/Sp embryos, either in the neural tube and dorsal root ganglia (E′) or in somites (K′).
Because a low level of transcript was detected using a Pax3 3′ probe in the Pax3Pax7-ILZ/Pax7-ILZ embryos (Supplementary Fig. S2A,B), possibly due to a cryptic promoter or splice site, we used Pax3- and Pax7-specific antibodies to verify that the replacement strategy had worked. In a preliminary experiment, we tested the Pax3 antibody on Pax3nLacZ/nLacZ embryos and found that, although a 3′UTR transcript was also observed (Supplementary Fig. S2C,D), no Pax3 protein was made (Supplementary Fig. S2E-J). This finding is consistent with the loss-of-function phenotype observed in the Pax3nLacZ/nLacZ-mutant embryos (Fig. 3E). We then performed coimmunostaining with the Pax3 and Pax7 antibodies, which recognize a-COOH region specific to each protein, on E10.5 Pax3Pax7-ILZ/Sp embryos, in which one Pax7 knock-in allele is expressed and there is no functional Pax3 allele. The results, presented in Figure 2C-K, confirm that no Pax3 protein is detected from the Pax3Pax7-ILZ allele, either in the neural tube and dorsal root ganglia (Fig. 2, cf. E′, and E,C′) or in the somites (Fig. 2, cf. K′, and K,I). Furthermore Pax7 is now detected at sites where Pax3, but not Pax7, is normally expressed, such as the dorsal-most region of the neural tube or the dorsal root ganglia of the peripheral nervous system (Fig. 2, cf. G′,H′, and F′) as well as in the somitic hypaxial dermomyotome (data not shown). From these data we conclude that the replacement strategy is working.
Figure 3.

Embryos in which Pax7 replaces Pax3 have defects in limb muscle formation, but other aspects of the Pax3 mutation are rescued. The number of Pax7, Pax3, or Pax3-mutant (-) alleles replacing the two Pax3 endogenous alleles is indicated below the panels. Expression and phenotypes at E11.5 in Pax3nLacZ/+ (A,F,K), Pax3Pax7-ILZ/+ (B,G,L), Pax3Pax7-ILZ/Pax7-ILZ (C,H,M), Pax3Pax7-ILZ/Sp (D,I,N), and Pax3nLacZ/nLacZ (E,J,O) embryos are revealed by X-Gal staining. Whole embryos are shown in A-E, thoracic regions in F-J, and anterior somites and forelimb in K-O. Pax7 can functionally compensate for Pax3 deficiencies in dorsal neural tube closure (A-E, red arrows), somite formation (F-J, black arrow), neural crest cells, and derivatives, such as melanocytes, cranial, and dorsal root ganglia (K-O, blue arrow). However, forelimb muscles are not present in Pax3Pax7-ILZ/Sp embryos (D,N, black arrow), similarly to Pax3nLacZ/nLacZ embryos (E,O, black arrow). Homozygote Pax3Pax7-ILZ/Pax7-ILZ embryos (C,M) have an intermediate phenotype with severe reduction in forelimb musculature compared with the control Pax3nLacZ/+ embryos (A,K). Hindlimb muscles are apparently less affected, although reduced in Pax3Pax7-ILZ/Sp embryos (D, see Fig. 5).
Expression of the Pax3Pax7-ILZ allele and resulting phenotypes
The Pax3Pax7-ILZ allele permit analysis of the expression of the Pax7 coding sequences targeted to Pax3 due to the presence of the IRESnLacZ reporter gene, as illustrated in Figure 3. Comparison of the Pax3Pax7-ILZ allele with the Pax3nLacZ reporter line, which contains an nLacZ reporter gene targeted to exon 1 of Pax3 (F. Relaix and M. Buckingham, in prep.) led us to conclude that expression of the Pax3Pax7-ILZ allele is identical (Fig. 3A,B). The results shown in Figure 3 demonstrate that Pax7 is able to replace Pax3 function in dorsal neural tube closure (avoiding exencephaly and spina bifida; Fig. 3A-E), in the formation of neural crest cell derivatives in the trunk, such as melanocytes, or cranial and spinal ganglia (Fig. 3K-O) and in somitogenesis, both in somite segmentation and in hypaxial and epaxial dermomyotome development (Fig. 3F-J; cf. Fig. 2K and Supplementary Fig. S2H). However, X-Gal staining of Pax3Pax7-ILZ/Sp embryos reveals a complete lack of forelimb muscles, whereas Pax3Pax7-ILZ/Pax7-ILZ embryos have a reduction (Fig. 3K-O). This suggests that Pax7 cannot compensate for Pax3 function in forelimb muscle development.
A hallmark of the Pax gene family is sensitivity to gene dosage (Tremblay and Gruss 1994). In the Pax3Pax7-ILZ/Sp embryos (in which one copy of Pax7 replaces both Pax3 alleles), there was no melanocyte phenotype, and the spina bifida phenotype was not observed at a higher frequency than in Pax3 heterozygote embryos. This suggests that the Pax7 allele is expressed at a comparable level to that of the endogenous Pax3 gene. This was confirmed by comparing the intensity of expression of the IRESnLacZ in the Pax3Pax7-ILZ/+ allele to that of the Pax3IRESnLacZ and Pax3nLacZ/+ alleles (Fig. 3; Relaix et al. 2003). We therefore conclude that the appendicular muscle defects observed in Pax3Pax7-ILZ/Sp and Pax3Pax7-ILZ/Pax7-ILZ embryos are due to Pax7 function rather than to a reduced level of expression of this allele.
Impaired muscle formation in the limbs of Pax3Pax7-ILZ/Pax7-ILZ embryos
To further investigate limb muscle defects when Pax7 replaces Pax3, we compared transverse sections of forelimbs from Pax3Pax7-ILZ/+ and Pax3Pax7-ILZ/Pax7-ILZ embryos at E11.5 (Fig. 4A,B). In homozygote Pax3Pax7-ILZ/Pax7-ILZ limbs, the distal ventral muscles are lacking (Fig. 4B, black arrow), whereas distal dorsal muscles are reduced (Fig. 4B, blue arrow). Furthermore, the proximal muscle masses are reduced and incorrectly patterned (Fig. 4A,B, arrowheads).
Figure 4.

Forelimb muscle development is impaired in Pax3Pax7-ILZ/Pax7-ILZ embryos. (A,B) X-Gal staining of transverse sections of Pax3Pax7-ILZ/+ (A) and Pax3Pax7-ILZ/Pax7-ILZ (B) embryos at E11.5. At this stage, the ventral distal muscles are lacking (black arrow), the dorsal distal muscle masses are reduced (blue arrow), and some proximal muscle masses are missing (arrowheads) in the Pax3Pax7-ILZ/Pax7-ILZ embryos (B) compared with the Pax3Pax7-ILZ/+ embryos (A), which are indistinguishable from controls. (C-H) Immunohistochemistry on proximal to distal transverse sections of forelimb from Pax3Pax7-ILZ/+ (C,E,G) or Pax3Pax7-ILZ/Pax7-ILZ (D,F,H) fetuses at E15.5 using an antibody recognizing striated muscle myosin heavy chains shows a strong reduction which is more pronounced distally, both in the dorsal (which are the most affected) and ventral muscle masses in the homozygote mutant (D,F,H) compared with the heterozygote control (C,E,G).
To evaluate the consequence of these defects at later stages, we analyzed transverse sections of the forelimb at E15.5 along the proximal to distal axis. Staining with an anti-myosin heavy chain antibody reveals major reduction and loss in muscle masses in the Pax3Pax7-ILZ/Pax7-ILZ fetuses compared with controls (Fig. 4C-H). This follows a proximal to distal gradient: Proximally, dorsal muscle masses (extensor muscles) are lacking or severely reduced and ventral masses (flexor muscles) are reduced (Fig. 4C,D); distally, all dorsal muscles masses and almost all ventral masses are lacking (Fig. 4G,H).
Forelimb muscles are absent in embryos in which only one Pax7-ILZ allele replaces Pax3 (Fig. 3D,N). In situ hybridization with a MyoD probe at E11.5 confirms that myogenic cells are absent from the forelimbs of Pax3Pax7-ILZ/Sp embryos (Fig. 5, cf. B and A), whereas muscle masses in the trunk or head express MyoD normally. At E13.5, this is also evident (Fig. 5D) when the nLacZ reporter gene in the forelimb is only expressed in cells of the peripheral nervous system, derived from neural crest, which is rescued by Pax7 (Fig. 5F). Patterning of the peripheral nervous system appears normal in the absence of any muscle masses in the forelimb of these mutant mice.
Figure 5.
