

Abstract
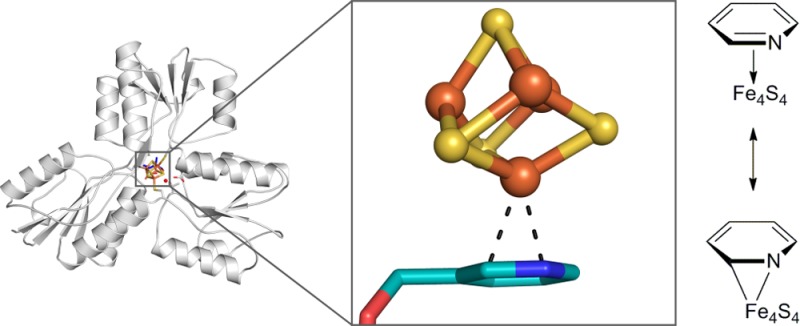
(E)-1-Hydroxy-2-methylbut-2-enyl 4-diphosphate reductase (IspH) is a [Fe4S4] cluster-containing enzyme involved in isoprenoid biosynthesis in many bacteria as well as in malaria parasites and is an important drug target. Several inhibitors including amino and thiol substrate analogues, as well as acetylene and pyridine diphosphates, have been reported. Here, we investigate the mode of binding of four pyridine diphosphates to Escherichia coli IspH by using X-ray crystallography. In three cases, one of the iron atoms in the cluster is absent, but in the structure with (pyridin-3-yl)methyl diphosphate, the most potent pyridine-analogue inhibitor reported previously, the fourth iron of the [Fe4S4] cluster is present and interacts with the pyridine ring of the ligand. Based on the results of quantum chemical calculations together with the crystallographic results we propose a side-on η2 coordination of the nitrogen and the carbon in the 2-position of the pyridine ring to the unique fourth iron in the cluster, which is in the reduced state. The X-ray structure enables excellent predictions using density functional theory of the 14N hyperfine coupling and quadrupole coupling constants reported previously using HYSCORE spectroscopy, as well as providing a further example of the ability of such [Fe4S4]-containing proteins to form organometallic complexes.
Introduction
Isoprenoids, including steroids and terpenes, constitute one of the largest and most diverse class of natural products. In all organisms they derive from the isoprene derivatives isopentenyl diphosphate (IPP, 1) and dimethylallyl diphosphate (DMAPP, 2).1 Two different biosynthetic pathways are known to produce both IPP and DMAPP: the mevalonate pathway present in mammals as well as some microorganisms, and the 1-deoxy-d-xylulose-5-phosphate (DXP) pathway found in most pathogenic bacteria including Mycobacterium tuberculosis, as well as the malaria parasite, Plasmodium falciparum.2,3 Due to use of the mevalonate pathway in humans, enzymes of the DXP pathway are important anti-infective drug targets.
The final step in the DXP pathway is the conversion of (E)-1-hydroxy-2-methylbut-2-enyl 4-diphosphate (HMBPP, 3) into a ∼6:1 mixture of IPP and DMAPP (Scheme 1).4 This reductive dehydroxylation reaction is catalyzed by HMBPP reductase (IspH), an oxygen-sensitive iron–sulfur enzyme.5,6 Despite the identification of several intermediate species of this reaction, the reaction mechanism is still the topic of lively debate.4,7−14
Scheme 1. Reaction Catalyzed by IspH.

IspH has a trefoil-like architecture with a [Fe4S4] cluster in the active site (Figure 1), serving both structural and catalytic roles.15,16 The decomposition of this oxygen-sensitive cofactor leads to partial loss of the tertiary structure and to complete loss of function,12 as observed also for aconitase17 and radical S-adenosyl-l-methionine (SAM)18,19 enzymes. Catalysis takes place at the unique iron site that is not coordinated by any cysteine side chain, unlike the three remaining iron sites.9 The HMBPP substrate binds directly to the unique fourth iron via the hydroxyl oxygen atom in the early stage of catalysis (Figure S1a in the Supporting Information).9 Recently, it was shown that several oxygen, nitrogen, and sulfur atom-containing substrate analogues are also able to coordinate to this unique iron site (Figure S1b,c in the Supporting Information). These observations are of great interest in the context of inhibitor development.20,21
Figure 1.
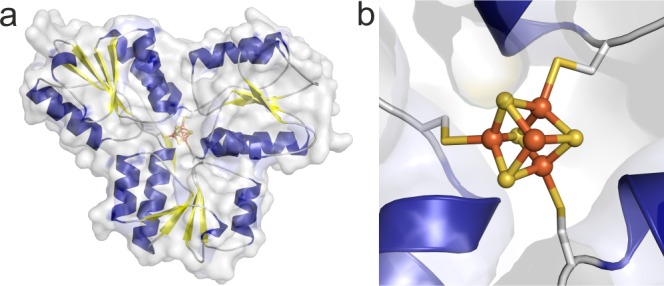
Structure of E. coli IspH. (a) Surface representation of the monomeric IspH with the surface in transparent gray and the cartoon model colored according to secondary structure elements (α-helices in blue, β-sheets in yellow and loops in gray). (b) The [Fe4S4] cluster in the active site is shown as a ball-and-stick model, with the iron atoms colored in orange and the sulfur atoms in gold. The cluster binds to the protein via three cysteine residues; other ligands can bind to the fourth iron site.
