

Abstract
We have developed a targeted lipidomics approach that makes it possible to directly analyze chiral eicosanoid lipids generated in cellular systems. The eicosanoids, including prostaglandins (PGs), thromboxanes (TXs), leukotrienes (LTs) and alcohols (HETEs), have been implicated as potent lipid mediators of various biological processes. Enzymatic formations of eicosanoids are regioselective and enantioselective, whereas reactive oxygen species (ROS)-mediated formation proceeds with no stereo-selectivity. To distinguish between enzymatic and non-enzymatic pathways of eicosanoid formation, it is necessary to resolve enantiomeric forms as well as regioisomers. High sensitivity is also required to analyze the eicosanoid lipids that are usually present as trace amounts (pM level) in biological fluids. A discovery of liquid chromatography-electron capture atmospheric pressure chemical ionization/mass spectrometry (LC- ECAPCI/MS) allows us to couple normal phase chiral chromatography without loss of sensitivity. Analytical specificity was obtained by the use of collision-induced dissociation (CID) and tandem MS (MS/MS). With combination of stable isotope dilution methodology, complex mixtures of regioisomeric and enantiomeric eicosanoids have been resolved and quantified in biological samples with high sensitivity and specificity. Targeted chiral lipidomics profiles of bioactive eicosanoid lipids obtained from various cell systems and their biological implications have been discussed.
Keywords: Bioactive lipids, Cellular systems, Cyclooxygenase, Eicosanoids, LC-ECAPCI/MS, Lipoxygenase, Targeted chiral lipidomics
INTRODUCTION
Lipidomics has been defined as the full characterization of lipid molecular species and of their biological roles with respect to expression of proteins involved in lipid metabolism and function, including gene regulation (1). It has evolved as a separate discipline within the field of metabonomics since lipids have extraordinary structural diversity and play such important roles in the pathophysiology of diseases (2, 3). Metabonomics, which is often used interchangeably with the term of metabolomics, provides a quantitative measurement of the multivariate metabolic responses of multicellular systems to pathophysiological stimuli or genetic manipulations (4). Now systems biology and functional genomics are trying to integrate metabonomic and lipidomic information with genomic, proteomic and transcriptomic information to improve our understanding of mechanisms for human disease (5, 6).
The rapid growth of lipidomics has primarily resulted from technological advances in mass spectrometry (MS). Gas chromatography (GC)-MS coupled with electron capture negative chemical ionization (ECNCI) was traditionally used for lipid analyses. Subsequently, the range of lipids that could be analyzed by MS was significantly expanded by discoveries of electrospray ionization (ESI) (7), atmospheric pressure chemical ionization (APCI) (8) and ECAPCI (9). Furthermore, by coupling liquid chromatography (LC) to MS, it has become possible to greatly increase the number of lipids that can be analyzed in a single experiment. Finally, the use of tandem mass spectrometry (MS/MS) can provide the detailed structural information necessary for characterization of novel lipids and the selectivity required for the determination of individual lipid species present in complex mixtures. Therefore, MS-based lipidomics methodology has become an essential tool for monitoring global differences in cellular lipid metabolism (2, 3). Such studies can also provide quantitative information on precursors to important bioactive lipids that play key roles in cellular functions (Scheme 1). However, the bioactive lipids themselves are present in only trace quantities and cannot be detected by global analyses. Analysis of trace amounts of bio-active lipids is further complicated by their physicochemical properties. Many of them bind to active sites on glassware and chromatography equipment, which results in specific losses of particular lipids. Fortunately, the use of stable isotope internal standards coupled with MS detection can prevent this problem because the stable isotopes act as carriers through complex work up and analysis procedures. This has led to the development of targeted approaches, which provide more detailed information than global analyses. However, the precision and accuracy of the data compensates for the more limited analytical targets.
Scheme 1.

Formation of bioactive ei-cosanoid lipids (see text for abbreviations).
Targeted chiral lipidomics by LC-ECAPCI/MS
Under conventional APCI conditions, the high-energy electrons from the corona discharge collide with the nitrogen sheath gas to form the N2•+ radical cations that initiate the ionization process (10). We reasoned that the low-energy electrons generated in the APCI source could also ionize suitable electron capturing molecules such as pentafluorobenzyl (PFB)-ester derivatives, through dissociative electron capture procedure (Fig. 1). The ionization process would then be analogous to ECNCI during GC-MS analysis (11). The initially formed radical anion would dissociate into a PFB radical and a carboxylate anion (Fig. 1). We discovered that ECAPCI indeed occurred in the APCI source and that ultrahigh sensitivity was obtained by analyzing the carboxylate anions derived from PFB ester derivatives of non-esterified oxidized lipids (9). Usually, a 25–100 fold increase in sensitivity for oxidized lipids is achieved by ECAPCI/MS compared with conventional APCI methods (9).
Fig. 1.

Mechanism for the formation of carboxylate anions by dissociative electron capture APCI/MS analysis of PFB-ester derivatives.
When 15(S)-hydroxyeicosatetraenoic acid (HETE)-PFB was analyzed under negative APCI conditions, an intense negative ion was observed at m/z 319, due to dissociation of a PFB radical from the initially formed radical anion, [M]•− (Fig. 2A) (3, 9). If conventional APCI had occurred, a negative ion corresponding to [M-H] would have been observed at m/z 499. In fact, when 15(S)-HETE-PFB was analyzed by positive APCI, the expected protonated molecule [MH]+ was observed at m/z 501. This meant that dissociative EC, a novel ionization process, had occurred in the source of the mass spectrometer under negative APCI conditions (Fig. 2A). We named the technique, ECAPCI (9). Importantly, the structural integrity of the thermally labile HETE was maintained during the EC process, which suggested that this technique would be useful for highly specific and sensitive analyses. Additional analytical specificity was conferred by the use of collision-induced dissociation (CID). Thus, CID of m/z 319, the [M-PFB]− ion for 15-HETE, resulted in the formation of product ion at m/z 219 that was diagnostic for 15-HETE through an α-hydroxy cleavage of the C-14 and C-15 bond at the vinylic position (Fig. 2B). This made it possible to readily distinguish 15(S)-HETE from the other HETE regioisomers and stereoisomers that are formed during lipid peroxidation of polyunsaturated fatty acids (Scheme 1). A similar α-hydroxy cleavage was observed for [2H8]-15(S)-HETE so that high sensitivity stable isotope dilution quantitative analyses could be conducted using multiple reaction monitoring (MRM) of the transitions m/z 319 → 219 for 15(S)-HETE and m/z 327 → 226 for [2H8]-15(S)-HETE used as the internal standard. Similar types of specific transitions were observed for many of the other oxidized lipids derived from arachidonic acid (AA) and linoleic acid (LA) (3, 12).
