

Abstract
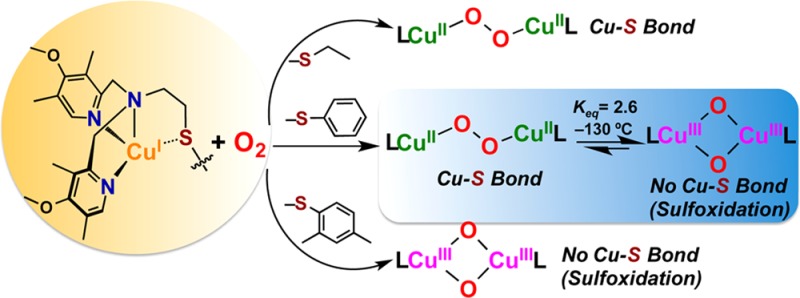
Current interest in copper/dioxygen reactivity includes the influence of thioether sulfur ligation, as it concerns the formation, structures, and properties of derived copper-dioxygen complexes. Here, we report on the chemistry of {L-CuI}2-(O2) species L = DMMESE, DMMESP, and DMMESDP, which are N3S(thioether)-based ligands varied in the nature of a substituent on the S atom, along with a related N3O(ether) (EOE) ligand. CuI and CuII complexes have been synthesized and crystallographically characterized. Copper(I) complexes are dimeric in the solid state, [{L-CuI}2](B(C6F5)4)2, however are shown by diffusion-ordered NMR spectroscopy to be mononuclear in solution. Copper(II) complexes with a general formulation [L-CuII(X)]n+ {X = ClO4–, n = 1, or X = H2O, n = 2} exhibit distorted square pyramidal coordination geometries and progressively weaker axial thioether ligation across the series. Oxygenation (−130 °C) of {(DMMESE)CuI}+ results in the formation of a trans-μ-1,2-peroxodicopper(II) species [{(DMMESE)CuII}2(μ-1,2-O22–)]2+ (1P). Weakening the Cu–S bond via a change to the thioether donor found in DMMESP leads to the initial formation of [{(DMMESP)CuII}2(μ-1,2-O22–)]2+ (2P) that subsequently isomerizes to a bis-μ-oxodicopper(III) complex, [{(DMMESP)CuIII}2(μ-O2–)2]2+ (2O), with 2P and 2O in equilibrium (Keq = [2O]/[2P] = 2.6 at −130 °C). Formulations for these Cu/O2 adducts were confirmed by resonance Raman (rR) spectroscopy. This solution mixture is sensitive to the addition of methylsulfonate, which shifts the equilibrium toward the bis-μ-oxo isomer. Further weakening of the Cu–S bond in DMMESDP or substitution with an ether donor in DMMEOE leads to only a bis-μ-oxo species (3O and 4O, respectively). Reactivity studies indicate that the bis-μ-oxodicopper(III) species (2O, 3O) and not the trans-peroxo isomers (1P and 2P) are responsible for the observed ligand sulfoxidation. Our findings concerning the existence of the 2P/2O equilibrium contrast with previously established ligand-CuI/O2 reactivity and possible implications are discussed.
Introduction
Copper ion-mediated dioxygen activation studies have focused on understanding the kinetics and thermodynamics of reactive intermediate formation and their subsequent diverse oxidative reactivity.1 Such work is inspired by the known utility of copper in oxidative transformation of organic substrates. Additionally, the probing of ligand-copper(I)-O2 chemistry is motivated by the presence of copper enzymes which mediate O2-processing.2 The detailed nature of the copper(s) active site environment (i.e., ligand type, coordination number and geometry, etc.) dictates the observed chemical reactivity, such as functioning as an O2-carrier, an oxygenase incorporating O atom(s) into a substrate, or an oxidase effecting substrate dehydrogenation chemistry.2 In chemical studies geared toward either practical oxidations or elucidation of fundamental aspects of copper-dioxygen chemistry, it is a chelating polydentate ligand which controls the (L)CuI/O2 reactivity.
The majority of the CuI–O2 literature on adduct formation and reactivity involves systems containing all nitrogen ligands, primarily bi-, tri-, or tetradentate entities.1 However, certain copper metalloproteins utilize sulfur-containing ligand residues, such as cysteine and methionine (Met) in “blue” copper electron-transfer proteins3 and in certain monooxygenases,4 (vide infra), (Figure 1).2
Figure 1.
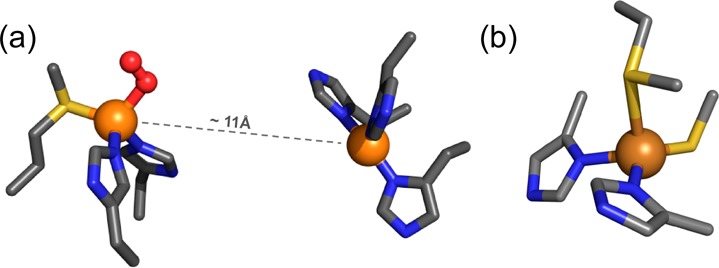
Copper enzymes/proteins with S-ligands: (a) Peptidylglycine-α-hydroxylating monooxygenase (PHM), C–H oxygenation, (b) Azurin, electron transfer.
Thioether S-ligation is important in the dioxygen activating monooxygenases peptidylglycine-α-hydroxylating monooxygenase (PHM) (Figure 1a)4 and dopamine β-monooxygenase (DβM).2a,4a These enzymes contain unique “non-coupled” binuclear copper centers, separated by ∼11 Å, one copper center with three histidine ligands (CuH), and the other with two histidines and one methionine (Met)(CuM). The CuM center is where O2-activation and substrate hydroxylation occur, while CuH receives reducing equivalents from ascorbate and is thought to serve as an electron transfer relay source to the CuM center as needed for monooxygenase activity.
Undoubtedly, the thioether ligand plays a major role in setting the electronic structure and coordination required for CuM O2-binding and activation leading to peptide pro-hormone (for PHM) oxidative N-dealkylation. However, the precise role of Met coordination and the actual PHM reaction mechanism have yet to be fully determined. A crystal structure obtained by Amzel and co-workers4b reveals dioxygen bound to CuM in an end-on superoxo fashion as depicted in Figure 1a. Computational or biochemical arguments suggest that this CuII–O2•– intermediate initiates the chemistry via substrate hydrogen-atom abstraction. Other O2-derived species have been suggested as the H atom abstracting intermediate; these include a cupric hydroperoxide (CuII–OOH) complex formed from reduction-protonation of the initially formed CuII–O2•– complex or a cupryl (CuIII=O ↔ CuII–O•) entity generated after even further reduction–protonation.5
Inspired by this biochemistry and with the motivation to elucidate the role of the Met ligand in PHM and DβM, a variety of ligand scaffolds with thioether donors have been synthesized, and their O2-reactivity has been interrogated. Within the N2S(thioether) family of ligands, copper(I) complexes with anionic ligands (Chart 1a)6 react to give bis-μ-oxo dicopper(III) products, however, thioether ligation was ruled out. CuI complexes with imidazolyl ligands (Chart 1b)7 were oxidized, but no discrete Cu/O2 intermediates formed. With tertiary amine donors (Chart 1c) we demonstrated that a μ-η2:η2-peroxodicopper(II) complex could be generated.8
Chart 1.
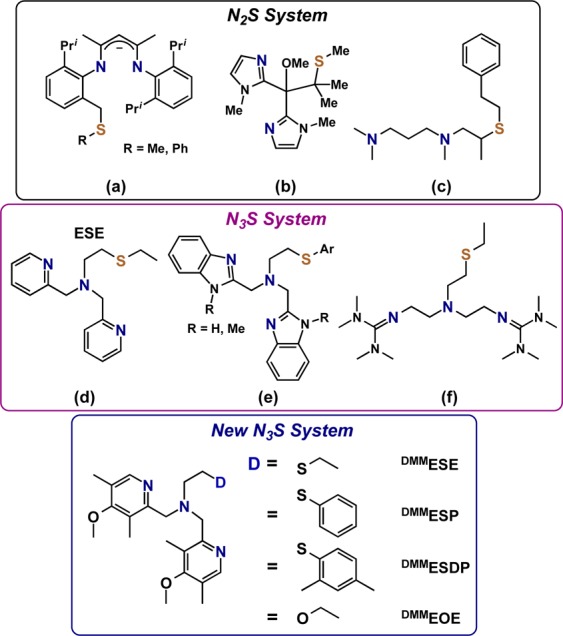
A number of N3S(thioether) type ligands have also been studied. In the case of two pyridyl and one amine donors (Chart 1d), we showed that CuI/O2 reactivity leads to a μ-1,2-trans-peroxo dicopper(II) species.9 In addition, evidence for a Cu(II)–S bond in this species was observed in the extended X-ray absorption fine structure (EXAFS) spectra. For a number of N3S(thioether) benzimidazole containing ligands (Chart 1e), Castilla and co-workers tested O2 reactivity with copper(I) derivatives, but no copper-dioxygen intermediates (as might be detected by spectroscopic interrogation) were observed during the complexes’ oxidation to copper(II).10 Depending on the identity if the Ar and R substituents, superoxide anion could be detected by a radical trapping agent.10 With guanidine donors (Chart 1f), Schindler and co-workers11 recently reported the formation of a side-on μ-η2:η2-peroxodicopper(II) complex that subsequently isomerized to an equilibrium mixture with a bis-μ-oxodicopper(III) product.