Since its key role in the biosynthesis of isoprenoids was discovered, IspH has become the subject of intensive research, particularly in regard to the development of new antimicrobial agents.22 Several studies have identified a variety of compounds that bind to the active site of IspH and inhibit its activity.10,23,24 Most of these molecules have a diphosphate group that binds in a similar way to IspH as does the HMBPP substrate. Structural, spectroscopic, and computational studies25 of IspH interacting with derivatives of the substrate 3 (Scheme 2), in which the hydroxyl group is replaced by an amino (4) or thiol (5) group, have shown that the heteroatoms coordinate to the unique iron site.20 Furthermore, crystallographic studies have revealed the promiscuous reactivity of IspH, hydrating acetylenes 6 and 7 to the aldehyde 8 and the ketone 9, respectively,21 with the enolate of 8 binding to the fourth iron and stabilizing the protein with respect to cluster decomposition in the presence of atmospheric oxygen. The importance of IspH as a new drug target and its versatile and flexible catalytic site thus provide motivation for the characterization of inhibitors that may be new drug leads.
Scheme 2. Structures of Compounds That Interact with IspH.

In addition to the linear compounds that, structurally, are closely related to 3, the pyridine derivatives 10–13 (Scheme 2) have also been shown to inhibit IspH enzymatic activity.23 Moreover, electron paramagnetic resonance (EPR) as well as X-band hyperfine sublevel correlation (HYSCORE) spectroscopic studies have indicated that 10 interacts with the unique iron of the [Fe4S4] cluster in the active site of IspH.26 What has, however, been unclear is just how the pyridine inhibitors bind into the active site.
In early work we used computational docking to propose that the pyridine inhibitors bound to reduced IspH as illustrated in Figure 2a. The aromatic ring in the inhibitor is located close to the fourth iron, but we speculated that most likely a Coulombic interaction between the pyridinium ring and the E126 carboxyl was important for ligand binding.23 In later studies we used HYSCORE spectroscopy (Figure 2b) to investigate the binding of 10 to 15N-labeled IspH finding that there was a large 14N hyperfine coupling (∼7 MHz) and that the nuclear quadrupole coupling constant (NQCC) was ∼3 MHz. These values are similar to those found for aromatic bases bound to Fe in both proteins as well as model systems,26 leading to the idea that 10 might bind end-on in the reduced protein, as shown in Figure 2c.
Figure 2.
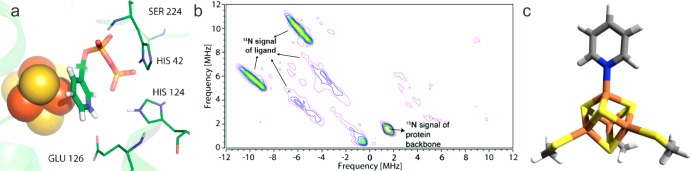
Predicted models and a 9 GHz 14N/15N HYSCORE spectrum for pyridine inhibitors binding to IspH. (a) Docking pose. Reprinted with permission from ref (23). Copyright 2010 American Chemical Society. (b) 9 GHz HYSCORE result for [14N] 10 binding to [15N]-labeled IspH. Reprinted with permission from ref (26). Copyright 2011 American Chemical Society. (c) Proposed end-on binding of pyridine to a [Fe4S4] cluster. Reprinted with permission from ref (26). Copyright 2011 American Chemical Society.
Here, we have investigated the binding of 10, as well as several analogues, to IspH by using X-ray crystallography. We also used density functional theory (DFT) methods to probe the nature of the bonding between the ligand and metal cluster, in addition to computing the HYSCORE observables: the hyperfine and NQCC values.
Results and Discussion
Crystallographic Structures
To investigate the mode of binding and inhibition we crystallized (pyridin-3-yl)methyl diphosphate (10) with Escherichia coli IspH and determined the structure to 1.7 Å resolution (PDB ID 4MUX); for details see Table S1 in the Supporting Information. The overall architecture of IspH in this complex is virtually identical to the previously reported structure of the protein bound to the substrate 3.9 The pyridine ring is located close to the apical iron of the cluster, however, this labile iron site is only present in the IspH–10 complex (Figure 3). Upon inspection of the apical iron, the difference electron density displays a negative peak at this site indicating that this site is partially occupied and, as previously noted, the unique fourth iron of the cluster readily dissociates when not stabilized by a ligand.12,21 Comprehensive investigation (Table S2 and Figure S2 in the Supporting Information) of the occupancy of the fourth iron (Fe3 in the PDB file) resulted in values of 0.45 for chain A and 0.51 for chain B indicating that there is a [Fe4S4] cluster in about 50% of the molecules in the IspH–10 crystal. The occupancy of the ligand, on the other hand, is 1. The position of the ligand 1o is very similar to that predicted earlier using computational docking23 with a root-mean-square deviation (rmsd) of 0.89 Å between the ligand’s heavy atoms in the docked and crystal structures, shown in Figure 3c.