Fig. 2.

(A) ECAPCI/MS analysis of 15(S)-HETE-PFB. (B) CID spectrum of the [M-PFB] negative ion at m/z 319 from 15(S)-HETE.
The resolution of enantiomeric forms of bioactive lipids is required in order to distinguish enzymatically generated lipids from those arising non-enzymatically through the action of reactive oxygen species (ROS). Typically, resolution of enantiomeric bioactive lipids has been conducted using normal phase chiral chromatography. In order to utilize the power of stable isotope labeled internal standards, it is necessary to use MS as the detection system. Unfortunately, both ESI and APCI have quite limited sensitivity for analyzing non-esterified bioactive lipids when normal-phase solvents are employed. In contrast, LC-ECAPCI/MS analyses of oxidized lipids PFB-derivatives can be conducted using normal phase solvents and chiral columns without loss of sensitivity. This makes it possible to separate complex mixtures of AA- and LA-derived lipid enantiomers and to quantify them with very high sensitivity using stable isotope dilution methodology (3, 12–14).
Chiral LC was conducted with isopropanol/methanol/hexane as a solvent system (Fig. 3). This was used in combination with LC-ECAPCI/MRM/MS for analysis of PFB-ester derivatives of 25 lipids and 12 heavy isotope analogue internal standards. It was possible to separate all the regioisomers and enantiomers by this technique. The PFB-esters of 9-HODE and 13-HODE were distinguished by the product ions at m/z 171 and 195, respectively (Fig. 3A); whereas, 5-, 8-, 11-, 12- and 15-HETE were distinguished by specific product ions at m/z 115, 155, 167, 179 and 219, respectively (Fig. 3B). The R-enantiomers generally eluted ahead of the S-enantiomers. The isomeric prostaglandin (PG) derivatives such as PGD2 and PGE2 as well as PG and isoprostane (iP) isomers such as PGF2α and iPF2α-III, respectively were also resolved under these conditions (Fig. 3C). Quantitation was performed by use of 12 heavy isotope internal standards. Detection limits for individual eicosanoids were found to be in the fg range.
Fig. 3.
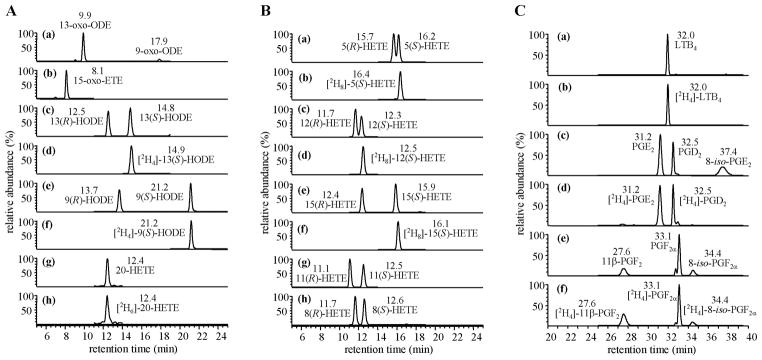
Chiral LC-MRM/ECAPCI/MS chromatograms from the analysis of eicosanoid lipids (50 pg each). MRM transitions are as follows: (A) (a) m/z 293 → 113, (b) m/z 317 → 273, (c) m/z 295 → 195, (d) m/z 299 → 198, (e) m/z 295 → 171, (f) m/z 299 → 172, (g) m/z 319 → 289, (h) m/z 325 → 295. (B) (a) m/z 319 → 115, (b) m/z 327 → 116, (c) m/z 319 → 179, (d) m/z 327 → 184, (e) m/z 319 → 219, (f) m/z 327 → 226, (g) m/z 319 → 167, (h) m/z 319 → 155. (C) (a) m/z 335 → 195, (b) m/z 339 → 197, (c) m/z 351 → 271, (d) m/z 355 → 275, (e) m/z 353 → 309, (f) m/z 357 → 313.
Bioactive eicosanoid lipids
Arachidonic acid (AA) is an important fatty acyl component of the lipidome. It is found esterified in sterol lipids and at the sn-2 position of glycerolipids and glycerophospholipids (especially phosphatidylethanolamine, phosphatidylcholine and phospha-tidylinositides) of cell membranes of the body and brain (15). AA is released by lipases such as a cytosolic phospholipase A2 (cPLA2, 85 kDa) and a low-molecular-weight secretory PLA2 (sPLA2, 14–18 kDa) (16). AA is oxidized to various bioactive lipophilic compounds called eicosanoids, including PGs, thromboxanes (TXs), leukotrienes (LTs), lipoxins, epoxides and alcohols such as HETEs (17). Many of the eicosanoids are potent lipid mediators of inflammation, asthma, fever, pain, hypertension, stroke and other pathologies (18). For example, cyclo-oxygenase (COX)-derived eicosanoids such as PGs and TXs have important roles as autacoids involved in the regulation of cardiovascular function (19) and tumor progression (20). In contrast, lipoxygenase (LO)-derived eicosanoids such as LTs have been implicated as important mediators of inflammation, asthma, cardiovascular disease and cancer (21). Cytochrome P-450 (P450)-derived epoxyeicosatrienoic acids (EETs) and their hydration products, the dihydroxyeicosatrienoic acids (DHETs), are vasodilators (22), whereas P450-derived 20-HETE is a vaso-constrictor (23). Some ROS-derived iPs have been implicated as mediators of vasoconstriction in different vascular beds (24). However, the intense interest in iPs stems primarily from their utility as biomarkers of oxidative stress (25).