In the investigations described in this present report, we explored the chemistry of three N3S(thioether) ligands. Salient features of these new ligands are that they all contain more electron-rich 4-methoxy-3,5-dimethylpyridyl (DMM) donors (relative to ESE, Chart 1d)12 and the ethyl linked thioether moieties. The thioether donor was tuned across a series of ligands by using a variety of substituents (either ethyl in DMMESE, phenyl in DMMESP, or 2,4-dimethylphenyl in DMMESDP, Chart 1). As will be detailed in this report, addition of dioxygen to the Cu(I) complex of DMMESE leads to the formation of a μ-1,2-trans-peroxodicopper(II) complex similar to the pyridine containing ESE. Weakening of the Cu–S interaction by substituting a phenyl or dimethylphenyl group (DMMESP and DMMESDP) stabilizes a copper(III) bis-μ-oxo isomer product as determined from UV–vis and resonance Raman (rR) spectroscopy. In the case of DMMESP, both Cu2O2 core isomers are present at equilibrium, the first example of such an equilibrium reaction in the presence of a thioether donor. The results have also been compared to those observed for the N3O ligand DMMEOE (Chart 1) that contains an ether donor, to further assess the effect the thioether donor has on the activation of dioxygen in these complexes.
Experimental Section
General
All materials used were commercially available analytical grade from Sigma-Aldrich chemicals and TCI. Acetone was distilled under an inert atmosphere over CaSO4 and degassed under argon prior to use. Diethyl ether was used after being passed through a 60 cm long column of activated alumina (Innovative Technologies) under argon. Acetonitrile was stored under N2 and purified via passage through 2 × 60 cm columns of activated alumina (Innovative Technologies Inc.). Inhibitor free tetrahydrofuran (THF) and 2-methyltetrahydrofuran (MeTHF) were purchased from Sigma-Aldrich and distilled under argon from sodium/benzophenone and degassed with argon prior to use. Pentane was freshly distilled from calcium hydride under an inert atmosphere and degassed prior to use. CuI(CH3CN)4(B(C6F5)4) was synthesized according to literature protocols,13 and its identity and purity were verified by elemental analysis and/or 1H NMR. Synthesis and manipulations of copper salts were performed according to standard Schlenk techniques or in an MBraun glovebox (with O2 and H2O levels below 1 ppm). Instrumentation: Bench-top low-temperature UV–vis experiments were carried out on a Cary Bio-50 spectrophotometer equipped with a liquid nitrogen chilled Unisoku USP-203-A cryostat using a 1 cm modified Schlenk cuvette. NMR spectroscopy was performed on Bruker 300 and 400 MHz instruments with spectra calibrated either to internal tetramethylsilane (TMS) standard or to a residual protio solvent. EPR measurements were performed on an X-Band Bruker EMX CW EPR controlled with a Bruker ER 041 XG microwave bridge operating at the X-band (∼9 GHz) in 5 mm quartz EPR tubes. ESI-Mass spectra were acquired using a Finnigan LCQDeca ion-trap mass spectrometer equipped with an electrospray ionization source (Thermo Finnigan, San Jose, CA). Single crystal X-ray diffraction was performed on suitable crystals of [{(DMMESE)CuI}2](B(C6F5)4)2 (12), [{(DMMESP)CuI}2](B(C6F5)4)2 (22), [{(DMMESDP)CuI}2](B (C6F5)4)2 (32), [(DMMESE)CuII(ClO4)](ClO4) (1a), [(DMMESP)CuII(H2O)](ClO4)2 (2a), and [(DMMESDP)CuII(H2O)](ClO4)2 (3a), which were mounted either on the tip of a glass fiber or on a loop with a tiny amount of Paratone-N oil and transferred to a N2 cold stream (100 K: 22, 110 K: 12, 32, 3a, 150 K: 1a, and 240 K: 2a). Data were collected with either a Xcalibur3 diffractometer [Mo Kα radiation (λ = 0.71073 Å)] or a SuperNova diffractometer [Cu Kα radiation (mirror optics, λ = 1.54178 Å)] from Agilent Technologies. Also, see the Supporting Information for further details (SI). Resonance Raman (rR) Measurements: A 1.5 mM stock solution of copper(I) complexes was prepared in MeTHF. A 500 μL aliquot of this copper(I) solution was added to the 5 mm NMR sample tube, capped with a septum, and chilled in a pentane/N2(l) bath. Oxygenation of the copper samples was achieved by slowly bubbling an excess of dioxygen through the solution using a Hamilton gastight syringe equipped with a three-way valve and needle outlet. Dioxygen, 16O2 (Airgas OX UHP-300) or 18O2 (Icon 6393), was added to an evacuated Schlenk flask fitted with a septum for the oxygenation reactions described above. Resonance Raman spectra were collected on a triple monochromator (Spex 1877 CP with 1200, 1800, and 2400 grooves/mm holographic spectrograph gratings) with a back-illuminated CCD (Princeton Instruments ST-135). Samples were placed in a liquid nitrogen finger dewar (Wilmad) in a ∼135° backscattering configuration, and excitation was provided by an argon ion laser (Innova Sabre 25/7) and a krypton ion laser (Coherent I90C–K). Data were collected for 5 min at 20 mW of power, and samples were hand spun for data collected at 380 nm. Peak positions were determined from fitting the experimental data with Gaussian transitions using Peakfit Version 4. For the DMMESE rR spectra, the CuI spectrum was subtracted from both the 16O2 and 18O2 spectra, and each transition was constrained to have an identical bandwidth.
Synthesis of Ligands
DMMESE
2-(Chloromethyl)-4-methoxy-3,5-dimethylpyridine hydrochloride (5.50 g, 24.76 mmol), 2-(ethylthio) ethylamine (1.24 g, 11.79 mmol), and potassium carbonate (17.10 g, 123.75 mmol) in CH3CN (100 mL) were placed in a 250 mL two-neck-flask. The mixture solvent was stirred for 4 days under Ar at room temperature. After removal of the solvent, crude yellow oil was dissolved in 100 mL dichloromethane and washed with water. After drying over MgSO4, the solution was filtered and removed by rotary evaporation. The resulting yellow oil was purified by column chromatography (silica gel, 100% ethyl acetate, Rf = 0.28) yielding a pale yellow oil (3.61 g, 76% yield). 1H NMR (400 MHz, CDCl3): δ 8.14 (s, 2H), 3.75 (s, 4H), 3.70 (s, 6H), 2.74–2.70 (t, 2H), 2.57–2.54 (t, 2H), 2.34–2.29 (q, 2H), 2.21(s, 6H), 2.11(s, 6H), 1.12–1.08 (t, 3H). 1H NMR (300 MHz, THF-d8): δ 8.14 (s, 2H), 3.77 (s, 4H), 3.74 (s, 6H), 2.73–2.70 (t, 2H), 2.64–2.58 (t, 2H), 2.37–2.31 (q, 2H), 2.24(s, 6H), 2.15(s, 6H), 1.16–1.12 (t, 3H). ESI-MS, m/z: 404.39 (M + H+).
DMMESP
In a 250 mL round-bottom flask, 2.21 g (20.060 mmol) thiophenol, 2-chloroethylamine hydrochloride (3.02 g, 26.04 mmol), and potassium carbonate (8.32 g, 60.20 mmol) were stirred in 120 mL methylene chloride at room temperature under Ar. After 48 h, the mixture solution was washed with water and then dried over Na2SO4. All solvent was removed by a reduced vacuum resulting in a crude 2-(phenylthio)ethanamine, and it was used directly for the subsequent reactions.14 Without further purification, a crude 2-(phenylthio)-ethanamine (3.073 g, 20.05 mmol) and 2-chloromethyl-4-methoxy-3,5-dimethyl-pyridine hydrochloride (8.910 g, 40.12 mmol), and potassium carbonate (11.09 g, 80.21 mmol) in CH3CN (120 mL) were placed in a 250 mL round-bottom flask, and then this mixture was stirred for 4 days under Ar at room temperature. After removal of the solvent, crude yellow oil was dissolved in 100 mL dichloromethane and washed with water. After drying over MgSO4, the solution was filtered and removed by rotary evaporation. The resulting yellow oil was purified by column chromatography (alumina, 25% ethyl acetate with hexane, Rf = 0.34) yielding a pale yellow oil (4.71 g, 52% yield). 1H NMR (400 MHz, CDCl3): δ 8.11 (s, 2H), 7.16–7.07 (m, 5H), 3.77 (s, 4H), 3.70 (s, 6H), 2.95–2.91 (t, 2H), 2.81–2.79 (t, 2H), 2.21 (s, 6H), 2.13 (s, 6H). ESI-MS, m/z: 452.42 (M + H+).