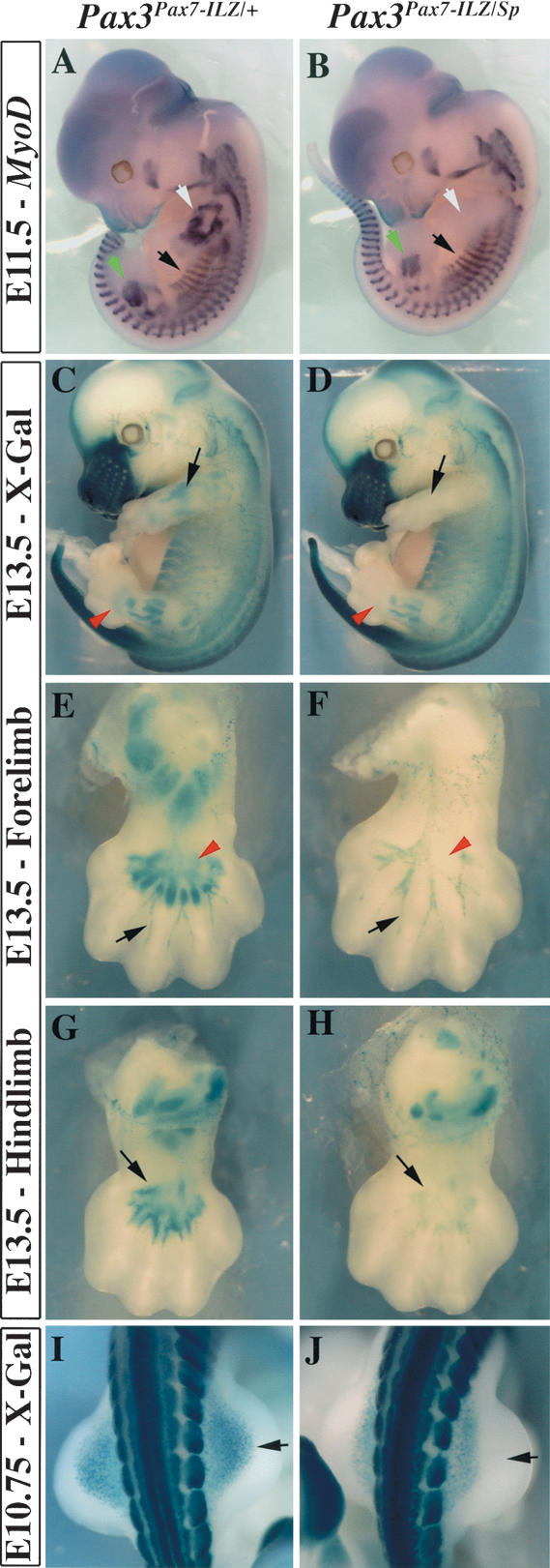
Distal hindlimb defects in Pax3Pax7-ILZ/Sp embryos. (A,B) MyoD in situ hybridization shows that myogenic cells are specifically lost in the forelimb of Pax3Pax7-ILZ/Sp embryos (B, white arrow), whereas hindlimb muscle masses are slightly reduced (green arrowhead). (A) Myotomal expression of MyoD (black arrow) like that in other trunk and head muscles is apparently not affected when compared to the control. (C,D) X-Gal staining of E13.5 Pax3Pax7-ILZ/+ (C) or Pax3Pax7-ILZ/Sp (D) embryos shows lack of forelimb muscles (black arrow) and reduced hindlimb muscles (red arrow) in a Pax3Pax7-ILZ/Sp embryo (D), compared with the control embryo (C). (E,F) X-Gal staining of the ventral forelimb of Pax3Pax7-ILZ/+ (E) or Pax3Pax7-ILZ/Sp (F) fetuses at E13.5 shows that, although muscles are completely absent from the Pax3Pax7-ILZ/Sp forelimb (red arrowhead), expression of Pax3 in the peripheral nervous system is not perturbed (black arrowhead). (G,H) X-Gal staining of Pax3Pax7-ILZ/+ (G) or Pax3Pax7-ILZ/Sp (H) in ventral hindlimbs at E13.5 shows loss of muscle masses, particularly evident for distal palm muscles in the mutant embryo (H, black arrow), compared with the control (G). (I,J) Dorsal view of the muscle progenitor cells that migrate into the hindlimb at E10.5 in Pax3Pax7-ILZ/+ (I) or Pax3Pax7-ILZ/Sp (J) embryos. Lack of distal cells in the mutant embryos is shown by a black arrow (J, cf. I).
Whereas Pax3Pax7-ILZ/Pax7-ILZ embryos do not have a hindlimb phenotype, muscle formation is abnormal in Pax3Pax7-ILZ/Sp embryos (Fig. 3A), as indicated by MyoD in situ hybridization at E11.5 (Fig. 5B). At E13.5 (Fig. 5D,H), hindlimb muscles masses are reduced and distal muscle masses of the palm are lacking in Pax3Pax7-ILZ/Sp fetuses (Fig. 5, cf. H and G).
One copy of Pax7 cannot replace Pax3 function in the delamination/migration of muscle progenitor cells
Limb muscles are formed by cells that delaminate from the hypaxial dermomyotome of somites facing the limbs and then migrate into the limb bud. We therefore investigated if the migration of these muscle progenitor cells is normal in the hindlimbs of Pax3Pax7-ILZ/Sp embryos, and found that distal cells are absent (Fig. 5I,J, black arrow). This correlates with the more pronounced effect of the mutant in the distal muscles of the limb.
Muscle progenitor cells initiate migration around the 20-somite stage (E9.5) to the forelimb and hypoglossal chord. After delamination, the cells rapidly invade the limb bud (∼22 somites in the mouse embryo; Fig. 6A,C). In Pax3Pax7-ILZ/Sp embryos, however, these cells leave the somite in the ventral region but do not migrate further into the limb field. At E10.25 (31 somites) in control embryos, migration to the forelimb is virtually complete (Fig. 6E,G). In Pax3Pax7-ILZ/Sp embryos, labeled cells are present ventrally, but myogenic progenitor cells have not invaded the limb bud (Fig. 6F,H). These results suggest that Pax7 can partially fulfill the Pax3 requirement for delamination, but one replacement allele of Pax7 cannot compensate for Pax3 function in migration (see Fig. 8, below).
Figure 6.