AA-derived hydroperoxides (HPETEs; hydroperoxyeicosatetraenoic acids) are formed enzymatically or non-enzymatically. HPETEs formed on esterified lipids are reduced to the corresponding HETEs by peroxiredoxin VI (26) or phospholipid hydroperoxide glutathione (GSH) peroxidase (27). They are subsequently released by lipases as the corresponding free HETEs (28). HPETEs formed from free AA are reduced to HETEs by GSH transferases and peroxidases (27, 28). The resulting free HETEs can then be converted to oxo-eicosatetraenoic acids (oxo-ETEs), which form GSH-adducts (29). Alternatively, esterified and free HPETEs undergo homolytic decomposition to highly reactive bifunctional electrophiles such as 4-oxo-2(E)- nonenal (ONE) and 4-hydroxy-2(E)-nonenal, which damage cellular DNA (30–32), RNA (33), proteins and peptides (34, 35).
ROS initiates a non-enzymatic HPETE formation by abstracting a bis-allylic hydrogen atom (36). Rearrangement of the resulting allylic radical and non-stereoselective addition of molecular oxygen result in the formation of 24 different HPETE isomers when AA was used as a substrate. In contrast, COX- and LO-mediated HPETE formation is highly stereoselective. For example, COX-2 converts AA to 11(R)-, 15(S)- and 15(R)- HPETE (29), whereas 15-LO-1 converts AA to 12(S)- and 15(S)- HPETE (17). A targeted chiral lipidomics approach has been successfully employed to analyze eicosanoid enantiomers and diastereomers with high sensitivity and specificity and elucidate the roles of eicosanoids in various cellular systems, which are detailed in the following sections.
COX-2-mediated metabolism of AA in rat intestinal epithelial cells
COX-mediated pathways of AA metabolism have been implicated as important mediators of carcinogenesis (37). It has been suggested that COX-2 is involved in cellular proliferation, angiogenesis, resistance to apoptosis and modulation of immunosuppression (38). These biological activities are thought to result primarily through the formation of PGs such as PGE2. However, little attention has been given to the potential for the formation of genotoxic bifunctional electrophile such as ONE (39, 40), which arises from homolytic decomposition of COX- 2-derived lipid hydroperoxides (30, 41). ONE can covalently modify DNA bases (42–44). We reasoned that using rat intestinal epithelial cells permanently expressing COX-2 gene (RIES cells), it would be possible to determine the major lipid hydroperoxide that is responsible for covalent modifications to cellular DNA. Furthermore, it would be possible to examine the effects of aspirin (a non-specific COX inhibitor) on lipid production and DNA modification.
RIES cells were grown to confluence and incubated for a further 24-h in media containing 0.1% FBS. Most of the serum was removed because of the potential interference in LC-MS assays from endogenous eicosanoids that are present. Serum- free media was separated from cells at the end of the incubation. Lipids and their heavy isotope internal standards were extracted from the media and converted to PFB derivatives, which were then subjected to a targeted chiral lipidomics analysis. Intact RIES cells were shown to produce 15(S)-HETE together with PGE2 (Fig. 4A) (30). Quantitatively, there was approximately two times as much PGE2 secreted as 15(S)-HETE. This implied that the major lipid hydroperoxide was 15(S)-HPETE, which undergoes homolytic decomposition to ONE, a precursor of heptanone-etheno-2′-deoxyguanosine (HεdGuo). However (as described below) in a subsequent study, we showed that 11(R)-HPETE was initially the most abundant of the COX- 2-derived HPETEs formed through metabolism of exogenously added AA (29), suggesting that it could make a major contribution to the formation of the etheno adducts that were identified in the DNA of RIES cells. The selective COX-2 inhibitor NS-398 was protective against cellular DNA damage. Biosyntheses of endogenously-derived PGE2 and 15(S)-HETE were completely inhibited by 110 μM NS-398 (Fig. 4B) (30). PGE2, 15(S)-HETE, 15(R)-HETE and 11(R)-HETE derived from exogenous AA were also inhibited by NS-398 (29). These studies suggest a potential mechanism of tumorigenesis, which involves DNA-adduct formation from COX-2-mediated lipid peroxidation rather than PG formation.
Fig. 4.

Targeted chiral lipidomics analysis of COX-2-derived AA and LA metabolites from (A) RIES cells, (B) RIES cells in the presence of NS-398 (110 μM). Reprinted with permission from Ref. (30).
It has been reported previously that aspirin can convert the COX-2 into a 15(R)-LO in vitro. Therefore, it was of interest to determine whether this could occur in the RIES cells, particularly in view of the potential for 15(R)-HPETE to undergo homolytic decomposition to the same genotoxins as the 15(S)- enantiomer. Aspirin at 200 μM completely inhibited the production of 15(S)-HETE and PGE2 (30). There was a concomitant increase in the formation of 15(R)-HETE as predicted by a previous in vitro study (45). This suggests that aspirin could in fact potentiate DNA damage through increased formation of 15(R)- HPETE, the precursor of 15(R)-HETE. However, the reduced PGG2 formation in the presence of aspirin would allow the COX-2 peroxidase activity to exclusively reduce the 15(R)- HPETE to 15(R)-HETE. This means that reducing equivalents would not be depleted so readily as in the absence of aspirin. Alternatively, aspirin has antioxidant properties (such as by increasing the peroxidase activity of COX-2) that might rapidly reduce the amount of 15(R)-HPETE that is formed.