DMMESDP
Following a similar methodology as described above, 2,4-dimethylbenzenethiol 2.20 g (15.92 mmol), 2-chloroethylamine hydrochloride 2.40 g (20.70 mmol), and potassium carbonate 6.60 g (60.20 mmol) were stirred in 100 mL methylene chloride in a 250 mL round-bottom flask at room temperature under Ar. After 5 days, the mixture solution was washed with water and then dried over Na2SO4. All solvent was removed by a reduced vacuum resulting in a crude 2-((2,4-dimethylpheny)thio)ethanamine, and it was used directly for the subsequent reactions.14 Without further purification, a crude 2-((2,4-dimethyl-pheny)-thio)ethanamine 1.52 g (8.38 mmol) and 2-chloromethyl-4-methoxy-3,5-dimethylpyridine hydrochloride (3.73 g, 16.77 mmol), and potassium carbonate (5.80 g, 41.97 mmol) in CH3CN (100 mL) were placed in a 250 mL round-bottom flask, and then this mixture was refluxed for 4 days under Ar. After removal of the solvent, crude yellow oil was dissolved in 100 mL dichloromethane and washed with water three times. After drying over MgSO4, the solution was filtered and removed by rotary evaporation. The resulting yellow oil was purified by column chromatography (alumina, 100% ethyl acetate, Rf = 0.67) yielding a pale yellow oil (2.36 g, 59% yield). 1H NMR (400 MHz, CDCl3): δ 8.11 (s, 2H), 6.93–6.81(m, 3H), 3.77 (s, 4H), 3.70 (s, 6H), 2.89–2.86 (t, 2H), 2.79–2.75 (t, 2H), 2.25–2.13 (s, 12H), 2.13 (s, 6H). ESI-MS, m/z: 480.50 (M + H+).
DMMEOE
2-(Chloromethyl)-4-methoxy-3,5-dimethylpyridine hydrochloride (2.26 g, 10.19 mmol), 2-ethoxyl ethanamine (0.41 g, 4.63 mmol), and K2CO3 (3.22 g, 23.30 mmol) in CH3CN (80 mL) were placed in a 100 mL round flask. The mixture solvent was stirred for 2 days under Ar at room temperature. After removal of the solvent, crude yellow oil was dissolved in 100 mL dichloromethane and washed with water. After drying over MgSO4, the solution was filtered and removed by rotary evaporation. The resulting yellow oil was purified by column chromatography over alumina (ethyl acetate:hexane = 2:1, Rf = 0.52) yielding a pale oil (1.52 g, 85% yield). 1H NMR (400 MHz, CDCl3): δ 8.14 (s, 2H), 3.77 (s, 4H), 3.70 (s, 6H), 3.47–3.44 (t, 2H), 3.38–3.33 (q, 2H), 2.72–2.70 (t, 2H), 2.16 (s, 6H), 2.08 (s, 6H), 1.20–1.10 (t, 3H). ESI-MS, m/z: 388.42 (M + H+).
Synthesis of Copper(I) Complexes
[(DMMESE)CuI](B(C6F5)4) (1)
In a 100 mL Schlenk flask in the glovebox, 258 mg (0.285 mmol) of [Cu(CH3CN)4](B(C6F5)4) was dissolved in 7 mL of THF. 115 mg (0.285 mmol) of DMMESE ligand dissolved in ∼5 mL of THF was added to the copper solution yielding a pale yellow solution. This yellow solution was allowed to stir for 10 min at which time ∼100 mL of degassed pentane was added to the solution. After 1 h, the supernatant was decanted, and the oil removed from the glovebox and dried under vacuum for 20 min affording 288 mg of a white powder (88% yield). Single crystals were obtained by topping of dry pentane into a solution of the complex in THF. 1H NMR (300 or 400 MHz, THF-d8): δ 8.41 (s, 2H), 4.34–4.28 (d, 2H), 4.10–4.04 (d, 2H), 3.81 (s, 6H), 3.03–3.00 (b, 4H), 2.42–2.37 (q, 2H), 2.29(s, 6H), 2.23(s, 6H), 1.27–1.22 (t, 3H). Elemental analysis: (C46H33BCuF20N3O2S) Calcd: C (48.20), H (2.90), N (3.67); Found: C (48.26), H (2.94), N (3.42).
[(DMMESP)CuI](B(C6F5)4) (2)
1H NMR (300 MHz, THF-d8): δ 8.34 (s, 2H), 7.48–7.31 (m, 5H), 4.25 (d, 2H), 4.09 (d, 2H), 3.73 (s, 6H), 3.50 (s, 2H), 2.99 (s, 2H), 2.24 (s, 6H), 2.17(s, 6H). Elemental analysis: (C50H33BCuF20N3O2S) Calcd: C (50.29), H (2.79), N (3.52); Found: C (50.39), H (2.92), N (3.27).
[(DMMESDP)CuI](B(C6F5)4) (3)
1H NMR (300 MHz, THF-d8): δ 8.21 (s, 2H), 7.14–6.90 (m, 5H), 4.22 (d, 2H), 4.01 (d, 2H), 3.73 (s, 6H), 3.42 (s, 2H), 3.02 (s, 2H), 2.41–2.18 (m, 18H). Elemental analysis: (C52H37BCuF20N3O2S) Calcd: C (51.10), H (3.05), N (3.44); Found: C (51.06), H (3.08), N (3.34).
[(DMMEOE)CuI](B(C6F5)4) (4)
1H NMR (300 MHz, THF-d8): δ 8.28 (s, 2H), 4.17–4.11 (d, 2H), 3.75 (s, 6H), 3.71–3.67 (d, 2H), 3.12–3.03 (m, 4H), 2.41–2.35 (d, 2H), 2.25(s, 6H), 2.22(s, 6H), 1.00–0.97 (t, 3H). Elemental analysis: (C46H33BCuF20N3O3) Calcd: C (48.89), H (2.94), N (3.72); Found: C (48.87), H (3.11), N (4.21).
Following a similar methodology as described above, dinuclear copper(I) complexes, [(N5)CuI2](B(C6F5)4)2, and [(XYL-H)CuI2]-(B(C6F5)4)2 were synthesized and characterized by NMR analyses (see SI).
Synthesis of Copper(II) Complexes
[(DMMESE)CuII(ClO4)](ClO4) (1a)
In a 100 mL Schlenk flask, 305 mg (0.756 mmol) of DMMESE ligand was dissolved in 20 mL dry acetone. 280 mg (0.756 mmol) of CuII(ClO4)2·6H2O was dissolved in 20 mL dry acetone, and then copper solution was added to DMMESE solution. After stirring for 10 min at room temperature, the complex was precipitated as a blue solid upon addition of diethyl ether (100 mL). The supernatant was decanted, and the resulting blue solid was washed two times with diethyl ether and dried under vacuum to afford 411 mg of a copper(II) complex (82% yield). Single crystals were obtained by vapor diffusion of diethyl ether into a solution of the complex in acetone. Elemental analysis: (C22H33Cl2CuN3O10S) Calcd: C (39.67), H (4.99), N (6.31); Found: C (38.95), H (5.09), N (5.98). EPR spectrum (Figure S18): X-band (2 mM, ν = 9.186 GHz) spectrometer in acetone at 70 K: g∥ = 2.263, A∥ = 168 G, g⊥ = 2.036.
[(DMMESP)CuII(H2O)](ClO4)2 (2a)
Elemental analysis: (C26H35Cl2CuN3O11S) Calcd: C (42.66), H (4.82), N (5.74); Found: C (42.69), H (4.98), N (5.69). EPR spectrum (Figure S18): X-band (2 mM, ν = 9.186 GHz) spectrometer in acetone at 70 K: g∥ = 2.263, A∥ = 168 G, g⊥ = 2.034.