Reduced delamination and aberrant migration of muscle progenitor cells correlate with defective c-met expression in Pax3Pax7-ILZ/Sp embryos. (A,B) X-Gal staining of E9.5 (22 somites) Pax3Pax7-ILZ/+ (A) or Pax3Pax7-ILZ/Sp (B) embryos shows a reduced number of muscle progenitor cells invading the forelimb bud of Pax3Pax7-ILZ/Sp embryos (B) compared with the control embryo (A). In contrast neural crest cells in the arches appear normal (black arrows). (C,D) Close-up views (boxed in A,B) of the somites at the forelimb level in E9.5 (22 somites) Pax3Pax7-ILZ/+ (C) or Pax3Pax7-ILZ/Sp (D) embryos, stained with X-Gal, show delamination but not migration from the hypaxial region of the somites in Pax3Pax7-ILZ/Sp embryos (D, black arrow) compared with the control embryo (C, black arrow). Neural crest cell migration from the dorsal neural tube is normal (yellow arrow). (E,F) Close-up views of the somites opposite to the forelimb in E10.25 (31 somites) Pax3nLacZ/+ (E) or Pax3Pax7-ILZ/Sp (F) embryos, stained with X-Gal, show that in the mutant embryos, labeled cells are present ventrally to the forelimb (white arrow), but muscle progenitor cells (black arrow) have not migrated in the limb. (G,H) X-Gal staining of a transverse section from Pax3nLacZ/+ (G) or Pax3Pax7-ILZ/Sp (H) embryos at E10.25 (31 somites) shows accumulation of muscle progenitor cells ventrally to the proximal forelimb of Pax3Pax7-ILZ/Sp embryos (H, arrow) probably corresponding to cells that will contribute to the diaphragm anlagen as in Pax3nLacZ/+ embryos (G, arrow). (I,J) At E10.5 the muscle progenitor cells of the hypoglossal chord are strongly reduced and migrate aberrantly in Pax3Pax7-ILZ/Sp embryos (J) compared with the control Pax3Pax7-ILZ/+ embryos (I). (K,L) X-Gal staining of Pax3Pax7-ILZ/+ (K) or Pax3Pax7-ILZ/Sp (L) embryos at E9.75 (26 somites) shows a loss of muscle progenitor cells populating the forelimb bud (FL) and hypoglossal chord (blue arrow) in Pax3Pax7-ILZ/Sp embryos (K) compared with the control embryo (L), whereas the hypaxial dermomyotome is intact in these embryos (yellow arrow). (M,N) In situ hybridization using a c-met probe shows severely reduced expression of c-met in the hypaxial domain of occipital/cervical somites (yellow arrowhead) and at the base of the limb bud (blue arrowhead), whereas epaxial expression in the somite is normal (black arrow), at E9.75 in Pax3Pax7-ILZ/Sp embryos (N) compared with Pax3Pax7-ILZ/+ control embryos (M). (O-P′) In situ hybridization using a c-met probe at E10.5 shows complete loss of c-met transcripts in the hypaxial dermomyotome of somites at the cervical level and in the forelimb in Pax3Pax7-ILZ/Sp embryos (P, black arrowhead) compared with Pax3Pax7-ILZ/+ control embryos (O, black arrowhead). Reduction in c-met expression is also observed in the hypaxial dermomyotome of thoracic somites in Pax3Pax7-ILZ/Sp embryos compared with Pax3Pax7-ILZ/+ control embryos (P, cf. O, red arrowhead). At this stage the epaxial expression of c-met is similar in Pax3Pax7-ILZ/Sp embryos compared with Pax3Pax7-ILZ/+ control embryos (P, cf. O, yellow arrowhead). In contrast to the forelimb c-met expression is just detectable in the muscle progenitor cells migrating to the hindlimbs of Pax3Pax7-ILZ/Sp embryos (P′, arrow) compared with Pax3Pax7-ILZ/+ control embryos (O′, arrow). (Q) Transactivation of the human c-met promoter by PAX3-FKHR, Pax3-VP16, and Pax7-VP16 in 293 cells. One microgram of test construct (or empty vector) was cotransfected with 0.2 μg c-metP-TK-nLacZ with 1 μg RSV-luciferase for normalization. Data show relative β-Gal activity 36 h after transfection. Fold induction was calculated with reference to the activity measured in cells transfected with the expression vector alone. Mean values are given and the standard deviation indicated for three independent determinations. (R-T′) Immunohistochemistry on transverse sections of thoracic somites from E10.5 Pax3nLacZ/+ (Pax3+/-; R,R″), Pax3nLacZ/nLacZ (Pax3-/-; S,S″), and Pax3Pax7-ILZ/Sp (T,T″) embryos using DAPI staining (R-T), antibodies recognizing β-Gal (R′-T′) or the active form of Caspase-3 (R″-T″). Apoptotic β-Gal-positive cells that have activated Caspase-3 are found in the hypaxial somite of Pax3nLacZ/nLacZ (S″, arrowhead, also in S,S′) but not in control Pax3nLacZ/+ or Pax3Pax7-ILZ/Sp embryos (R″,T″). Note the presence of an intact hypaxial epithelial dermomyotome in Pax3Pax7-ILZ/Sp embryos (T,T′) compared with Pax3nLacZ/nLacZ (S,S′).
Figure 8.

Requirement for Pax3 function at several steps of limb muscle formation. Successive stages are depicted schematically on the left on a transverse section at the forelimb level. Defects at these different stages (formation of the hypaxial dermomyotome, delamination, migration and proliferation of muscle progenitor cells, and formation of proximal and distal, dorsal and ventral, muscle masse) with different Pax3 alleles are indicated (+, normal; -, absent or reduction is indicated). (MPC) Myogenic progenitor cells; (NT) neural tube; (Nc) notochord; (Sc) sclerotome.
Muscles of the pharynx and some tongue muscles are also derived from long-range migrating muscle progenitor cells. This stream of cells (hypoglossal chord) originates from the cervical and occipital somites and migrates through the pharyngeal region between E9.75 and E11.5. Pax3 is also required for its formation (Tajbakhsh and Buckingham 2000). At E10.5, Pax3Pax7-ILZ/Sp embryos have a severely reduced hypoglossal chord (Fig. 6, cf. J and I). This is consistent with the limb results, which suggest that Pax7 is not functionally equivalent to Pax3 for the development of muscles that require long-range migration of muscle progenitor cells. Identical results were obtained for the cells migrating to the diaphragm anlagen at the level of the ventral forelimb (data not shown). Impairment of diaphragm development probably underlies the perinatal lethality of Pax3Pax7-ILZ/Sp and Pax3Pax7-ILZ/Pax7-ILZ mouse lines (from 12 and 15 litters, respectively, there were no live neonates).
Expression of c-met is impaired in Pax3Pax7-ILZ/Sp embryos
Pax3 controls delamination and migration of muscle progenitor cells through the direct transcriptional control of c-met, which encodes a receptor tyrosine kinase essential for these processes (Bladt et al. 1995; Epstein et al. 1996; Yang et al. 1996; Relaix et al. 2003). We therefore investigated whether c-met expression is detected in embryos in which Pax7 replaces Pax3. At E9.75, in Pax3Pax7-ILZ/Sp embryos, c-met is activated in the epaxial dermomyotome but not in the hypaxial dermomyotome of cervical somites (Fig. 6M,N). At E10.5, cmet expression is not detectable in the forelimb of Pax3Pax7-ILZ/Sp embryos and is reduced in the hypaxial somites at the thoracic level (Fig. 6O,P, red arrowhead), whereas epaxial expression of c-met appears normal (Fig. 6O,P, yellow arrowhead). In hindlimbs, in which some migration of myogenic progenitor cells occurs in Pax3Pax7-ILZ/Sp embryos, c-met expression is detectable, although severely reduced (Fig. 6O′-P′). Unlike Pax3-mutant mice, in which the hypaxial dermomyotome is missing, the Pax3Pax7-ILZ/Sp allele rescues this phenotype (Fig. 6K,L), indicating that Pax7 can substitute for the antiapoptotic function of Pax3 (Borycki et al. 1999; Pani et al. 2002), as shown by labeling with an antibody to the activated form of Caspase-3, which marks apoptotic cells (Fig. 6R″-T″). This is in contrast to the effect on c-met activation. The oncogenic gain-of-function form of Pax3, PAX3-FKHR, has been shown previously to transactivate the human c-met promoter (Epstein et al. 1996). We therefore investigated whether Pax7 was able to transactivate the human c-met promoter region, because the putative Pax3-binding sites are not conserved in the mouse sequence. Pax3 and Pax7 act in vivo as transcriptional activators (see Fig. 1R-T; Relaix et al. 2003); however, both proteins on their own are very poor transactivators, probably reflecting a requirement for coactivators. We found that Pax7-VP16 was able to transactivate the c-met reporter gene better than was PAX3-FKHR or Pax3-VP16 (Fig. 6Q). This indicates that the inability of Pax7 to activate c-met in the hypaxial somite is probably due to the lack of Pax7-specific coactivator(s). We conclude that genes involved in the migration of myogenic progenitor cells are regulated by Pax3 independently of other Pax3/7 functions, probably through interaction with Pax3-specific coactivators.
The role of Pax3 in patterning limb muscles and in cell proliferation
Pax3Pax7-ILZ/Sp embryos fail to develop forelimb muscles, and homozygous Pax3Pax7-ILZ/Pax7-ILZ embryos present an intermediate phenotype, with a reduction of limb musculature (Figs. 3, 4). In these embryos, the onset of the migratory process is apparently normal, but the number of delaminating cells is reduced compared with that of control embryos (Fig. 7A,B). By E10.5, when migration to the forelimb is complete, in Pax3Pax7-ILZ/Pax7-ILZ embryos the number of muscle precursor cells located in the forelimb is severely reduced as revealed by whole-mount X-Gal staining (Fig. 7, cf. D and C, arrow). Histological examination at this stage reveals an 80% reduction of myogenic progenitor cells (Fig. 7, cf. F and E), which tend to accumulate in the dorsal and ventral-most position in the forelimb bud at this stage (Fig. 7E,F). This deficit leads to a reduction in muscle masses by E11.5 (Figs. 7G,H, 4A,B). However, strikingly, the muscle cells are organized in a pattern that mimics normal muscle development (Figs. 7G,H [arrows], 3K-M). Notably, distal palm muscles are absent (Fig. 4), although it is clear that proximal muscle masses are also affected (Fig. 7H). The overall muscle pattern in Pax3Pax7-ILZ/Pax7-ILZ embryos is very penetrant and almost identical in all embryos examined at this stage.
Figure 7.