Previous studies of COX-2 mediated AA metabolism have been conducted primarily with recombinant enzymes (46) or in cells where the enzyme activity is extremely high (47). There had been no comprehensive studies of cellular COX-2- mediated AA metabolism. Therefore, a full characterization of eicosanoid biosynthesis was undertaken in the RIES cells in view of the potential for further metabolism of HPETEs and HETEs to bioactive molecules. We reasoned that this would provide a more complete picture of the role that COX-2-mediated AA metabolism plays in tumorigenesis.
When intact RIES cells were treated with calcium ionophore A-23187 (CI, 1 μM) for 1-h, 11(R)-HETE was the most abundant metabolite, followed by PGE2, 15(S)-HETE and 15-oxo- ETE (29). The ratio of PGE2 to 15(S)-HETE (2.9:1) was similar to that observed in unstimulated cells (1.9:1) after a 24-h incubation. Incubation for a further 23-h after the CI was removed resulted in a substantial increase in PGE2 concentrations while HETEs and 15-oxo-ETE concentrations decreased to almost undetectable levels. When RIES cells were treated with increasing concentrations of AA for 24-h, a similar metabolic profile was observed. However, the PGE2 to 15(S)-HETE ratio increased substantially as the AA concentration increased. Incubation of the RIES cells with 10 μM AA revealed that maximal concentrations of 11(R)-HETE, 15(S)-HETE and 15-oxo-ETE occurred after 10 min of incubation when the 15(S)-HETE concentrations were approximately twice that of PGE2 (29). There was a gradual decrease in the concentration of HETEs and 15-oxo-ETE over time, whereas PGE2 concentrations increased steadily until they reached a maximum after 24-h of incubation. The ratio of PGE2 to 15(S)-HETE was then approximately 20:1. This means that when cellular COX-2 levels are modestly induced, 15(S)- HETE and PGE2 are both major metabolites. This study also suggests that the use of CI or exogenously added AA and different incubation times can significantly affect HETE formation as well as the ratio of 15(S)-HETE to PGE2.
15(S)-HETE and 15-oxo-ETE concentrations declined in the cell media during prolonged incubations (29). 15(S)-HETE was shown to undergo metabolism primarily to 15-oxo-ETE, which was further metabolized to a GSH-adduct (15-OEG). The efficient formation of 15-oxo-ETE in the RIES cells suggested that there was a specific dehydrogenase present in the cells. In subsequent studies (Wei et al. unpublished), we showed that 15-hy-droxyprostaglandin dehydrogenase (15-PGDH) was responsible for the formation of 15-oxo-ETE in the RIES cells. 15-OEG was further metabolized in the extracellular milieu to a cysteinylglycine- adduct (15-OEC) analogous to the γ-glutamyltranspeptidase (GGTP)-mediated formation of the potent bronchoconstrictor, LTD4 in the 5-LO pathway (Scheme 1). Thus, we have established for the first time that 15-oxo-ETE can be formed biosynthetically from AA, that 15(S)-HETE is its immediate precursor, and that 15-oxo-ETE forms a GSH-adduct. However, we cannot rule out the possibility that 15(S)-HPETE and 15(R)-HETE can also be converted to 15-oxo-ETE in the RIES cells (29).
5-LO-mediated metabolism of AA in human lymphoblastoid CESS cells
5-LO metabolizes AA into 5(S)-HPETE which is either reduced to 5(S)-HETE or serves as a precursor to the formation of leukotrienes (LTs) (Scheme 1) (48). The conversion of AA to 5(S)- HPETE by 5-LO is critically dependent upon the presence of FLAP, the 5-LO-activating protein (49). 5-LO-mediated LT formation is thought to play a critical role in inflammation and allergic disorders (50) since 5-LO and FLAP are expressed primarily in inflammatory cells (48). In addition, 5-LO-derived AA metabolites LTC4 and LTD4 have been implicated as mediators of atherogenesis and heart disease (Scheme 1) (48). Our previous in vitro studies characterized ONE as one of major products arising from the homolytic decomposition of 5-LO- derived 5(S)-HPETE (31). Cellular 5-LO synthesizes lipid hydro-peroxides on the nuclear membrane like COX-2. Therefore, it was highly possible that 5-LO could also mediate the formation of lipid hydroperoxide-derived endogenous DNA-adducts in cells. CESS cells, a human lymphoblastic cell line expressing both 5-LO and FLAP, was employed to elucidate the relationship of 5-LO mediated-lipid peroxidation and DNA-adduct formation. Targeted chiral lipidomics methodology was used to monitor the concomitant formation of lipid hydroperoxides in the presence of different enzyme stimulator or inhibitors.
Western blot analyses revealed that the lymphoblastoid CESS cell line constitutively express both 5-LO and COX-1 (Fig. 5) (51). There was a substantial and stereoselective formation of 5(S)-HETE and LTB4 upon stimulation with CI, which confirmed the presence of a functional 5-LO activity (Fig. 6). Interestingly, almost equimolar amounts of 5(S)-HETE and LTB4 were generated. This suggested that sufficient 5(S)-HETE could have been generated to undergo homolytic decomposition to ONE. MK886, a FLAP inhibitor, inhibited 97% of 5(S)-HETE production and 99% of LTB4 production. Low levels of PGs together with 11(R)-, 15(S)- and 15(R)-HETE were formed by COX-1 expressed in the CESS cells. With aspirin treatment, 11- and 15-HETEs productions were reduced and became racemic (51).
Fig. 5.

Chromatograms from targeted lip-idomics analysis using LC-ECAPCI/MRM/ MS for analysis of lipid metabolites from CESS cells. Reprinted with permission from Ref. (51).
Fig. 6.

Chromatograms from targeted lip-idomics analysis using LC-ECAPCI/MRM/ MS for analysis of lipid metabolites from CESS cells treated with A23187 (1.0 μM). Reprinted with permission from Ref. (51).