[(DMMESDP)CuII(H2O)](ClO4)2 (3a)
Elemental analysis: (C28H39Cl2CuN3O11S) Calcd: C (44.24), H (5.17), N (5.53); Found: C (44.27), H (5.48), N (5.54). EPR spectrum (Figure S18): X-band (2 mM, ν = 9.186 GHz) spectrometer in acetone at 70 K: g∥ = 2.262, A∥ = 168 G, g⊥ = 2.034.
[(DMMEOE)CuII(H2O](ClO4)2 (4a)
Elemental analysis: (C22H35Cl2CuN3O12) Calcd: C (39.56), H (5.28), N (6.29); Found: C (40.03), H (5.43), N (6.00). EPR spectrum (Figure S18): X-band (2 mM, ν = 9.186 GHz) spectrometer in acetone at 70 K: g∥ = 2.265, A∥ = 170 G, g⊥ = 2.036.
The description of experimental procedures for generating and handling the copper-dioxygen adducts in order to carry out UV–vis, IR, CV, ESI-MS, and NMR analyses and the reactivity study where examination of ligand thioether sulfoxidation is probed are further described in the Supporting Information (SI).
Results and Discussion
Synthesis and Characterization of Copper(I) Complexes, 1, 2, and 3
Reaction of ligand DMMESE and [CuI(CH3CN)4](B(C6F5)4) under Ar in THF gives a white powder, formulated as [(DMMESE)CuI](B(C6F5)4), 1, on the basis of elemental analysis. Attempts to grow crystals of 1 in THF/pentane lead to the isolation of a dinuclear complex, [{(DMMESE)CuI}2](B(C6F5)4)2 (designated as compound 12) shown in Figure 2. X-ray data and details of the structure determination for 12 are provided in the SI. Selected bond lengths and bond angles of this complex are listed in Figure 2. Complex 12 is found as a centrosymmetric dimer in the solid state, where each copper ion is coordinated by one ligand via the three N-donors and by the other ligand via its thioether atom, thus giving the overall dinuclear structure. Each copper center is found in a distorted tetrahedral arrangement in which the three nitrogen atoms and one S atom from the adjacent copper site are the vertices of a distorted tetrahedron.
Figure 2.

Representations of the dimeric Cu(I) complexes described in the crystal structures of (a) [{(DMMESE)CuI}2](B(C6F5)4)2 (12), (b) [{(DMMESP)CuI}2](B(C6F5)4)2 (22), and (c) [{(DMMESDP)CuI}2](B(C6F5)4)2 (32). Selected bond distances (Å) and bond angles (deg) are also listed. The H atoms and counterions were omitted for clarity.
Copper(I) complexes containing at least one thioether ligand reveal Cu–S bond distances between 2.2 and 2.44 Å.7a,8,9b,15 Accordingly, the Cu–S distance of 2.1891(5) Å in the dimer 12 is fairly typical (but suggesting relatively strong binding of the thioether arm). Following very similar procedures, [{(DMMESP)CuI}2](B(C6F5)4)2 (22) and [{(DMMESDP)CuI}2](B(C6F5)4)2 (32) were also isolated and crystallized (Figure 2). Their coordination environments are quite similar to that of [{(DMMESE)CuI}2](B(C6F5)4)2 (12). In fact, this type of copper(I) complex dimerization in the solid state has been observed previously for a number of three- or four-coordinate complexes, with a thioether donor and even with all nitrogen donor ligands.6,9b,10,16,17
While the three ligand-copper(I) complexes are dimeric in the solid state, we postulate that in solution these Cu(I) complexes are mononuclear species. Support for this conclusion was obtained from the NMR spectroscopic technique diffusion ordered spectroscopy (DOSY). This utilizes pulsed field gradients to measure the translational diffusion of molecules.18 Our DOSY measurement gives a log D (diffusion constant) of −9.08 for the [(DMMESE)CuI]+ (1) complex in THF. From this value, we calculate a hydrodynamic diameter of 1.13 × 10–9 m, which agrees closely (1.08 × 10–9 m) with that obtained from X-ray structural parameters for the mononuclear copper(II) complex [(DMMESE)CuII(ClO4)]+ (1a), see SI.19 Further, our diffusion constant for 1 closely matches that determined via DOSY measurements for the well-known mononuclear Cu species [(TMPA)CuI(CH3CN)](B(C6F5)4),20 however log D varies considerably from that determined for the discrete dicopper complexes [(N5)(CuI)2](B(C6F5)4)2 and [(XYL-H)CuI2](B(C6F5)4)2, see SI including Table S1.
Additionally, variable-temperature (VT) 1H NMR experiments on (1) in THF-d8, acetone-d6, or DMF-d7 (25 °C to −60 °C) show minimal temperature dependence resulting from chemical dynamics (see Figures S7–S11 for details). This is in contrast to systems where the dimer is in equilibrium with the monomer, resulting in significant chemical shift changes with temperature.6,16 Further, DMF and acetone are reasonably good ligands for copper(I),21 and even weak solvent coordination would be expected to break up any dimeric structure.22
Synthesis and Characterization of Copper(II) Complexes
To experimentally probe the effect of the thioether substituent on the Cu(II)–S interaction, which would also be expected to occur in copper-dioxygen adducts where the copper is either CuII or CuIII (vide infra), copper(II) perchlorate complexes (Chart 2) were synthesized for each ligand. [(DMMESE)CuII(ClO4)](ClO4) (1a), [(DMMESP)CuII(H2O)](ClO4)2 (2a), and [(DMMESDP)CuII(H2O)](ClO4)2 (3a) were generated by simple combination of ligand and Cu(ClO4)2·6H2O in acetone. The structures of 1a, 2a, and 3a were determined by single crystal X-ray diffraction, which reveals that all three compounds are mononuclear complexes with a distorted square pyramidal (SP) coordination (τ = 0.39 for 1a, τ = 0.38 for 2a, and τ = 0.12 for 3a; τ = 0.00 for idealized SP geometries), see Figure 3.23 The thioether donor groups are found in an axial position, and the Cu–S bond distances are 2.536 Å in 1a, 2.603 Å in 2a, and 2.691 Å for 3a; similar to the 2.68 Å Cu–S(methionine) bond length observed in an oxidized form of PHM. The shortest Cu–S bond occurs for complex 1a with its donating ethyl S-substituent. For 2a and 3a, the aryl substituent is a poorer donor electronically (then an ethyl group), while steric factors are clearly relevant, even with comparing the Cu–S bond lengths 2a directly with 3a, the latter with its o-methylaryl substituent. We carried out cyclic voltammetry experiments on 1a, 2a, and 3a (see the SI). A slightly more negative E1/2 value observed for 1a compared to 2a and 3a indicates that the ethylthio arm of 1a is at least a marginally better donor than the thiophenyl-type arms in 2a and 3a. Overall, however, it is difficult to tease out the differences in electronic vs steric factors in these and related complexes.24
Chart 2.
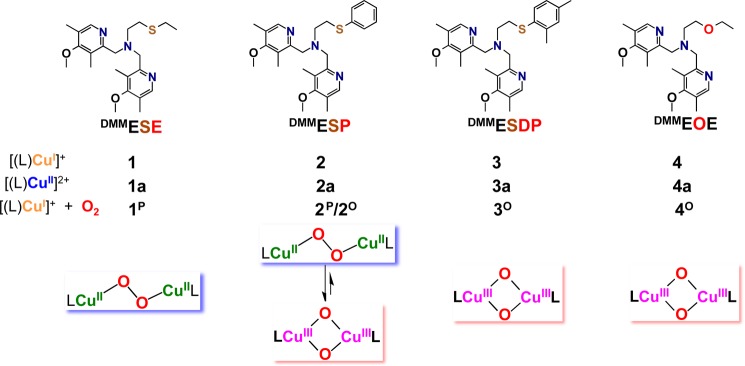
Figure 3.

Representations of the monomeric Cu(II) complexes described in the crystal structures of (a) [(DMMESE)CuII(ClO4)](ClO4) (1a), (b) [(DMMESP)CuII(H2O)](ClO4)2 (2a), and (c) [(DMMESDP)CuII(H2O)](ClO4)2 (3a). Selected bond distances (Å) and bond angles (deg) are also listed. The H atoms (except for those of the water ligands found in 2a and 3a) and noncoordinating counterions were omitted for clarity.
The EPR spectra for all of these complexes in solution are of the axial type (Figure S18) indicating that these mononuclear Cu(II) complexes are found in a distorted SP environment. This observation that the S(thioether) atom binds axially in these copper(II) complexes suggests that such binding can occur in the copper-dioxygen adducts described below.