Impaired muscle progenitor cell proliferation and patterning of muscle masses in Pax3Pax7-ILZ/Pax7-ILZ embryos. (A,B) X-Gal staining of the anterior somites of Pax3Pax7-ILZ/+ (A) or Pax3Pax7-ILZ/Pax7-ILZ (B) embryos at E9.75 (26 somites) shows reduced numbers of muscle progenitor cells invading the forelimb (red arrow) and the hypoglossal chord (black arrow) in Pax3Pax7-ILZ/Pax7-ILZ embryos (B) compared with the control embryo (A). However, anterior neural crest cell migration (blue arrowhead) appears normal. (C,D) X-Gal staining of the forelimb of Pax3Pax7-ILZ/+ (C) or Pax3Pax7-ILZ/Pax7-ILZ (D) embryos at E10.5 shows severe reduction of muscle precursor cells once the migration process is achieved (arrow). (E,F) X-Gal staining of transverse sections (plane of section shown in C,D) of the forelimb of Pax3Pax7-ILZ/+ (E) or Pax3Pax7-ILZ/Pax7-ILZ (F) embryos at E10.5 shows that the muscle progenitor cells are reduced by ∼80% and accumulate in the dorsal and ventral-most part of the limb bud (black and red arrowheads, respectively). (G,H) X-Gal staining of the forelimb of Pax3Pax7-ILZ/+ (G) or Pax3Pax7-ILZ/Pax7-ILZ (H) embryos at E11.5. Muscle masses that are reduced in the mutant embryo are indicated by a black arrow, whereas muscle masses that are undetectable are labeled by an asterisk. (I,J) In situ hybridization using a MyoD probe on the forelimb of Pax3Pax7-ILZ/+ (I) or Pax3Pax7-ILZ/Pax7-ILZ (J) embryos at E11.5. Although muscle masses are reduced in size and number, expression of MyoD is detectable in the mutant forelimb (H) compared with the control (G). (K-N) Immunohistochemistry on transverse sections of the forelimb (K-N) and dorsal root ganglia (K′-N′) of E11.5 Pax3Pax7-ILZ/+ (K,K′,M,M′) and Pax3Pax7-ILZ/Sp (L,L′,N,N′) embryos using antibodies recognizing desmin (pink; with DAPI staining in blue; K-L′) and the active form of Caspase-3 which marks apoptotic cells (bright green; with desmin in red, M-N′). At this stage, whereas apoptotic cells can be found in the dorsal root ganglia of both Pax3Pax7-ILZ/+ (M′) and Pax3Pax7-ILZ/Sp (N′) embryos, no apoptosis is detected in the limb muscle masses of Pax3Pax7-ILZ/+ (M) and Pax3Pax7-ILZ/Sp (N) embryos. (O) Proliferation of muscle precursor cells is reduced in the forelimb of Pax3Pax7-ILZ/Pax7-ILZ compared with Pax3Pax7-ILZ/+ embryos at E11.5. Coimmunostaining with an antibody that recognizes the phosphorylated form of histone H3 Ser 10 in β-Gal-positive cells gave an indication of cell proliferation. The mean percentage of β-Gal/phosphorylated histone H3 (Ser 10) double-positive cells as well as the standard deviation are indicated for four independent determinations.
In the mouse forelimb, myogenesis is initiated around E10.5 by the activation of Myf5, followed a few hours later by MyoD (Tajbakhsh et al. 1996). Because MyoD is a potential Pax3 target (Tajbakhsh et al. 1997; Relaix et al. 2003), we performed MyoD in situ hybridization, which revealed that MyoD expression is normal in the remaining muscle masses in Pax3Pax7-ILZ/Pax7-ILZ embryos (Fig. 7I,J).
The reduction in limb musculature observed in the Pax3Pax7-ILZ/Pax7-ILZ embryos is due to a reduction in the migration of muscle progenitor cells, but it may also reflect a reduction in their proliferation or, alternatively, an increase in apoptosis. We found no significant apoptosis in the limb muscle masses of E11.5 Pax3Pax7-ILZ/Pax7-ILZ embryos compared with the control (Fig. 7K-N). We therefore examined the proliferation of these cells in Pax3Pax7-ILZ/Pax7-ILZ embryos compared with control embryos by scoring the percentage of β-galactosidase (β-Gal)-positive cells (which marks all the muscle progenitor cells at this stage) within the forelimb that are in mitosis, by labeling with an antibody directed against the Ser 10 phosphorylated form of histone H3. We found that at E11.5 the forelimbs of Pax3Pax7-ILZ/Pax7-ILZ embryos contained 2.9% of β-Gal/H3-P double-positive cells, versus 4.7% in the control Pax3Pax7-ILZ/+ embryos (Fig. 7O). From this, we conclude that the proliferative capacity of muscle progenitor cells is reduced by 40% when two Pax7 alleles replace a single Pax3 allele.
In addition to the role of Pax3 in the hypaxial dermomyotome, which is replaced by Pax7, Pax7 only partially fulfills the function of Pax3 in the delamination, migration, and proliferation of limb muscle progenitor cells and in the establishment of individual muscle masses (Fig. 8).
Discussion
Pax3 and Pax7 proteins are functionally equivalent in somite, neural tube, and neural crest development
Although Pax3 and Pax7 are expressed in partially overlapping domains in the mouse embryo, mice mutated in each of these genes have very contrasting phenotypes. To investigate whether these are caused by differences in temporal expression or biochemical activity of the Pax3 and Pax7 proteins, we replaced Pax3 by a Pax7 coding sequence, using gene targeting in the mouse. Our data show that Pax7, generated from one Pax3 allele, can functionally compensate for Pax3 at all sites where it is expressed, with the exception of the muscle progenitor cells, which migrate from the somites to form distant muscle masses such as those in the limbs. Therefore, differences in spatiotemporal expression probably account for phenotypic differences between these two mouse mutants in the dorsal neural tube, in neural crest and its derivatives, and in somites and trunk musculature. The function of Pax3/7 in the dorsal neural tube, neural crest, and somite probably existed before the Pax3/7 gene duplication at the onset of vertebrate evolution.
Of the three other Pax subfamilies in vertebrates—Pax1/9, Pax4/6, and Pax2/5/8—a previous report of gene replacement of Pax2 by Pax5 showed that these transcription factors have maintained equivalent biochemical functions, because Pax5 was able to compensate functionally for all Pax2 functions in the mouse (Bouchard et al. 2000). Moreover, replacement of closely related genes in families of transcription factors that play important regulatory roles in development—such as the bHLH transcription factors myogenin and Myf5 (Wang et al. 1996), or MesP1 and MesP2 (Saga 1998), the homeobox transcription factors Otx1 and Otx2 (Suda et al. 1999), or En1 and En2 (Hanks et al. 1995), the components of the AP-1 transcription factor Jun and JunB (Passegue et al. 2002), or Fra-1 and c-Fos (Fleischmann et al. 2000)—has been reported to suggest conservation of function.
However, gene replacement of more distantly related transcription factors, such as the homeobox-containing proteins Emx2 or Otx2 (Suda et al. 2001) or the proneural genes Mash1 and Ngn2 (Parras et al. 2002), demonstrated that these proteins are not functionally equivalent, and that antiquity in evolution and divergence of biochemical functions are directly linked.
Pax3 and Pax7 function in neural crest and nervous system development
Pax7-mutant mice have subtle embryonic abnormalities in facial ossification that almost disappear on a mixed genetic background. Defects in anterior neural crest cells are believed to account for this phenotype (Mansouri et al. 1996b). Splotch mice have no facial phenotype, although, in contrast, a function for Pax3 has been described in human facial development, in which PAX3 mutations lead to Waardenburg syndrome types I and III, a rare (1/40,000) disorder characterized clinically by the association of craniofacial dysmorphism, pigmentation defects, and severe sensorineural congenital hearing impairment (Tassabehji et al. 1993). This congenital disorder is likely to be caused by defective function of the embryonic neural crest. In contrast, no human developmental syndrome has been associated so far with mutations in PAX7.
Spontaneous mutations in the Pax3 locus were first identified and analyzed 50 years ago (Auerbach 1954), and these deficiencies have been well documented in the mouse embryo since then. Our Pax3nlacZ allele illustrates the deficiencies of the mutant embryo in many sites where Pax3 is expressed: for example, in the developing neural tube, where Pax7 and Pax3 expression overlaps in the alar plate. At this level, Pax3 transcription is initiated at the neural plate stage and precedes that of Pax7, which begins after neural tube closure (Jostes et al. 1990; Goulding et al. 1991). In addition, Pax7 is never expressed in the roof plate. As a consequence, Pax3-deficient mice exhibit neural tube defects such as spina bifida and, with a lower frequency, exencephaly (Fig. 4). Migrating neural crest cells in the head and body also express Pax3 (Fig. 6A,C) as do neural crest cell derivatives, such as melanocytes or the dorsal root ganglia. In the absence of Pax3, there is almost no neural crest cell migration from the dorsal neural tube in the trunk, which leads to the almost complete absence of the peripheral nervous system and melanocytes (Tremblay et al. 1995). The severity of the neural crest cell defects increases along the rostrocaudal axis (Auerbach 1954), although the molecular basis remains unclear. Because Pax7 is expressed in anterior neural crest cells (Mansouri et al. 1996b), it is possible that an anteroposterior gradient of Pax7 expression underlies this phenotype. Our demonstration that Pax7 can replace Pax3 in the dorsal neural tube and neural crest points to a common function for the two proteins and suggests its pre-existence in the nervous system before the Pax3/7 gene duplication at the onset of vertebrate evolution rather than coevolution. This may also apply to the neural crest function because the Amphi-Pax3/7 is expressed in a cellular population comparable to the premigratory neural crest cells of vertebrates (Holland et al. 1999).