HεdGuo was quantified in the CESS cell DNA. CI stimulation of the CESS cells resulted in a 3-fold increase in HεdGuo over basal levels. MK886 decreased the production of HεdGuo by almost 90% to a level that was not significantly different from the basal levels. In addition, aspirin had no detectable effect on HεdGuo formation. This confirmed that HεdGuo formation in CESS cell DNA arose primarily from 5-LO-mediated lipid peroxidation (51). Therefore, this novel finding provided additional insight into previous observations that increased 5-LO activity is associated with cancers and cardiovascular diseases (52, 53).
15-LO-1-mediated metabolism of AA in human monocytes and mouse RAW macrophages
15-LOs can convert both free and esterified AA (54) to HPETEs in contrast to 5-LOs, which strongly prefer free AA as substrate (51). Type 1 human 15-LO (15-LO-1) is a cytoplasmic enzyme responsible for converting AA to 15(S)-HPETE and a small amount of 12(S)-HPETE (55). 15-LO-1 is up-regulated in athero-sclerotic lesions and localized at sites of macrophage accumulation (56). It has been suggested that several pro- versus anti-angiogenesis, inflammatory and atherosclerosis functions are mediated by 15-LO-derived metabolites (57, 58). These conflicting observations were thought to result from the different biological effects of many lipid mediators generated by 15-LO-1 pathway, which have not yet been fully elucidated. Therefore, our targeted chiral lipidomics approach was employed to identify and quantify AA metabolites formed through the actions of 15-LO-1 and to explore their potential pharmacological roles in angiogenesis, an important mediator of tumorigenesis.
The expression of 15-LO-1 in human monocytes is induced by cytokine IL-4. When the IL-4 stimulated human monocytes were treated with exogenous AA or with CI, 15(S)-HETE and 15-oxo-ETE were major metabolites (59). It has been reported that 15-LO-1 translocates to plasma membrane in a calcium- dependent manner and that the translocation activates the oxygenase activity of the enzyme (60). It has been also demonstrated that the CI stimulation of IL-4-treated monocytes generates predominantly esterified 15-HETE (61). These studies suggested that CI treatment of the monocytes would result in the formation of esterified 15(S)-HPETE after 15-LO-1 translocated to the plasma membrane. The resulting esterified 15(S)-HPETE would be reduced to 15(S)-HETE and a significant amount of free 15(S)-HETE would then be released by cPLA2. Indeed, CI treatment of R15L cells (mouse RAW macrophages that stably express human 15-LO-1) led to the production of 15(S)-HETE and 15-oxo-ETE, which confirms that they are metabolites of endogenously-derived AA (59). These data also suggested that 15(S)-HETE was the predominant precursor of 15-oxo-ETE through 15-LO-1 mediated metabolism of AA. This contrasts with COX-2-mediated formation of 15-oxo-ETE, which could be derived from 15(S)-HETE, 15(R)-HETE or 15(S)- HPETE (31). Pre-treatment of R15L cells with CDC, a LO inhibitor, resulted in a significant reduction in 15-oxo-ETE and 15(S)-HETE levels. Thus, these data provide further evidence that 15(S)-HETE and 15-oxo-ETE are 15-LO-1-specific metabolites of AA. Furthermore, 15(S)-HETE was found to be rapidly converted into 15-oxo-ETE in the R15L cells. The specific 15-PGDH inhibitor (CAY10397) significantly decreased the production of 15-oxo-ETE in the macrophages treated with either AA or 15(S)-HETE. There was a concomitant increase in 15(S)-HETE concentrations after inhibition of 15-oxo-ETE formation. These data clearly show that 15-oxo-ETE arose through the oxidation of 15(S)-HETE by murine 15-PGDH, which was known to catalyze NAD+-mediated oxidation of 15(S)-hydroxyl group of PGs and lipoxins (62).
15-oxo-ETE was rapidly cleared from the R15L cells, with a half-life of only 11 min, indicating that it underwent further metabolism. As suggested in our previous study (29), 15-oxo- ETE is also metabolized in the R15L cells to a 15-OEG, which is then hydrolyzed by GGT to a 15-OEC. 15-oxo-ETE was also shown to inhibit the proliferation of human vascular vein en-dothelial cells (HUVECs) by suppressing DNA synthesis. Thus, in the absence of 15-PGDH, as is found in human colon cancer (63), no 15-oxo-ETE would be formed and the anti-angiogenesis activity on ECs would be lost. This could then lead to increased tumor growth and metastasis. Therefore, these studies provide a pharmacological basis for the design of new targeted therapeutic strategies to inhibit angiogenesis.
Summary and future perspectives
The high sensitivity and specificity of targeted chiral lipidomics approach using ECAPCI/MS has proved to be particularly useful in our studies on AA metabolism in various cellular systems. We were able to show the importance of endogenous 15(S)-HPETE formation in unstimulated RIES cells as reflected by 15(S)-HETE concentrations (31). The HεdGuo DNA-adduct found in the DNA of unstimulated RIES cells was shown originally to arise from COX-2-mediated AA metabolism to 15(S)- HPETE (31). However, subsequent studies have suggested that COX-2-mediated AA metabolism to 11(R)-HPETE could also make a significant contribution to HεdGuo DNA-adduct formation in the RIES cells (29). In contrast, 5-LO-mediated formation of 5(S)-HPETE was responsible for the generation of HεdGuo DNA-adduct in human lymphoblastoid CESS cells (51). 15-Oxo-ETE was found to be a COX-2-derived metabolite in RIES cells (29). More recently, it was also found to be a major AA-derived 15-LO-1 metabolite in human monocytes. It arose from 15-PGDH-mediated metabolism of 15(S)-HETE in the human monocytes and was shown to inhibit EC proliferation (59). The isomeric 5-LO-derived 5-oxo-ETE arises through an analogous pathway involving 5(S)-hydroxyeicosanoid dehydrogenase (5-HEDH). It is a potent chemoattractant for eosinophils and has similar effects on neutrophils, basophils and monocytes (64). It promotes the survival of tumor cells and blocks the induction of apoptosis by 5-LO inhibitors through activation of the Gi/o-coupled OXE receptor (64). Further metabolism of 5-oxo-ETE and 15-oxo-ETE results in the formation of GSH-adducts (FOG7 and 15-OEG) and their GGTP-derived cysteinylglycine metabolites that could potentially also have important pharmacological effects in vivo, analogous to the more widely studied sulfidopeptide LTs-LTC4 and LTD4 (Scheme 1).