Oxygenation Reactions of Cu(I) complexes. [(DMMESE)CuI]+ (1) + O2(g)
Dioxygen reacts with [(DMMESE)CuI]+ (1) in MeTHF at −130 °C, forming the low-temperature stable intense indigo blue species (1P)(see Chart 2). The UV–vis spectrum of 1P is characterized by three transitions at 445 (2150 M–1 cm–1), 521 (8640 M–1 cm–1), and 615 nm (10 850 M–1 cm–1, Figure S23) and is similar to other trans-peroxo species such as [{(TMPA)CuII}2(μ-1,2-O22–)]2+.25 Yet, the lower energy charge transfer (CT) transition at 615 nm for 1P is more intense than the higher energy transition. This intensity ratio was previously observed in [{(ESE)CuII}2(μ-1,2-O22–)]2+ (Chart 1d, Figure 4b) and was ascribed to a geometric distortion from the trigonal bipyramidal coordination environment in TMPA (Figure 4a) toward a SP geometry in ESE. This geometric distortion would increase the overlap of the π*ν orbital with the Cu(II) hole, resulting in a higher intensity CT transition.9b The similar intensity ratio of the CT transitions in 1P and [{(ESE)CuII}2(μ-1,2-O22–)]2+ indicate it also possesses a distorted SP geometry, similar to the coordination environment observed in the X-ray structure of [(DMMESE)-CuII(ClO4)](ClO4) (1a) (vide supra).
Figure 4.
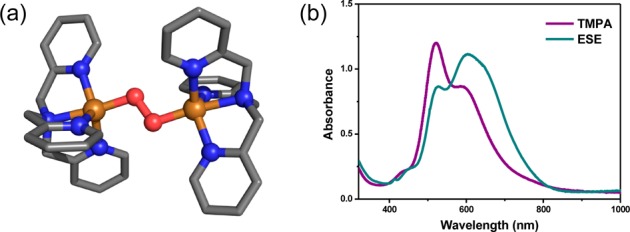
(a) Structure of [{(TMPA)CuII}2(μ-1,2-O22–)]2+ obtained from X-ray crystallography and (b) low-temperature UV–vis absorption spectra of [{(TMPA)CuII}2(μ-1,2-O22–)]2+ (purple) and [{(ESE)-CuII}2(μ-1,2-O22–)]2+ (blue).9b,25
[(DMMESP)CuI]+ (2) + O2(g)
Upon oxygenation in MeTHF at −130 °C, a trans-peroxodicopper(II) species [{(DMMESP)CuII}2(μ-1,2-O22–)]2+ (2P) with UV–vis features at 504 and 644 nm was initially formed. Over time (∼30 min) these features decay, and a new species (2O) with an absorption feature at 388 nm forms (Figure 5).
Figure 5.
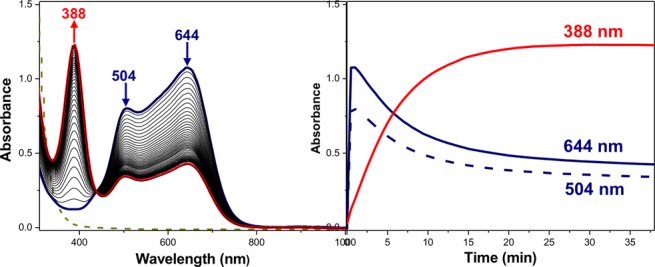
UV–vis spectra and time profile of the equilibrium between 2P and 2O in MeTHF at −130 °C.
However, the absorption features of 2P do not completely convert to 2O (Figure 5), indicating an equilibrium between these complexes (Figure 6a, upper), with Keq = [2O]/[2P] = 2.6 at −130 °C.26 The rate constant for the trans-peroxo to bis-μ-oxo interconversion can be well estimated from the data in Figure 5, ktrans→bis-oxo = 9.6 s–1 (−130 °C). Further evidence that the low-temperature combination of 2P and 2O represents a dynamic equilibrium mixture can be readily seen from examination of a series of spectra we recorded for the 2 + O2 reaction at a variety of temperatures, in some cases then also warming. Those spectra and explanatory comments are given in Figures S19–S20.
Figure 6.
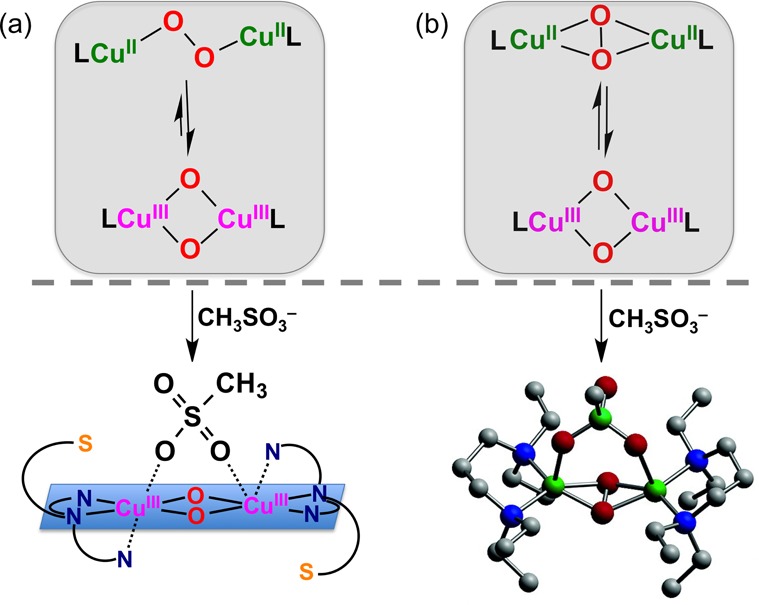
Equilibrium between (a) a μ-1,2-trans-peroxodicopper(II) and bis-μ-oxodicopper(III) species; with 1 equiv CH3SO3–, the mixture converts to a pure bis-μ-oxodicopper(III) complex, and (b) Stack and co-workers’ mixture of side-on μ-η2:η2-peroxodicopper(II) and bis-μ-oxodicopper(III) species reacts with chelating anions to give a clean side-on μ-η2:η2-peroxodicopper(II) complex.
We have previously characterized a similar trans-peroxo/bis-μ-oxo equilibrium with the all nitrogen tetradentate donor (BQPA) ligand (BQPA = bis(2-quinolylmethyl)(2-pyridylmethy1)-amine). Similarly, [(BQPA)CuI]+ reacts with dioxygen to first form a [CuII2(μ-1,2-O22–)]2+ species that converts to a [CuIII2(μ-O)2]2+ complex, ktrans→bis-oxo = 0.16 s–1 (−90 °C); Keq =3.2 (−90 °C).27 The observation of a trans-peroxo/bis-μ-oxo equilibrium in these systems differs from what has typically been presumed, that only a side-on μ-η2:η2-peroxodicopper(II) could be in (rapid) equilibrium with a bis-μ-oxodicopper(III) complex (Figure 6b, upper).1
The [CuIII2(μ-O)2]2+/[CuII2(μ-1,2-O22–)]2+ equilibrium with DMMESP (Figure 5 and 6a) is also sensitive to the addition of the coordinating anion methylsulfonate. A spectroscopically pure (UV–vis) bis-μ-oxo species 2O-(CH3SO3–), λmax = 386 nm (13 000) (Figure S22), is obtained with one equiv of (nBu4N+)(CH3SO3–) (Figure 6a, lower part).28 Stack and co-workers29 previously demonstrated that addition of methylsulfonate or other chelating anions converts a mixture of side-on μ-η2:η2-peroxodicopper(II) and bis-μ-oxodicopper(III) species with bidentate ligands to an anion bridged side-on peroxo species (Figure 6b). Masuda and co-workers30 also observed conversion of a [CuIII2(μ-O2–)2]2+ complex to a side-on peroxo species bridged by a benzoate donor with addition of the latter, indicating coordinating anions stabilize the side-on peroxo isomer relative to the bis-μ-oxo.
[(DMMESDP)CuI]+ (3) + O2(g)
The reaction of [(DMMESDP)CuI](B(C6F5)4) (3) with O2 only gives a bis-μ-oxodicopper(III) species (UV–vis criterion), [{(DMMESDP)CuIII}2-(O2–)2]2+ (3O) (λmax = 388 nm, 13 000 M–1 cm–1), see Figure S23. This species (3O) is stable for hours at −130 °C with only minimal decomposition occurring (UV–vis criterion). The increased size of the dimethylphenyl group weakens the Cu–S interaction (as evident from the longer Cu(II)–S bond in the structure of 3a) with the result that DMMESP acts like as a tridentate ligand, which favors the bis-μ-oxo dicopper(III) isomer. A steric clash due to the dimethylphenyl substituent likely also contributes to a destabilization of a trans-μ-1,2-peroxo dicopper(II) analogue; in the structure of [{(TMPA)CuII}2(μ-1,2-O22–)]2+ (Figure 4), the ligand arms are interdigitated.