Pax3 and Pax7 functions in somatogenesis and myogenesis
In addition to its role in the nervous system and in neural crest, Pax3 has important functions in somitogenesis. This role has been acquired during evolution because the primitive Pax3/7 gene is not expressed in mesoderm. Again, it is in Amphioxus that this function is observed (Holland et al. 1999). Our results demonstrate that Pax7 can efficiently substitute for Pax3 during somite segmentation and in the development and maintenance of the dermomyotome. Furthermore, myogenesis proceeds normally in the trunk in the absence of Pax3. This therefore suggests that during vertebrate evolution, Pax7 has retained the biochemical activity that is now mainly exercised by Pax3 during somitogenesis in the mouse.
Pax3 also has a critical function in appendicular muscle formation and in that of other muscles, such as those derived from the hypoglossal chord, that also depend on the long-range migration of muscle progenitor cells from the hypaxial somite. Pax7 is not expressed in these migrating cells and is only transcribed later as limb muscles develop. It was therefore not clear whether the Pax3-mutant phenotype reflected the lack of Pax7 in these cells. The fact that the hypaxial dermomyotome is lost in the mutant further complicates any interpretation. However, when Pax7 replaces Pax3, the hypaxial dermomyotome, which is the source of the migratory cells, is present, and in this situation it is now possible to analyze the role of Pax7 in these myogenic progenitor cells. The results demonstrate that Pax7 is not equivalent to Pax3 in the context of limb muscle formation, and this also applies to other muscles that depend on long-range migration, such as the diaphragm or those derived from the hypoglossal chord.
Both Pax3 and Pax7 genes may generate more than one transcript and protein isoform (see Ziman and Kay 1998; Barber et al. 1999), although it has not been shown that these are functionally significant. Because a sequence encoding a single isoform of Pax7 was inserted into the Pax3 locus, it is formally possible that other isoforms of Pax7 may be able to perform specific functions such as those required for limb muscle formation. The isoform that was targeted to Pax3 lacks the first of three putative ATGs, and thus potentially lacks 10 N-terminal amino acid residues. It has been proposed that the first 90 N-terminal amino acids of Pax3 may contribute to repressor activity (Chalepakis et al. 1994), but transactivation experiments showed no difference between the activity of this isoform of Pax7 and Pax3 (data not shown), suggesting that the repression domain does not include these potential N-terminal residues of the Pax7 protein. In addition, we have previously shown that Pax3 acts genetically as a transcriptional activator during limb muscle formation (Relaix et al. 2003). The stability of the Pax7 transcript or protein produced from the knock-in allele may be different from that of endogenous Pax3, leading to altered relative protein levels, which might affect limb muscle formation if this is more sensitive to dose than other sites of myogenesis. However, the nLacZ reporter gene expression and the antibody staining shown here would suggest that any differences in mRNA or protein levels between the Pax3nLacZ or the Pax3Pax7-ILZ alleles are probably minor.
Pax3 controls several steps in limb muscle development
Pax3Pax7-ILZ/Sp embryos (with one copy of Pax7 replacing both Pax3 alleles) and Pax3Pax7-ILZ/Pax7-ILZ embryos (with two copies of Pax7 replacing both Pax3 alleles) have graded defects in limb formation, showing that Pax7 acts as a hypomorph of Pax3, revealing previously hidden functions for Pax3 in this process (Fig. 8). Defects in hypoglossal chord formation also clearly illustrate this hypomorphic activity of the Pax7 replacement allele.
As a result of early apoptosis, Pax3-deficient embryos lack the hypaxial dermomyotome from which the migrating myogenic progenitor cells delaminate. The role of Pax3 in maintaining this structure is similar to the situation in the dorsal neural tube, where Pax3 has a role in blocking p53-mediated apoptosis (Pani et al. 2002). Because the hypaxial somite is intact in Pax3Pax7-ILZ/Sp or Pax3Pax7-ILZ/Pax7-ILZ embryos, Pax7 clearly rescues this early function, suggesting that, similarly to Pax3 (Borycki et al. 1999; Relaix et al. 2003), it may target anti-apoptotic genes.
In the presence of an intact hypaxial dermomyotome, myogenic progenitor cells expressing two Pax7 alleles in place of Pax3 will form apparently normal skeletal musculature in the hindlimb, whereas in the forelimb this is already compromised. In the presence of only one Pax7 replacement allele, myogenesis in the hindlimb is also defective and there is no muscle formation in the fore limb. This anterior/posterior difference may be related to the developmental timing of limb formation, with a higher threshold for Pax3/7 activity initially. In addition, embryonic limbs differ in the expression of genes such as Tbx4 and 5, expressed in fore- and hindlimbs, respectively (Gibson-Brown et al. 1996). Such differences in the regulatory environment may affect the behavior of myogenic cells. The Hox code is another potential modulator of myogenesis on the anterior/posterior axis (Alvares et al. 2003).
Myogenic progenitor cells delaminate from the epithelium of the dermomyotome and then migrate into the limb field. Some delamination occurs, even in situations in which there is no subsequent migration to the limb, as seen in Pax3Pax7-ILZ/Sp embryos. There is therefore uncoupling between the two processes. When limb muscle formation occurs but is compromised, migration of muscle progenitor cells appears to be retarded, which may explain why the most distal palm muscles are lacking in the hindlimbs of Pax3Pax7-ILZ/Sp embryos. However, in this situation some proximal muscles are also no longer detectable, and there is an overall reduction in both delamination and migration. A key effector of these processes is the tyrosine kinase receptor, Met (Yang et al. 1996). In Pax3Pax7-ILZ/Sp embryos, c-met expression is barely detectable hypaxially. A few cells are still able to delaminate from the hypaxial dermomyotome, suggesting that some residual Met signaling activity is still present. However, the myogenic progenitor cells are unable to migrate to invade the forelimb bud in these embryos, indicating that a threshold level of Met is not reached and suggesting that in the absence of efficient Met signaling the cells that have delaminated cannot enter the limb. These results further confirm the role of Pax3 in the transcriptional activation of c-met (Epstein et al. 1996; Relaix et al. 2003) and demonstrate that Pax7 performs this function much less efficiently. In addition, our data show that a Pax7-VP16 protein can transactivate a c-met reporter construct as well if not better than the equivalent Pax3 fusion protein, indicating that it binds efficiently. Because Pax3 and Pax7 alone are very poor transcriptional activators, the functional difference between Pax3 and Pax7 in c-met activation probably lies in the recruitment of specific coactivator(s). Mutants for the Gab1 adapter protein, an important mediator of Met signaling, display limb muscle phenotypes similar to those observed in Pax3Pax7-ILZ/Pax7-ILZ embryos (Sachs et al. 2000), suggesting that reduced c-met expression mainly accounts for the limb muscle phenotype. Surprisingly, c-met expression in the epaxial domain of the somite is maintained in the presence of only one replacement allele of Pax7. In Pax3-mutant mice the epaxial, similarly to the hypaxial, extremity of the dermomyotome is lost, and it is therefore not possible to conclude about Pax3 regulation of this site of c-met expression. However, in a gain-of-function mutant due to the presence of PAX3-FKHR, c-met is also up-regulated in this domain (Relaix et al. 2003). Epaxial expression may also depend on transcriptional regulators other than Pax3. The role of Met signaling in the epaxial somite is not clear, because the mutants do not have epaxial defects (Bladt et al. 1995; Dietrich et al. 1999; Relaix et al. 2003). This expression may be an evolutionary remnant of an epaxial function of Met lost in amniotes, required, for example, in dorsal fin formation of fish.