Using LC-ECAPCI/MRM/MS procedures and stable isotope dilution it is now possible to quantify up to 60 eicosanoids in a single analytical run. However, recent developments in LC-MS instrumentation provide even more improved sensitivity and increased number of MRM transitions that can be monitored. It is anticipated that in the future it will be able to extend the current targeted chiral lipidomics methodology to the analysis of P450 metabolites - EETs and DHETs. In addition, this methodology can be readily modified to include oxidized lipids derived from polyunsaturated fatty acids (PUFAs) other than AA and LA such as eicosapentaenoic acid and docosahexaenoic acid. The ability to quantify hundreds of bioactive lipids including eicosanoids in a single analysis would provide an excellent screening tool that helps to further define their roles in diverse pathophysiological processes.
Acknowledgments
We acknowledge the support of NIH grants RO1CA091016 and P30ES013508.
References
- 1.Lagarde M, Geloen A, Record M, Vance D, Spener F. Lipidomics is emerging. Biochim Biophys Acta. 2003;1634:61. doi: 10.1016/j.bbalip.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Han X, Gross RW. Global analyses of cellular lipidomes directly from crude extracts of biological samples by electrospray ionization mass spectrometry: a bridge to lipidomics. J Lipid Res. 2003;44:1071–1079. doi: 10.1194/jlr.R300004-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Lee SH, Williams MV, DuBois RN, Blair IA. Targeted lipidomics using electron capture atmospheric pressure chemical ionization mass spectrometry. Rapid Comm Mass Spec. 2003;17:2168–2176. doi: 10.1002/rcm.1170. [DOI] [PubMed] [Google Scholar]
- 4.Nicholson JK, Wilson ID. Opinion: understanding ‘global’ systems biology: metabonomics and the continuum of metabolism. Nat Rev Drug Discov. 2003;2:668– 676. doi: 10.1038/nrd1157. [DOI] [PubMed] [Google Scholar]
- 5.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 6.Hood L, Galas D. The digital code of DNA. Nature. 2003;421:444–448. doi: 10.1038/nature01410. [DOI] [PubMed] [Google Scholar]
- 7.Fenn JB, Mann M, Meng CK, Wong SF, White-house CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 8.Dzidic I, Carroll DI, Stillwell RN, Horning EC. Atmospheric pressure ionization (API) mass spectrometry: formation of phenoxide ions from chlorinated aromatic compounds. Anal Chem. 1975;47:1308–1312. doi: 10.1021/ac60358a077. [DOI] [PubMed] [Google Scholar]
- 9.Singh G, Xu K, Gutierrez A, Blair IA. Liquid chromatography/electron capture atmospheric pressure chemical ionization/mass spectrometry: analysis of pentafluorobenzyl derivatives of biomolecules and drugs in the attomole range. Anal Chem. 2000;72:3007–3013. doi: 10.1021/ac000374a. [DOI] [PubMed] [Google Scholar]
- 10.Carroll DI, Dzidic I, Stillwell RN, Haegele KD, Horning EC. Atmospheric pressure ionization mass spectrometry: corona discharge ion source for use in liquid chromatograph-mass spectrometer-computer analytical system. Anal Chem. 1975;47:2369–2373. [Google Scholar]
- 11.Hunt DF, Stafford GC, Crow FW, Russell JW. Pulsed positive negative-Ion chemical ionization mass-spectrometry. Anal Chem. 1976;48:2098–2105. [Google Scholar]
- 12.Lee SH, Williams MV, Blair IA. Targeted chiral lipidomics analysis. Prostaglandins Other Lipid Mediat. 2005;77:141–157. doi: 10.1016/j.prostaglandins.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Lee SH, Blair IA. Targeted chiral lip-idomics analysis by liquid chromatography electron capture atmospheric pressure chemical ionization mass spectrometry (LC-ECAPCI/MS) Methods Enzymol. 2007;433:159–174. doi: 10.1016/S0076-6879(07)33009-7. [DOI] [PubMed] [Google Scholar]
- 14.Mesaros C, Lee SH, Blair IA. Targeted quantitative analysis of eicosanoid lipids in biological samples using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009 Mar 17; doi: 10.1016/j.jchromb.2009.03.011. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stafforini DM, Sheller JR, Blackwell TS, Sapirstein A, Yull FE, McIntyre TM, Bonventre JV, Prescott SM, Roberts LJ. Release of free F2-isoprostanes from esterified phospholipids is catalyzed by intracellular and plasma platelet-activating factor acetylhydrolases. J Biol Chem. 2006;281:4616–4623. doi: 10.1074/jbc.M507340200. [DOI] [PubMed] [Google Scholar]
- 16.Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68–69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 17.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu T. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;49:23–50. doi: 10.1146/annurev.pharmtox.011008.145616. [DOI] [PubMed] [Google Scholar]
- 19.Iniguez MA, Cacheiro-Llaguno C, Cuesta N, Diaz-Munoz M, Fresno M. Prostanoid function and cardiovascular disease. Arch Physiol Biochem. 2008;114:201–209. doi: 10.1080/13813450802180882. [DOI] [PubMed] [Google Scholar]
- 20.Wang MT, Honn KV, Nie D. Cyclooxygenases, prostanoids, and tumor progression. Cancer Metastasis Rev. 2007;26:525–534. doi: 10.1007/s10555-007-9096-5. [DOI] [PubMed] [Google Scholar]