The influence of the Cu–S interaction on the oxygenated products was further probed in comparison to a ligand lacking the thioether donor atom. Oxygenation of the Cu(I) complex (4) of DMMEOE, possessing the same three N-donors but having an ether O atom replacing the sulfur (Charts 1 and 2), leads to the formation of a new species with prominent optical absorption at 382 nm (20 000 M–1 cm–1, Chart 2), nearly identical in λmax and absorptivity to the bis-μ-oxo complexes 2O and 3O. The further comparison of the UV–vis spectrum of 4O with those of 2O and 3O and other literature examples15e,31 clearly shows this is a bis-μ-oxodicopper(III) complex, [{(DMMEOE)CuIII}2(O2–)2}2+ (4O). This suggests that as the fourth donor is weakened, the bis-μ-oxo isomer is stabilized, and the ligand behaves like a N3 donor. Since the oxygen atom of the ether arm in DMMEOE has an extremely weak to nonexistent interaction with the copper, this also strongly suggests that the thioether is coordinated to the Cu(II) trans-peroxo complexes 1P and 2P, similar to [{(ESE)CuII}2(μ-1,2-O22–)]2+ where a Cu(II)–S bond was characterized by EXAFS spectroscopic data.9a
Resonance Raman Spectroscopic Characterization of 1P, 2P, 2O, 2O-(CH3SO3–), and 3O
Resonance Raman spectroscopy confirms the assignment of 1P and 2P as trans-peroxo isomers and 2O, 2O-(CH3SO3–), and 3O as bis-μ-oxo isomers. Laser excitation of oxygenated samples of 2 that were frozen after ∼1 min contains mostly 2P and yields rR spectra (Figure 7a) with four isotope sensitive bands that are consistent in energy and isotope shift with the νO–O (16O2: 830 and 810 cm–1; 18O2: 784 and 772 cm–1) and the symmetric νCu–O (16O2: 545 and 531 cm–1; 18O2: 518 and 500 cm–1) of previously characterized end-on peroxo species.1a The presence of two νO–O is similar to the rR spectra of [{(BQPA)CuII}2(O22–)]2+ and results from two end-on peroxo isomers in both systems.27 These two species likely arise from the asymmetric coordination environment present in DMMESP, which yields two different arrangements of the ligand around the CuII2(O22–) core in the interdigitated dimer. In one isomer, the thioether ligands would be anti to each other and in the other syn (Scheme 1). Also present in the rR spectra is an isotope insensitive vibration at 415 cm–1. A band at similar energy and intensity is observed in the rR spectra of [{(TMPA)CuII}2(O22–)]2+ and has been previously assigned as the Cu–Namine stretch.25b The rR spectra of 1P are also characteristic of an end-on peroxo species (Figure S24) and are consistent with the presence of both syn and anti trans-peroxo isomers.
Figure 7.
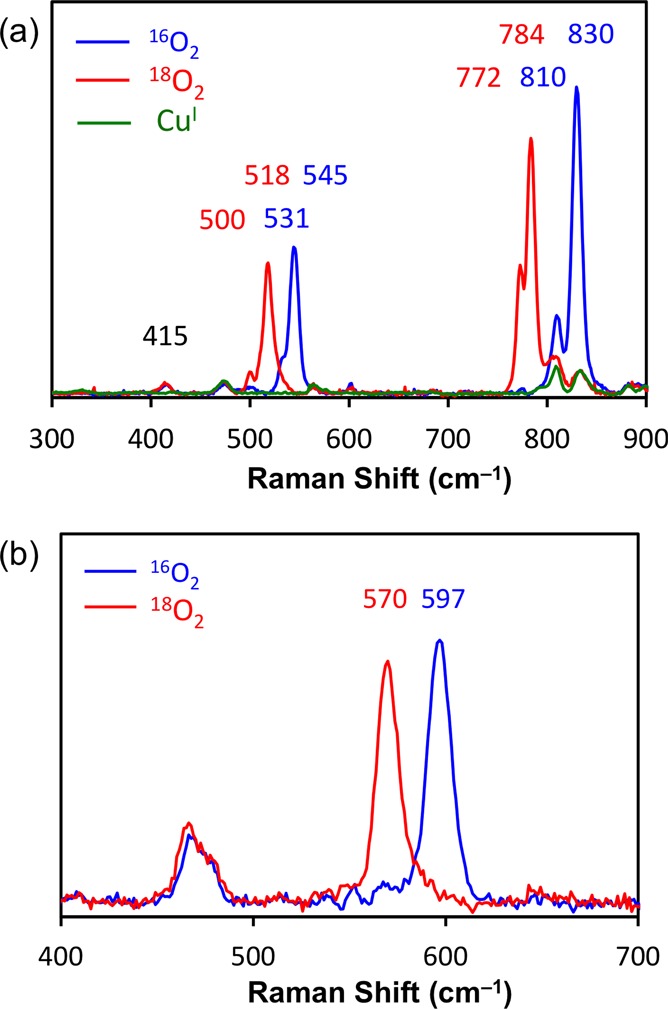
Resonance Raman spectra of 2P (a) with 647 nm excitation and 2O (b) with 380 nm excitation in MeTHF collected at 77 K; see text.
Scheme 1.
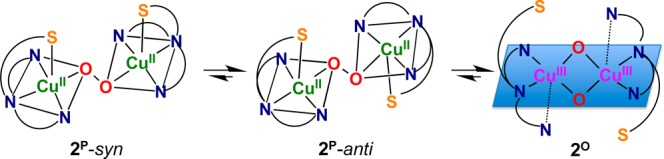
Laser excitation of an oxygenated sample of [(DMMESP)CuI]+ (2) that was frozen after ∼90 min and contains mostly 2O produces one isotope sensitive stretch (16O2: 597 cm–1; 18O2: 570 cm–1, Figure 7b) that is similar to the rR spectra of the structurally characterized [{Me2tpaCuIII}2(μ-O2–)2]2+ complex34 and the bis-μ-oxo isomer of BQPA.27 This vibration has been previously assigned as the symmetric Cu2(μ-O2–)2 core-breathing mode. The addition of dioxygen and 1 equiv of tetrabutylammonium methanesulfonate (CH3SO3–) (per copper dimer) to a solution of the CuI salt 2 results in an absorption spectrum that only contains features arising from a bis-μ-oxo species (vide infra). The rR spectra of this complex is indistinguishable (2 cm–1 resolution) from the rR spectrum of 2O, which was synthesized with a B(C6F5)4– counterion (Figure S25). Additionally, similar rR spectra were observed for 3O (Figure S26). This further indicates that while 1P and 3O are pure trans-peroxo and bis-μ-oxo species, respectfully, the oxygenated products of 2 in the presence of B(C6F5)4– (and not CH3SO3–) are an equilibrium mixture of these two isomers.
The combination of the absorption and rR data indicate that the coordination environment of 1P and 2P is distorted toward SP (Scheme 1). Not only is this coordination geometry observed in the perchlorate crystal structures 1a and 2a but also the lower energy π*v CT transition in both 1P and 2P is more intense than the higher energy π*σ CT transition. This is in contrast to the structure of [{(TMPA)CuII}2(μ-1,2-O22–)]2+ (Figure 4a) where a trigonal bipyramidal coordination geometry (τ = 0.9) is observed.25a,32 The ligand environment is rather flexible since 2P can readily equilibrate to give bis-μ-oxo species (2O), adopting a CuIII2(μ-O)2 core with a square planar geometry (Scheme 1) analogous to the crystallographically characterized complex [{(Me2tpa)CuIII}2(O2–)2]2+ where Me2tpa has two of the three pyridyl donors (of TMPA) possessing ortho (6-position) methyl groups.33 In this case, the methylpyridines are axial (Cu···N = 2.5 Å). By analogy, the apical amine and one pyridyl ligand arm are equatorial in 2O and 3O, with the other pyridyl and thiophenyl arms interacting axially with the Cu(III) ion. However, due to the potential lability of this interaction, it is possible that 2O and 3O do not possess even a weak, axial thioether–Cu(III) interaction as we have depicted in Scheme 1.