Myogenic progenitor cells in the limb proliferate, and we show that the effects of a reduction in the number of these cells that reach the limb bud is compounded by a defect in proliferation (but not apoptosis) in the presence of a Pax7 replacement allele. Met plays a role in tumorigenesis and proliferation (Danilkovitch-Miagkova and Zbar 2002; Trusolino and Comoglio 2002). In skeletal muscle it has been implicated in the proliferation of fetal, but not embryonic, myoblasts (Maina et al. 1996). In addition to c-met, Pax3 probably targets other genes involved in cell cycle regulation, as suggested by microarray analysis on cultured cells (Khan et al. 1999; Mayanil et al. 2001).
When limb muscle masses are reduced, on Pax3 replacement by Pax7, differentiation appears to proceed normally, with activation of MyoD, a Pax3 target (Tajbakhsh et al. 1997; Relaix et al. 2003). It has been suggested that Pax3 itself is not required for muscle differentiation in the limb, based on transplantation experiments of myogenic progenitor cells from Sp-mutant embryos into the chick wing bud where they formed muscle (Daston et al. 1996). It is striking that although they are reduced in size, the overall patterning of muscle masses is retained, as seen in the hindlimbs of Pax3Pax7-ILZ/Sp mice. Some muscles are, however, lost. Loss of distal muscles may reflect a failure of migrating progenitor cells to reach these locations, as discussed. More surprising is the apparent loss of specific proximal muscles. It is possible that this reflects a reduction in muscle progenitor cells at this site, due to migration and proliferation defects, below a threshold level required for a community effect, leading to skeletal muscle formation (Cossu and Borello 1999; Buckingham 2003).
Pax3/7 function in myogenic progenitor cells and evolutionary history of vertebrate appendicular muscles
In the vertebrate clade, the ancestral mechanism for appendicular muscle development involves the direct extension of the epithelial (dermo)myotome into the fin or limb bud. Subsequently, a mechanism for the delamination and migration of muscle progenitor cells (Haines and Currie 2001) may have evolved in teleost fishes, just prior to tetrapod radiation (Neyt et al. 2000). However, as pointed out by Galis (2001), there is a highly mosaic distribution of these characters in the vertebrate clade as well as the presence of an intermediate mode (with myotomal extension then short range migration) in some reptiles and amphibians, suggesting that evolution of vertebrate appendicular muscle formation is complex. Our observation that Pax7 only partially fulfills the functions of Pax3 in limb muscle formation, whereas it performs the role of Pax3 in somitogenesis and trunk myogenesis as well as in the neural tube and neural crest, is consistent with the later evolution of this myogenic mode and demonstrates the central importance of Pax3 in this development. Two possible hypotheses can be formulated for the hypomorphic Pax7 activity in appendicular muscle development: (1) either this Pax3 function evolved independently after Pax3/7 duplication, reusing biochemical functions already partially present in the Pax3/7 protein (e.g., the ancestral function in myotomal ventral body wall formation or myotomal extension into the fin or limb bud), or (2) this function was already present in the Pax3/7 protein before gene duplication and was partially lost during independent Pax7 evolution in vertebrates. The acquisition of molecular properties such as interaction with cofactors that distinguish Pax3 from Pax7 and the ancestral Pax3/Pax7 sequence was therefore probably an essential prerequisite for the evolution of vertebrate appendicular muscle.
Materials and methods
Targeting vectors and mice
The targeting construct is derived from one which was previously reported (Relaix et al. 2003). Cloning details are available on request. Briefly, the Pax3DsRed(Pax7-ILZ) allele contains 2.4 kb of 5′ genomic region, replacing the coding sequence of exon 1 and 4 kb of 3′ sequence containing exons 2-4. The genomic sequences surround a Floxed Dsred-FRT-Puro cassette followed by 1.8 kb mouse Pax7 cDNA (kindly provided by M. Rudnicki [Ottawa Health Research Institute, Ottawa, Canada], accession no. NM_011039; we confirmed by sequencing that the Pax7 sequence corresponds to that originally published [Seale et al. 2000], which starts with two consecutive ATGs, as putative translational start sites), then an IRESnLacZ cassette surrounded by FRT sites. In addition, a PGK-DTA cassette encoding the A subunit of the Diphtheria toxin gene (Meilhac et al. 2003) was inserted 5′ of the constructs to allow negative selection in ES cells. The targeting vector was electroporated in CK35 ES cells (Kress et al. 1998). ES cells were selected and screened for recombination events by Southern blot analysis using EcoRV digests and a 5′-flanking probe and were verified by using 3′ and internal probes (Fig. 2). Targeted ES cells were recovered with a 0.5%-1% frequency and injected into blastocysts to generate chimaeras. Germline-transmitted alleles were identified by the classical Splotch heterozygote phenotype (lack of melanocyte colonization of the belly), and by PCR or by Southern blotting. Cre transgenic mice have been described previously (Lallemand et al. 1998). Generation of the P34TKZ line was reported previously (Relaix et al. 2003).
X-Gal staining, histology, immunohistochemistry, whole-mount in situ hybridization, and whole-mount immunohistochemistry
Mouse embryos were collected after natural overnight matings; for staging, fertilization was considered to take place at 6 a.m. For X-Gal staining, dissected embryos were fixed for 10-30 min (depending on the stage) with 4% paraformaldehyde (PAF) in PBS, on ice. Embryos were rinsed twice with PBS and then stained with X-Gal (Roche), using 0.4 mg/mL X-Gal in 2 mM MgCl2, 0.02% NP-40, 0.1 M PBS (pH 7.5), 20 mM K4Fe(CN)6, and 20 mM K3Fe(CN)6 for 4-16 h at 37°C, with shaking. Embryos were rinsed in PBS and postfixed overnight in 4% PAF. For histological analysis, sections (12 μm) were prepared from X-Gal colored embryos and stained with eosin according to standard procedures. Genotyping for whole-mount in situ hybridization was carried out by PCR or X-Gal staining in X-Gal + 0.2% PAF for 30 min following 1-2-h fixation in 4% PAF, on ice. When light color had developed (20′-30′), embryos were rinsed in PBS and postfixed overnight in 4% PAF. Whole-mount in situ hybridization with digoxigenin-labeled riboprobes was performed as described (Tajbakhsh et al. 1997). The MyoD riboprobe was as previously described (Tajbakhsh et al. 1997). The c-met probe was kindly provided by C. Birchmeier (Max Delbrück Center for Molecular Medicine, Berlin, Germany; Bladt et al. 1995). The Pax7 probe was obtained from M. Rudnicki (Seale et al. 2000). Fluorescent coimmunohistochemistry was carried out as described previously (Relaix et al. 2003), using the following antibodies: polyclonal anti-β-galactosidase (Molecular Probe. Diluted 1:200), polyclonal anti-MHC (obtained from G. Cossu, Stem Cell Research Institute, H.S. Raffaele, Milan, Italy, 1:200), monoclonal anti-phospho histone H3 (Cell Signaling, 1:100), monoclonal anti-β-Gal (Sigma, 1:200), monoclonal anti-Pax7 (Developmental Studies Hybridoma Bank, 1:100), monoclonal anti-Pax3 (kindly provided by M. Bronner-Fraser, California Institute of Technology, Pasadena, CA, 1:100), monoclonal antidesmin (Dako, 1:200), and polyclonal anti-active Caspase 3 (Pharmingen, 1:250).
Transient transfection experiments
The human c-met promoter (3 kb) was amplified by PCR from mouse genomic DNA and cloned in front of the TK-nLacZpA plasmid (Relaix et al. 2003). Transient transfections, β-Gal, and luciferase assays were performed as described previously (Relaix et al. 2003).
Acknowledgments
We thank Catherine Bodin for excellent histology work, Dominique Michel for technical assistance, and the Buckingham laboratory for helpful discussions. The work in M.B.'s laboratory was supported by the Pasteur Institute and the CNRS, and by grants from the AFM and the AC Integrative Biology program of the MJER. F.R. is supported by Inserm.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.301004.
Supplemental material is available at http://www.genesdev.org.