- 21.Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 22.Natarajan R, Reddy MA. HETEs/EETs in renal glomerular and epithelial cell functions. Curr Opin Pharmacol. 2003;3:198–203. doi: 10.1016/s1471-4892(03)00015-8. [DOI] [PubMed] [Google Scholar]
- 23.Miyata N, Roman RJ. Role of 20-hydroxyeico-satetraenoic acid (20-HETE) in vascular system. J Smooth Muscle Res. 2005;41:175–193. doi: 10.1540/jsmr.41.175. [DOI] [PubMed] [Google Scholar]
- 24.Cracowski JL, Durand T. Cardiovascular pharmacology and physiology of the isoprostanes. Fundam Clin Pharmacol. 2006;20:417–427. doi: 10.1111/j.1472-8206.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- 25.Pratico D, Rokach J, Lawson J, FitzGerald GA. F2-isoprostanes as indices of lipid peroxidation in inflammatory diseases. Chem Phys Lipids. 2004;128:165–171. doi: 10.1016/j.chemphyslip.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med. 2005;38:1422–1432. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Kühn H, Borchert A. Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic Biol Med. 2002;33:154–172. doi: 10.1016/s0891-5849(02)00855-9. [DOI] [PubMed] [Google Scholar]
- 28.Chaitidis P, Schewe T, Sutherland M, Kühn H, Nigam S. 15-Lipoxygenation of phospholipids may precede the sn-2 cleavage by phospholipases A2: reaction specificities of secretory and cytosolic phospholipases A2 towards native and 15-lipoxygenated arachidonoyl phospholipids. FEBS Lett. 1998;434:437–441. doi: 10.1016/s0014-5793(98)01024-2. [DOI] [PubMed] [Google Scholar]
- 29.Lee SH, Rangiah K, Williams MV, Wehr AY, DuBois RN, Blair IA. Cyclooxygenase-2- mediated metabolism of arachidonic acid to 15-oxo-eico-satetraenoic acid by rat intestinal epithelial cells. Chem Res Toxicol. 2007;20:1665–1675. doi: 10.1021/tx700130p. [DOI] [PubMed] [Google Scholar]
- 30.Lee SH, Williams MV, DuBois RN, Blair IA. Cyclooxygenase-2-mediated DNA damage. J Biol Chem. 2005;280:28337–28346. doi: 10.1074/jbc.M504178200. [DOI] [PubMed] [Google Scholar]
- 31.Jian W, Lee SH, Arora JS, Silva Elipe MV, Blair IA. Unexpected formation of etheno-2′-deoxygua-nosine adducts from 5(S)-hydroperoxyeicosatetraenoic acid: evidence for a bis-hydroperoxide intermediate. Chem Res Toxicol. 2005;18:599–610. doi: 10.1021/tx049693d. [DOI] [PubMed] [Google Scholar]
- 32.Blair IA. DNA adducts with lipid peroxidation products. J Biol Chem. 2008;283:15545–15549. doi: 10.1074/jbc.R700051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu P, Lee SH, Wehrli S, Blair IA. Characterization of a lipid hydroperoxide-derived RNA adduct in rat intestinal epithelial cells. Chem Res Toxicol. 2006;19:809– 817. doi: 10.1021/tx0600189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oe T, Arora JS, Lee SH, Blair IA. A novel lipid hydroperoxide-derived cyclic covalent modification to histone H4. J Biol Chem. 2003;278:42098–42105. doi: 10.1074/jbc.M308167200. [DOI] [PubMed] [Google Scholar]
- 35.Lee SH, Goto T, Oe T. A novel 4-oxo-2(E)- nonenal-derived modification to angiotensin II: oxidative decarboxylation of N-terminal aspartic acid. Chem Res Toxicol. 2008;21:2237–2244. doi: 10.1021/tx800316v. [DOI] [PubMed] [Google Scholar]
- 36.Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- 37.Jones R, del-Alvarez LA, Alvarez OR, Broaddus R, Das S. Arachidonic acid and colorectal carcinogenesis. Mol Cell Biochem. 2003;253:141–149. doi: 10.1023/a:1026060426569. [DOI] [PubMed] [Google Scholar]
- 38.Wang D, Dubois RN. Cyclooxygenase-2: a potential target in breast cancer. Semin Oncol. 2004;31:64–73. doi: 10.1053/j.seminoncol.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 39.Lee SH, Blair IA. Characterization of 4- oxo-2-nonenal as a novel product of lipid peroxidation. Chem Res Toxicol. 2000;13:698–702. doi: 10.1021/tx000101a. [DOI] [PubMed] [Google Scholar]
- 40.Lee SH, Oe T, Blair IA. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science. 2001;292:2083–2086. doi: 10.1126/science.1059501. [DOI] [PubMed] [Google Scholar]
- 41.Williams MV, Lee SH, Pollack M, Blair IA. Endogenous lipid hydroperoxide-mediated DNA-adduct formation in min mice. J Biol Chem. 2006;281:10127–10133. doi: 10.1074/jbc.M600178200. [DOI] [PubMed] [Google Scholar]
- 42.Rindgen D, Nakajima M, Wehrli S, Xu K, Blair IA. Covalent modifications to 2′-deoxyguanosine by 4-oxo-2-nonenal a novel product of lipid peroxidation. Chem Res Toxicol. 1999;12:1195–1204. doi: 10.1021/tx990034o. [DOI] [PubMed] [Google Scholar]
- 43.Lee SH, Rindgen D, Bible RA, Hajdu E, Blair IA. Characterization of 2′-deoxyadenosine adducts derived from 4-oxo-2-nonenal, a novel product of lipid peroxidation. Chem Res Toxicol. 2000;13:565–574. doi: 10.1021/tx000057z. [DOI] [PubMed] [Google Scholar]
- 44.Pollack M, Oe T, Lee SH, Silva Elipe MV, Arison BH, Blair IA. Characterization of 2′-deoxy-cytidine adducts derived from 4-oxo-2-nonenal, a novel lipid peroxidation product. Chem Res Toxicol. 2003;16:893– 900. doi: 10.1021/tx030009p. [DOI] [PubMed] [Google Scholar]