Ligand Substrate Reactivity of 1P, 2P, and 3O Species
To seek further insight into Cu-dioxygen bonding and reactivity in the presence of a thioether donor, the copper(I) complexes were oxygenated at slightly warmer temperatures to probe for the possibility of ligand sulfoxidation chemistry. [(DMMESP)CuI](B(C6F5)4) (2) was bubbled with dioxygen gas in MeTHF at −120 °C, forming as described above a mixture of [{(DMMESP)CuII}2(μ-1,2-O22–)]2+ (2P) and [{(DMMESP)CuIII}2(μ-O2–)2]2+ (2O). Further hour-long incubation at −120 °C led to decomposition of both intermediates resulting in a color change from dark indigo blue to green. Workup of this reaction product solution at low temperature (see SI) and analysis of the organic products, the ligands, and any oxidized species showed that approximately one out of two DMMESP thioether-amine (N3S) ligands had undergone sulfoxidation as determined by TLC, NMR spectroscopy, and ESI-MS spectrometry, with a ∼ 40% measured yield obtained (Scheme 2).
Scheme 2.
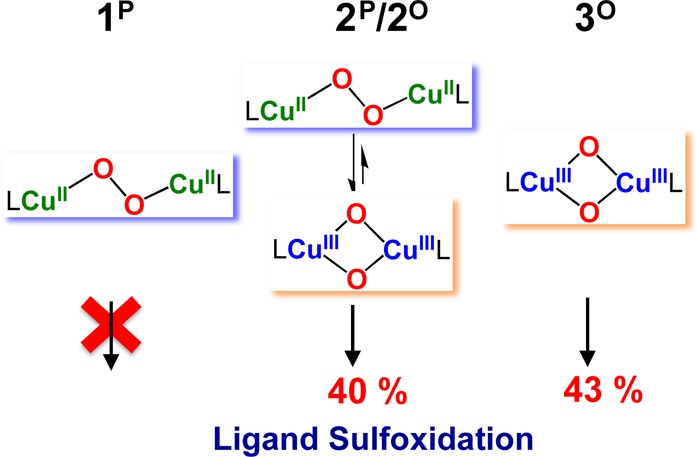
These results suggest that a monooxygenase-type reaction occurred with the 2P/2O mixture, as we previously observed in the room temperature oxygenation of [(ESE)CuI]+.9b Here, two electrons provided by two copper(I) complexes lead to a peroxo or bis-μ-oxodicopper product which is capable of only one oxo-transfer (net) reaction, giving a sulfoxide; the theoretical yield is a maximum of 50%. By comparison, under identical conditions, similar reactivity studies were carried out for the trans-peroxo species, [{(DMMESE)CuII}2(O22–)]2+ (1P) and bis-μ-oxo species, [{(DMMESDP)CuIII}2(O2–)2}2+ (3O). The same behavior was observed for 3O leading to ∼43% ligand sulfoxidation. However, 1P showed no ligand sulfoxidation at −120 °C (Scheme 2) indicating that the bis-μ-oxodicopper(III) isomer is responsible for the ligand sulfoxidation, as we had previously hypothesized with ESE (Chart 1d).9b Interestingly, when an excess of triphenylphosphine (PPh3) was added to the 2P/2O mixture and allowed to stand for 2–3 h, low-temperature work-up showed that no sulfoxidation occurred and rather O=PPh3 formed. Thus, the ligand (intramolecular) sulfoxidation was suppressed in favor of a more facile intermolecular oxidation of PPh3.
Summary and Conclusion
The oxygenation of copper(I) salts of a series of N3S DMM derivatized ligands indicates that the nature of the oxygenated product can be tuned by modulating the Cu–S interaction. While a trans-peroxo isomer is favored for DMMESE, an increase in steric bulk (and/or decrease in donor strength) at the thioether donor causes a weakening of the Cu–S interaction resulting in the stabilization of the bis-μ-oxo isomer observed for DMMESP and DMMESDP with their S-aryl substituents. In the case of DMMESP, an equilibrium mixture of the two species is observed, the first example supported by a ligand with a thioether donor. The addition of the coordinating anion CH3SO3– stabilized the bis-μ-oxo, relative to the trans-peroxo isomer, in contrast to the effect of coordinating anions on the side-on peroxo/bis-μ-oxo equilibrium observed in other systems. Additionally, only ligands with an accessible bis-μ-oxo isomer oxidize the thioether to the corresponding sulfoxide at low temperatures indicating this isomer is the oxidant responsible for this electrophilic reactivity. The observation of this trans-peroxo/bis-μ-oxo equilibrium in the present N3S and a previous N4 system27 shifts a previous paradigm which assumed that a bis-μ-oxo dicopper(III) complex generation occurs from initially formed side-on μ-η2:η2-peroxodicopper(II) species. These systems demonstrate a new strategy to reversibly cleave an O–O bond, which has new potential implications for the development of a synthetic, copper-based water splitting catalyst.34
Acknowledgments
The authors acknowledge support of this research from the National Institutes of Health (GM28962 from the National Institute of General Medical Sciences to K.D.K. and R01DK031450 from the National Institute of Diabetes and Digestive and Kidney Diseases to E.I.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
Details concerning X-ray crystallographic analyses including cif files, variable-temperature NMR, DOSY NMR, UV–vis, IR and rR spectroscopies, cyclic voltammetry, ESI mass spectrometry, and the procedures for carrying out the sulfoxidation reactivity. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Mirica L. M.; Ottenwaelder X.; Stack T. D. P. Chem. Rev. 2004, 104, 1013–1045. [DOI] [PubMed] [Google Scholar]; b Lewis E. A.; Tolman W. B. Chem. Rev. 2004, 104, 1047–1076. [DOI] [PubMed] [Google Scholar]; c Hatcher L. Q.; Karlin K. D. J. Biol. Inorg. Chem. 2004, 9, 669–683. [DOI] [PubMed] [Google Scholar]
- a Solomon E. I.; Heppner D. E.; Johnston E. M.; Ginsbach J. W.; Cirera J.; Qayyum M.; Kieber-Emmons M. T.; Kjaergaard C. H.; Hadt R. G.; Tian L. Chem. Rev. 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Peterson R. L.; Kim S.; Karlin K. D. In Comprehensive Inorganic Chemistry II; 2nd ed.; Elsevier: Amsterdam, 2013; pp 149–177. [Google Scholar]
- Solomon E. I.; Szilagyi R. K.; George S. D.; Basumallick L. Chem. Rev. 2004, 104, 419–458. [DOI] [PubMed] [Google Scholar]
- a Klinman J. P. J. Biol. Chem. 2006, 281, 3013–3016. [DOI] [PubMed] [Google Scholar]; b Prigge S. T.; Eipper B.; Mains R.; Amzel L. M. Science 2004, 304, 864–867. [DOI] [PubMed] [Google Scholar]
- a Crespo A.; Marti M. A.; Roitberg A. E.; Amzel L. M.; Estrin D. A. J. Am. Chem. Soc. 2006, 128, 12817–12828. [DOI] [PubMed] [Google Scholar]; b Yoshizawa K.; Kihara N.; Kamachi T.; Shiota Y. Inorg. Chem. 2006, 45, 3034–3041. [DOI] [PubMed] [Google Scholar]
- Aboelella N. W.; Gherman B. F.; Hill L. M. R.; York J. T.; Holm N.; Young V. G.; Cramer C. J.; Tolman W. B. J. Am. Chem. Soc. 2006, 128, 3445–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhou L.; Powell D.; Nicholas K. M. Inorg. Chem. 2006, 45, 3840–3842. [DOI] [PubMed] [Google Scholar]; b Zhou L.; Powell D.; Nicholas K. M. Inorg. Chem. 2007, 46, 7789–7799. [DOI] [PubMed] [Google Scholar]; c Zhou L.; Nicholas K. M. Inorg. Chem. 2008, 47, 4356–4367. [DOI] [PubMed] [Google Scholar]
- Park G. Y.; Lee Y.; Lee D.-H.; Woertink J. S.; Sarjeant A. A. N.; Solomon E. I.; Karlin K. D. Chem. Commun. 2010, 46, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hatcher L. Q.; Lee D.-H.; Vance M. A.; Milligan A. E.; Sarangi R.; Hodgson K. O.; Hedman B.; Solomon E. I.; Karlin K. D. Inorg. Chem. 2006, 45, 10055–10057. [DOI] [PubMed] [Google Scholar]; b Lee D.-H.; Hatcher L. Q.; Vance M. A.; Sarangi R.; Milligan A. E.; Narducci Sarjeant A. A.; Incarvito C. D.; Rheingold A. L.; Hodgson K. O.; Hedman B.; Solomon E. I.; Karlin K. D. Inorg. Chem. 2007, 46, 6056–6068. [DOI] [PubMed] [Google Scholar]
- Castillo I.; Ugalde-Saldivar V. M.; Rodriguez Solano L. A.; Sanchez Eguia B. N.; Zeglio E.; Nordlander E. Dalton Trans. 2012, 41, 9394–9404. [DOI] [PubMed] [Google Scholar]
- Hoppe T.; Josephs P.; Kempf N.; Wölper C.; Schindler S.; Neuba A.; Henkel G. Z. Anorg. Allg. Chem. 2013, 639, 1504–1511. [Google Scholar]
- Electrochemical studies employing cyclic volatammetry have been carried out on the copper(II) complexes of ESE, DMMESE, DMMESP, and DMMESDP, which show more negative potentials for the DMM conaining ligands, thus confirming they are stronger donors compared to what is found in ESE. See SI for details and descriptions.