References
- Alvares L.E., Schubert, F.R., Thorpe, C., Mootoosamy, R.C., Cheng, L., Parkyn, G., Lumsden, A., and Dietrich, S. 2003. Intrinsic, Hox-dependent cues determine the fate of skeletal muscle precursors. Dev. Cell 5: 379-390. [DOI] [PubMed] [Google Scholar]
- Auerbach R. 1954. Analysis of the developmental effects of a lethal mutation in the house mouse. J. Exper. Zool. 127: 305-329. [Google Scholar]
- Barber T.D., Barber, M.C., Cloutier, T.E., and Friedman, T.B. 1999. PAX3 gene structure, alternative splicing and evolution. Gene 237: 311-319. [DOI] [PubMed] [Google Scholar]
- Barr F.G. 2001. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 20: 5736-5746. [DOI] [PubMed] [Google Scholar]
- Baumgartner S., Bopp, D., Burri, M., and Noll, M. 1987. Structure of two genes at the gooseberry locus related to the paired gene and their spatial expression during Drosophila embryogenesis. Genes & Dev. 1: 1247-1267. [DOI] [PubMed] [Google Scholar]
- Bladt F., Riethmacher, D., Isenmann, S., Aguzzi, A., and Birchmeier, C. 1995. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 376: 768-771. [DOI] [PubMed] [Google Scholar]
- Bober E., Franz, T., Arnold, H.H., Gruss, P., and Tremblay, P. 1994. Pax-3 is required for the development of limb muscles: A possible role for the migration of dermomyotomal muscle progenitor cells. Development 120: 603-612. [DOI] [PubMed] [Google Scholar]
- Borycki A.G., Li, J., Jin, F., Emerson, C.P., and Epstein, J.A. 1999. Pax3 functions in cell survival and in pax7 regulation. Development 126: 1665-1674. [DOI] [PubMed] [Google Scholar]
- Bouchard M., Pfeffer, P., and Busslinger, M. 2000. Functional equivalence of the transcription factors Pax2 and Pax5 in mouse development. Development 127: 3703-3713. [DOI] [PubMed] [Google Scholar]
- Buckingham M. 2003. How the community effect orchestrates muscle differentiation. Bioessays 25: 13-16. [DOI] [PubMed] [Google Scholar]
- Buckingham M., Bajard, L., Chang, T., Daubas, P., Hadchouel, J., Meilhac, S., Montarras, D., Rocancourt, D., and Relaix, F. 2003. The formation of skeletal muscle: From somite to limb. J. Anat. 202: 59-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalepakis G., Jones, F.S., Edelman, G.M., and Gruss, P. 1994. Pax-3 contains domains for transcription activation and transcription inhibition. Proc. Natl. Acad. Sci. 91: 12745-12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy I.M. and Rando, T.A. 2002. The regulation of notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell 3: 397-409. [DOI] [PubMed] [Google Scholar]
- Cossu G. and Borello, U. 1999. Wnt signaling and the activation of myogenesis in mammals. EMBO J. 18: 6867-6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilkovitch-Miagkova A. and Zbar, B. 2002. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J. Clin. Invest. 109: 863-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daston G., Lamar, E., Olivier, M., and Goulding, M. 1996. Pax-3 is necessary for migration but not differentiation of limb muscle precursors in the mouse. Development 122: 1017-1027. [DOI] [PubMed] [Google Scholar]
- Davis R.J., D'Cruz, C.M., Lovell, M.A., Biegel, J.A., and Barr, F.G. 1994. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 54: 2869-2872. [PubMed] [Google Scholar]
- Dearden P.K., Donly, C., and Grbic, M. 2002. Expression of pair-rule gene homologues in a chelicerate: Early patterning of the two-spotted spider mite Tetranychus urticae. Development 129: 5461-5472. [DOI] [PubMed] [Google Scholar]
- Dietrich S., Abou-Rebyeh, F., Brohmann, H., Bladt, F., Sonnenberg-Riethmacher, E., Yamaai, T., Lumsden, A., Brand-Saberi, B., and Birchmeier, C. 1999. The role of SF/HGF and c-Met in the development of skeletal muscle. Development 126: 1621-1629. [DOI] [PubMed] [Google Scholar]
- Epstein D.J., Vogan, K.J., Trasler, D.G., and Gros, P. 1993. A mutation within intron 3 of the Pax-3 gene produces aberrantly spliced mRNA transcripts in the splotch (Sp) mouse mutant. Proc. Natl. Acad. Sci. 90: 532-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J.A., Shapiro, D.N., Cheng, J., Lam, P.Y., and Maas, R.L. 1996. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc. Natl. Acad. Sci. 93: 4213-4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann A., Hafezi, F., Elliott, C., Reme, C.E., Ruther, U., and Wagner, E.F. 2000. Fra-1 replaces c-Fos-dependent functions in mice. Genes & Dev. 14: 2695-2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz T. 1989. Persistent truncus arteriosus in the Splotch mutant mouse. Anat. Embryol. (Berl) 180: 457-464. [DOI] [PubMed] [Google Scholar]
- Franz T., Kothary, R., Surani, M.A., Halata, Z., and Grim, M. 1993. The Splotch mutation interferes with muscle development in the limbs. Anat. Embryol. (Berl) 187: 153-160. [DOI] [PubMed] [Google Scholar]
- Galili N., Davis, R.J., Fredericks, W.J., Mukhopadhyay, S., Rauscher III, F.J., Emanuel, B.S., Rovera, G., and Barr, F.G. 1993. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 5: 230-235. [DOI] [PubMed] [Google Scholar]
- Galis F. 2001. Evolutionary history of vertebrate appendicular muscle. Bioessays 23: 383-387. [DOI] [PubMed] [Google Scholar]
- Gibson-Brown J.J., Agulnik, S.I., Chapman, D.L., Alexiou, M., Garvey, N., Silver, L.M., and Papaioannou, V.E. 1996. Evidence of a role for T-box genes in the evolution of limb morphogenesis and the specification of forelimb/hindlimb identity. Mech. Dev. 56: 93-101. [DOI] [PubMed] [Google Scholar]
- Goulding M., Lumsden, A., and Paquette, A.J. 1994. Regulation of Pax-3 expression in the dermomyotome and its role in muscle development. Development 120: 957-971. [DOI] [PubMed] [Google Scholar]
- Goulding M.D., Chalepakis, G., Deutsch, U., Erselius, J.R., and Gruss, P. 1991. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 10: 1135-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruss P. and Walther, C. 1992. Pax in development. Cell 69: 719-722. [DOI] [PubMed] [Google Scholar]
- Haines L. and Currie, P.D. 2001. Morphogenesis and evolution of vertebrate appendicular muscle. J. Anat. 199: 205-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks M., Wurst, W., Anson-Cartwright, L., Auerbach, A.B., and Joyner, A.L. 1995. Rescue of the En-1 mutant phenotype by replacement of En-1 with En-2. Science 269: 679-682. [DOI] [PubMed] [Google Scholar]
- Hobert O. and Ruvkun, G. 1999. Pax genes in Caenorhabditis elegans: A new twist. Trends Genet. 15: 214-216. [DOI] [PubMed] [Google Scholar]
- Holland L.Z., Schubert, M., Kozmik, Z., and Holland, N.D. 1999. AmphiPax3/7, an amphioxus paired box gene: Insights into chordate myogenesis, neurogenesis, and the possible evolutionary precursor of definitive vertebrate neural crest. Evol. Dev. 1: 153-165. [DOI] [PubMed] [Google Scholar]
- Jostes B., Walther, C., and Gruss, P. 1990. The murine paired box gene, Pax7, is expressed specifically during the development of the nervous and muscular system. Mech. Dev. 33: 27-37. [DOI] [PubMed] [Google Scholar]
- Khan J., Bittner, M.L., Saal, L.H., Teichmann, U., Azorsa, D.O., Gooden, C.G., Pavan, W.J., Trent, J.M., and Meltzer, P.S. 1999. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc. Natl. Acad. Sci. 96: 13264-13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress C., Vandormael-Pournin, S., Baldacci, P., Cohen-Tannoudji, M., and Babinet, C. 1998. Nonpermissiveness for mouse embryonic stem (ES) cell derivation circumvented by a single backcross to 129/Sv strain: Establishment of ES cell lines bearing the Omd conditional lethal mutation. Mamm. Genome 9: 998-1001. [DOI] [PubMed] [Google Scholar]
- Lallemand Y., Luria, V., Haffner-Krausz, R., and Lonai, P. 1998. Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res 7: 105-112. [DOI] [PubMed] [Google Scholar]
- Le Douarin N. and Kalcheim, C. 1999. The Neural crest. Cambridge University Press, New York.