- 45.Schneider C, Brash AR. Stereospecificity of hydrogen abstraction in the conversion of arachidonic acid to 15R-HETE by aspirin-treated cyclooxygenase-2. J Biol Chem. 2000;275:4743–4746. doi: 10.1074/jbc.275.7.4743. [DOI] [PubMed] [Google Scholar]
- 46.O’Neill GP, Mancini JA, Kargman S, Yergey J, Kwan MY, Falgueyret JP, Abramovitz M, Kennedy BP, Ouellet M, Cromlish W. Overexpression of human prostaglandin G/H synthase-1 and -2 by recombinant vaccinia virus: inhibition by nonsteroidal anti-inflammatory drugs and biosynthesis of 15-hydroxyeicosatetraenoic acid. Mol Pharmacol. 1994;45:245–254. [PubMed] [Google Scholar]
- 47.Holtzman MJ, Turk J, Shornick LP. Identification of a pharmacologically distinct prostaglandin H synthase in cultured epithelial cells. J Biol Chem. 1992;267:21438–21445. [PubMed] [Google Scholar]
- 48.Lotzer K, Funk CD, Habenicht AJ. The 5-lipoxygenase pathway in arterial wall biology and atherosclerosis. Biochim Biophys Acta. 2005;1736:30–37. doi: 10.1016/j.bbalip.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 49.Woods JW, Evans JF, Ethier D, Scott S, Vickers PJ, Hearn L, Heibein JA, Charleson S, Singer II. 5-Lipoxygenase and 5-lipoxygenase-activating protein are localized in the nuclear envelope of activated human leukocytes. J Exp Med. 1993;178:1935–1946. doi: 10.1084/jem.178.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- 51.Jian W, Lee SH, Williams MV, Blair IA. 5-Lipoxygenase-mediated Endogenous DNA Damage. J Biol Chem. 2009;284:16799–16807. doi: 10.1074/jbc.M109.011841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupta S, Srivastava M, Ahmad N, Sakamoto K, Bostwick DG, Mukhtar H. Lipoxygenase-5 is overexpressed in prostate adenocarcinoma. Cancer. 2001;91:737–743. doi: 10.1002/1097-0142(20010215)91:4<737::aid-cncr1059>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 53.Hennig R, Ding XZ, Tong WG, Schneider MB, Standop J, Friess H, Buchler MW, Pour PM, Adrian TE. 5-Lipoxygenase and leukotriene B(4) receptor are expressed in human pancreatic cancers but not in pancreatic ducts in normal tissue. Am J Pathol. 2002;161:421–428. doi: 10.1016/S0002-9440(10)64198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kühn H, O’Donnell VB. Inflammation and Immune Regulation by 12/15-Lipoxygenases. Prog Lipid Res. 2006;45:334–356. doi: 10.1016/j.plipres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 55.Bryant RW, Bailey JM, Schewe T, Rapoport SM. Positional specificity of a Rreticulocyte lipoxygenase. Conversion of arachidonic acid to 15-S-hydroperoxy-eicosatetraenoic acid. J Biol Chem. 1982;257:6050– 6055. [PubMed] [Google Scholar]
- 56.Kühn H, Chan L. The role of 15-lipoxygenase in atherogenesis: pro- and antiatherogenic actions. Curr Opin Lipidol. 1997;8:111–117. doi: 10.1097/00041433-199704000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Viita H, Markkanen J, Eriksson E, Nurminen M, Kinnunen K, Babu M, Heikura T, Turpeinen S, Laidinen S, Takalo T, Ylä-Herttuala S. 15-Lipoxygenase-1 prevents vascular endothelial growth factor A- and placental growth factor-induced angiogenic effects in rabbit skeletal muscles via reduction in growth factor MRNA levels, NO bioactivity, and downregulation of VEGF receptor 2 expression. Circ Res. 2008;102:177–184. doi: 10.1161/CIRCRESAHA.107.155556. [DOI] [PubMed] [Google Scholar]
- 58.Wittwer J, Hersberger M. The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot Essent Fatty Acids. 2007;77:67–77. doi: 10.1016/j.plefa.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 59.Wei C, Zhu P, Shah SJ, Blair IA. 15-Oxoeicosatetraenoic acid, a metabolite of macrophage 15-hydroxyprostaglandin dehydrogenase that inhibits endothelial cell proliferation. Mol Pharmacol. 2009 Jun 17; doi: 10.1124/mol.109.057489. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brinckmann R, Schnurr K, Heydeck D, Rosenbach T, Kolde G, Kühn H. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood. 1998;91:64–74. [PubMed] [Google Scholar]
- 61.Maskrey BH, Bermudez-Fajardo A, Morgan AH, Stewart-Jones E, Dioszeghy V, Taylor GW, Baker PR, Coles B, Coffey MJ, Kühn H, O’Donnell VB. Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J Biol Chem. 2007;282:20151–20163. doi: 10.1074/jbc.M611776200. [DOI] [PubMed] [Google Scholar]
- 62.Tai HH, Tong M, Ding Y. 15-Hydroxypros-taglandin dehydrogenase (15-PGDH) and lung cancer. Prostaglandins Other Lipid Mediat. 2007;83:203–208. doi: 10.1016/j.prostaglandins.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, Musiek ES, Milne GL, Katkuri S, Dubois RN. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005;280:3217–3223. doi: 10.1074/jbc.M411221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grant GE, Rokach J, Powell WS. 5-Oxo-ETE and the OXE receptor. Prostaglandins Other Lipid Mediat. 2009 May 18; doi: 10.1016/j.prostaglandins.2009.05.002. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]