- Liang H.-C.; Kim E.; Incarvito C. D.; Rheingold A. L.; Karlin K. D. Inorg, Chem. 2002, 41, 2209–2212. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Xu Y.-J.; He H.-Y.; Mehta S. J. Org. Chem. 2001, 66, 5590–5594. [DOI] [PubMed] [Google Scholar]
- a Karlin K. D.; Dahlstrom P. L.; Stanford M. L.; Zubieta J. J. Chem. Soc. Chem. Commun. 1979, 465–466. [Google Scholar]; b Karlin K. D.; Hayes J. C.; Hutchinson J. P.; Zubieta J. Inorg. Chim. Acta 1983, 78, L45–L46. [Google Scholar]; c Ambundo E. A.; Deydier M.-V.; Grall A. J.; Aguera-Vega N.; Dressel L. T.; Cooper T. H.; Heeg M. J.; Ochrymowycz L. A.; Rorabacher D. B. Inorg. Chem. 1999, 38, 4233–4242. [Google Scholar]; d Ohta T.; Tachiyama T.; Yoshizawa K.; Yamabe T.; Uchida T.; Kitagawa T. Inorg. Chem. 2000, 39, 4358–4369. [DOI] [PubMed] [Google Scholar]; e Lee Y.; Lee D.-H.; Park G. Y.; Lucas H. R.; Narducci Sarjeant A. A.; Kieber-Emmons M. T.; Vance M. A.; Milligan A. E.; Solomon E. I.; Karlin K. D. Inorg. Chem. 2010, 49, 8873–8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennari M.; Tegoni M.; Lanfranchi M.; Pellinghelli M. A.; Giannetto M.; Marchio L. Inorg. Chem. 2008, 47, 2223–2232. [DOI] [PubMed] [Google Scholar]
- a Sorrell T. N.; Borovik A. S. J. Am. Chem. Soc. 1987, 109, 4255–4260. [Google Scholar]; b Diez J.; Gamasa M. P.; Panera M. Inorg. Chem. 2006, 45, 10043–10045. [DOI] [PubMed] [Google Scholar]; c Mandal S.; Mukherjee R. Inorg. Chim. Acta 2006, 359, 4019–4026. [Google Scholar]
- Morris K. F.; Johnson C. S. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar]
- a Garcia-Simon C.; Garcia-Borras M.; Gomez L.; Garcia-Bosch I.; Osuna S.; Swart M.; Luis J. M.; Rovira C.; Almeida M.; Imaz I.; Maspoch D.; Costas M.; Ribas X. Chem.—Eur. J. 2013, 19, 1445–1456. [DOI] [PubMed] [Google Scholar]; b Ribas X.; Dias J. C.; Morgado J.; Wurst K.; Almeida M.; Parella T.; Veciana J.; Rovira C. Angew. Chem., Int. Ed. 2004, 43, 4049–4052. [DOI] [PubMed] [Google Scholar]; c Valentini M.; Ruegger H.; Pregosin P. S. Helv. Chim. Acta 2001, 84, 2833–2853. [Google Scholar]
- Zhang C. X.; Kaderli S.; Costas M.; Kim E.-i.; Neuhold Y.-M.; Karlin K. D.; Zuberbühler A. D. Inorg. Chem. 2003, 42, 1807–1824. [DOI] [PubMed] [Google Scholar]
- a Munakata M.; Kitagawa S.; Emori T. J. Chem. Soc., Chem. Comm. 1991, 1244–1245. [Google Scholar]; b Naskar J. P.; Hati S.; Datta D.; Tocher D. A. Chem. Commun. 1997, 1319–1320. [Google Scholar]; c Munakata M.; Kuroda-Sowa T.; Maekawa M.; Nakamura M.; Akiyama S.; Kitagawa S. Inorg. Chem. 1994, 33, 1284–1291. [Google Scholar]
- Carbon monoxide adducts of ligand–copper(I) complexes 1–3 have been generated in situ and characterized by IR spectroscopy and 1H NMR spectroscopy (for 1). In these adducts, the data indicate that the complexes are mononuclear and that the thioether ligands are not ligated, leaving four-coordinate N3(CO) copper(I) ligation.
- Addison A. W.; Rao T. N.; Reedijk J.; van Rijn J.; Verschoor G. C. J. Chem. Soc., Dalton Trans. 1984, 1349–1356. [Google Scholar]
- a Blackwell I.; William C.; Bunich D.; Concolino T. E.; Rheingold A. L.; Rabinowich D. Inorg. Chem. Commun. 2000, 3, 325–327. [Google Scholar]; b Harper K. C.; Bess E. N.; Sigman M. S. Nat. Chem. 2012, 4, 366–374. [DOI] [PubMed] [Google Scholar]
- a Jacobson R. R.; Tyeklár Z.; Karlin K. D.; Liu S.; Zubieta J. J. Am. Chem. Soc. 1988, 110, 3690–3692. [Google Scholar]; b Baldwin M. J.; Ross P. K.; Pate J. E.; Tyeklár Z.; Karlin K. D.; Solomon E. I. J. Am. Chem. Soc. 1991, 113, 8671–8679. [Google Scholar]
- For this Keq calculation, we assumed the molar absorptivity of 2O to be 13 000 M–1 cm–1, based on the corresponding values obtained for 2O-(CH3SO3–) or 3O.
- Kieber-Emmons M. T.; Ginsbach J. W.; Wick P. K.; Lucas H. R.; Helton M. E.; Lucchese B.; Suzuki M.; Zuberbühler A. D.; Karlin K. D.; Solomon E. I. Angew. Chem., Int. Ed. 2014, 53, 4935–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In other attempts to perturb the equilbrium, we added strong base ligands to early on formed [{(DMMESP)CuII}2(μ-1,2-O22–)]2+ (2P), including 1,5-dicyclohexylimidazole, 1-methylimidazole, or pyridine, thinking that we could prevent formation of a the analog [{(DMMESP)CuIII}2(μ-O2–)2]2+ (2O), by providing a strong N4 ligand environment, as in [{(TMPA)CuII}2(μ-1,2-O22–)]2+. However, conversion of 2P to 2O still occurred. Also, to 2P and 2P/2O mixtures, we added TMPA or an analog tris((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)amine, attempting to displace the N3S(thioether) ligand DMMESP, but this also did not occur.
- Ottenwaelder X.; Rudd D. J.; Corbett M. C.; Hodgson K. O.; Hedman B.; Stack T. D. P. J. Am. Chem. Soc. 2006, 128, 9268–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funahashi Y.; Nishikawa T.; Wasada-Tsutsui Y.; Kajita Y.; Yamaguchi S.; Arii H.; Ozawa T.; Jitsukawa K.; Tosha T.; Hirota S.; Kitagawa T.; Masuda H. J. Am. Chem. Soc. 2008, 130, 16444–16445. [DOI] [PubMed] [Google Scholar]
- Maiti D.; Woertink J. S.; Narducci Sarjeant A. A.; Solomon E. I.; Karlin K. D. Inorg. Chem. 2008, 47, 3787–3800. [DOI] [PubMed] [Google Scholar]
- Tyeklár Z.; Jacobson R. R.; Wei N.; Murthy N. N.; Zubieta J.; Karlin K. D. J. Am. Chem. Soc. 1993, 115, 2677–2689. [Google Scholar]
- Hayashi H.; Fujinami S.; Nagatomo S.; Ogo S.; Suzuki M.; Uehara A.; Watanabe Y.; Kitagawa T. J. Am. Chem. Soc. 2000, 122, 2124–2125. [Google Scholar]
- a Barnett S. M.; Goldberg K. I.; Mayer J. M. Nat. Chem. 2012, 4, 498–502. [DOI] [PubMed] [Google Scholar]; b Chen Z.; Meyer T. J. Angew. Chem., Int. Ed. 2013, 52, 700–703. [DOI] [PubMed] [Google Scholar]; c Zhang M.-T.; Chen Z.; Kang P.; Meyer T. J. J. Am. Chem. Soc. 2013, 135, 2048–2051. [DOI] [PubMed] [Google Scholar]; d Zhang T.; Wang C.; Liu S. B.; Wang J. L.; Lin W. B. J. Am. Chem. Soc. 2014, 136, 273–281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.