- Li X. and Noll, M. 1994. Evolution of distinct developmental functions of three Drosophila genes by acquisition of different cis-regulatory regions. Nature 367: 83-87. [DOI] [PubMed] [Google Scholar]
- Lu J.R., Bassel-Duby, R., Hawkins, A., Chang, P., Valdez, R., Wu, H., Gan, L., Shelton, J.M., Richardson, J.A., and Olson, E.N. 2002. Control of facial muscle development by MyoR and capsulin. Science 298: 2378-2381. [DOI] [PubMed] [Google Scholar]
- Maina F., Casagranda, F., Audero, E., Simeone, A., Comoglio, P.M., Klein, R., and Ponzetto, C. 1996. Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell 87: 531-542. [DOI] [PubMed] [Google Scholar]
- Mansouri A. and Gruss, P. 1998. Pax3 and Pax7 are expressed in commissural neurons and restrict ventral neuronal identity in the spinal cord. Mech. Dev. 78: 171-178. [DOI] [PubMed] [Google Scholar]
- Mansouri A., Hallonet, M., and Gruss, P. 1996a. Pax genes and their roles in cell differentiation and development. Curr. Opin. Cell. Biol. 8: 851-857. [DOI] [PubMed] [Google Scholar]
- Mansouri A., Stoykova, A., Torres, M., and Gruss, P. 1996b. Dysgenesis of cephalic neural crest derivatives in Pax7-/- mutant mice. Development 122: 831-838. [DOI] [PubMed] [Google Scholar]
- Marcelle C., Wolf, J., and Bronner-Fraser, M. 1995. The in vivo expression of the FGF receptor FREK mRNA in avian myoblasts suggests a role in muscle growth and differentiation. Dev. Biol. 172: 100-114. [DOI] [PubMed] [Google Scholar]
- Mayanil C.S., George, D., Freilich, L., Miljan, E.J., Mania-Farnell, B., McLone, D.G., and Bremer, E.G. 2001. Microarray analysis detects novel Pax3 downstream target genes. J. Biol. Chem. 276: 49299-42309. [DOI] [PubMed] [Google Scholar]
- Meilhac S.M., Kelly, R.G., Rocancourt, D., Eloy-Trinquet, S., Nicolas, J.E., and Buckingham, M.E. 2003. A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development 130: 3877-3889. [DOI] [PubMed] [Google Scholar]
- Neyt C., Jagla, K., Thisse, C., Thisse, B., Haines, L., and Currie, P.D. 2000. Evolutionary origins of vertebrate appendicular muscle. Nature 408: 82-86. [DOI] [PubMed] [Google Scholar]
- Noll M. 1993. Evolution and role of Pax genes. Curr. Opin. Genet. Dev. 3: 595-605. [DOI] [PubMed] [Google Scholar]
- Pani L., Horal, M., and Loeken, M.R. 2002. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: Implications for Pax-3-dependent development and tumorigenesis. Genes & Dev. 16: 676-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parras C.M., Schuurmans, C., Scardigli, R., Kim, J., Anderson, D.J., and Guillemot, F. 2002. Divergent functions of the proneural genes Mash1 and Ngn2 in the specification of neuronal subtype identity. Genes & Dev. 16: 324-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegue E., Jochum, W., Behrens, A., Ricci, R., and Wagner, E.F. 2002. JunB can substitute for Jun in mouse development and cell proliferation. Nat. Genet. 30: 158-166. [DOI] [PubMed] [Google Scholar]
- Relaix F., Polimeni, M., Rocancourt, D., Ponzetto, C., Schäfer, B.W., and Buckingham, M. 2003. The transcriptional activator PAX3-FKHR rescues the defects of Pax3 mutant mice but induces a myogenic gain of function phenotype with ligand-independent activation of Met signaling in vivo. Genes & Dev. 17: 2950-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki M.A., Braun, T., Hinuma, S., and Jaenisch, R. 1992. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell 71: 383-390. [DOI] [PubMed] [Google Scholar]
- Sachs M., Brohmann, H., Zechner, D., Muller, T., Hulsken, J., Walther, I., Schaeper, U., Birchmeier, C., and Birchmeier, W. 2000. Essential role of Gab1 for signaling by the c-Met receptor in vivo. J. Cell. Biol. 150: 1375-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saga Y. 1998. Genetic rescue of segmentation defect in MesP2-deficient mice by MesP1 gene replacement. Mech. Dev. 75: 53-66. [DOI] [PubMed] [Google Scholar]
- Schubert F.R., Tremblay, P., Mansouri, A., Faisst, A.M., Kammandel, B., Lumsden, A., Gruss, P., and Dietrich, S. 2001. Early mesodermal phenotypes in splotch suggest a role for Pax3 in the formation of epithelial somites. Dev. Dyn. 222: 506-521. [DOI] [PubMed] [Google Scholar]
- Seale P., Sabourin, L.A., Girgis-Gabardo, A., Mansouri, A., Gruss, P., and Rudnicki, M.A. 2000. Pax7 is required for the specification of myogenic satellite cells. Cell 102: 777-786. [DOI] [PubMed] [Google Scholar]
- Seo H.C., Saetre, B.O., Havik, B., Ellingsen, S., and Fjose, A. 1998. The zebrafish Pax3 and Pax7 homologues are highly conserved, encode multiple isoforms and show dynamic segment-like expression in the developing brain. Mech. Dev. 70: 49-63. [DOI] [PubMed] [Google Scholar]
- Suda Y., Nakabayashi, J., Matsuo, I., and Aizawa, S. 1999. Functional equivalency between Otx2 and Otx1 in development of the rostral head. Development 126: 743-757. [DOI] [PubMed] [Google Scholar]
- Suda Y., Hossain, Z.M., Kobayashi, C., Hatano, O., Yoshida, M., Matsuo, I., and Aizawa, S. 2001. Emx2 directs the development of diencephalon in cooperation with Otx2. Development 128: 2433-2450. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S. and Buckingham, M. 2000. The birth of muscle progenitor cells in the mouse: Spatiotemporal considerations. Curr. Top. Dev. Biol. 48: 225-268. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S., Rocancourt, D., and Buckingham, M. 1996. Muscle progenitor cells failing to respond to positional cues adopt non-myogenic fates in myf-5 null mice. Nature 384: 266-270. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S., Rocancourt, D., Cossu, G., and Buckingham, M. 1997. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell 89: 127-138. [DOI] [PubMed] [Google Scholar]
- Tassabehji M., Read, A.P., Newton, V.E., Patton, M., Gruss, P., Harris, R., and Strachan, T. 1993. Mutations in the PAX3 gene causing Waardenburg syndrome type 1 and type 2. Nat. Genet. 3: 26-30. [DOI] [PubMed] [Google Scholar]
- Tremblay P. and Gruss, P. 1994. Pax: Genes for mice and men. Pharmacol. Ther. 61: 205-226. [DOI] [PubMed] [Google Scholar]
- Tremblay P., Kessel, M., and Gruss, P. 1995. A transgenic neuroanatomical marker identifies cranial neural crest deficiencies associated with the Pax3 mutant Splotch. Dev. Biol. 171: 317-329. [DOI] [PubMed] [Google Scholar]
- Tremblay P., Dietrich, S., Mericskay, M., Schubert, F.R., Li, Z., and Paulin, D. 1998. A crucial role for Pax3 in the development of the hypaxial musculature and the long-range migration of muscle precursors. Dev. Biol. 203: 49-61. [DOI] [PubMed] [Google Scholar]
- Trusolino L. and Comoglio, P.M. 2002. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2: 289-300. [DOI] [PubMed] [Google Scholar]
- Wada H., Holland, P.W., and Satoh, N. 1996. Origin of patterning in neural tubes. Nature 384: 123. [DOI] [PubMed]
- Wada H., Holland, P.W., Sato, S., Yamamoto, H., and Satoh, N. 1997. Neural tube is partially dorsalized by overexpression of HrPax-37: The ascidian homologue of Pax-3 and Pax-7. Dev. Biol. 187: 240-252. [DOI] [PubMed] [Google Scholar]
- Wang Y., Schnegelsberg, P.N., Dausman, J., and Jaenisch, R. 1996. Functional redundancy of the muscle-specific transcription factors Myf5 and myogenin. Nature 379: 823-825. [DOI] [PubMed] [Google Scholar]
- Xue L., Li, X., and Noll, M. 2001. Multiple protein functions of paired in Drosophila development and their conservation in the Gooseberry and Pax3 homologs. Development 128: 395-405. [DOI] [PubMed] [Google Scholar]
- Yang X.M., Vogan, K., Gros, P., and Park, M. 1996. Expression of the met receptor tyrosine kinase in muscle progenitor cells in somites and limbs is absent in Splotch mice. Development 122: 2163-2171. [DOI] [PubMed] [Google Scholar]
- Ziman M.R. and Kay, P.H. 1998. Differential expression of four alternate Pax7 paired box transcripts is influenced by organ- and strain-specific factors in adult mice. Gene 217: 77-81. [DOI] [PubMed] [Google Scholar]
- Ziman M.R., Fletcher, S., and Kay, P.H. 1997. Alternate Pax7 transcripts are expressed specifically in skeletal muscle, brain and other organs of adult mice. Int. J. Biochem. Cell. Biol. 29: 1029-1036. [DOI] [PubMed] [Google Scholar]