Figure 3.
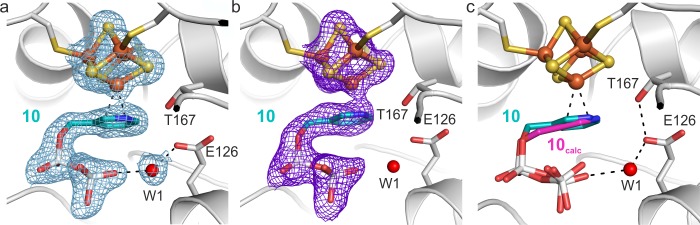
X-ray structure of IspH bound to (pyridin-3-yl)methyl diphosphate, 10. (a) A 2FO – FC electron density map (blue mesh, contoured at 1.0σ) is presented for the [Fe4S4] cluster, the ligand, and the solvent molecules in the first coordination sphere. The protein is shown as cartoon and the iron–sulfur cluster as a ball-and-stick model; the ligand and amino acid side chains are shown as stick models; the water molecule is shown as a red sphere, and the dotted lines indicate hydrogen or coordinative bonds. (b) A FO – FC electron density map (purple mesh, contoured at 3.0 σ) obtained by omitting the [Fe4S4] cluster and the ligand from the refinement. (c) Comparison between X-ray and docking results from ref (26).
The crystal structure of 10 bound to IspH reveals two main points of interest. First, there are two pyridine ring atoms that are very close (2.3–2.4 Å) to the fourth iron. Second, there is evidence for a continuous electron density between the fourth iron and the pyridine nitrogen. Taken together, these results suggest that 10 might coordinate to and stabilize both oxidized (as crystallized) and reduced clusters (the cluster is likely reduced in the X-ray beam, as observed with 3),9 raising of course the question: how does 10 bind to the fourth iron? While it is not possible to unequivocally assign the two ligand atoms that are close to the fourth Fe to C or N (since the ring could be flipped), the resonance structures shown in Scheme 3 seem likely since they permit η2 interactions with the cluster. In addition, as we shall describe below, the 180 degree ring-flipped structures cannot account for the HYSCORE results.
Scheme 3. Proposed Binding Mode of the Pyridine Ring of 10 to the [Fe4S4] Cluster in the IspH–10 Complex.

While it is possible that the aromatic ring in 10 might simply be forming a van der Waals complex, the following observations suggest that the pyridine ring is covalently bound to the cluster: (i) the short metal–ligand distances; (ii) the continuous electron density; (iii) the stabilization of the fourth iron; and (iv) the higher inhibition activity with 10 as compared to 11–13, reported previously with Aquifex aeolicus IspH.23 Based on our X-ray diffraction results we propose a η2 side-on metal–ligand coordination in the IspH–10 complex. This type of bonding is seen with pyridine ligands coordinating to other metals such as Ta,27,28 and similar bonding has also been proposed to occur in Rh-catalyzed reactions.29
An interaction of the fourth iron with a nitrogen-containing ligand has been observed previously with the substrate analogue 4.20 Superposition of the IspH–10 and IspH–4 complexes (Figure S3 in the Supporting Information) shows that the positions of the nitrogen atoms are similar, even though the core structure of the molecules and their bonding show significant differences. Both compounds permit one water molecule to occupy the remaining space in the active site, and they are both able to stabilize the apical iron of the [Fe4S4] cluster, supporting the idea that the ability to interact with the fourth iron in the cluster is key for enhanced IspH inhibition.
This intriguing X-ray structure raised several questions: Which orientation does the pyridine ring adopt in the molecules with a [Fe4S4] cluster? What impact do substituents at the pyridine ring have? Is the structure consistent with previous spectroscopic observations? To further investigate these questions we determined the structures of E. coli IspH in complex with (pyridin-4-yl)methyl diphosphate (11), (pyridin-2-yl)methyl diphosphate (12), and (6-chloropyridin-3-yl)methyl diphosphate (13). Compounds 11–13 are also pyridine analogues carrying a methylene diphosphate substituent in the 3- (10, 13), 4- (11), or 2- (12) position with 13 being the 6-Cl analogue of 10 (Scheme 2). The structures of the IspH–ligand complexes were solved to 1.8 Å resolution (11, PDB ID 4MUY), 1.9 Å resolution (12, PDB ID 4MV0), and 1.9 Å resolution (13, PDB 1D 4MV5). All four species (10–13) bind in a similar manner to the SXN motif in the IspH active site with the diphosphate groups located in the same pocket as seen with diverse diphosphates.
Unlike the IspH–10 complex with a [Fe4S4] cluster, the complexes of 11–13 (Figure 4) contain only a [Fe3S4] cluster. In a formal sense it is not known whether the [Fe3S4] cluster is selectively crystallized in the 11–13 complexes, or whether the fourth iron is simply lost during crystallization of the oxidized protein with the cluster being stabilized by binding to 10. Two pieces of information suggest that the latter possibility is more likely. First, in four crystal structures of IspH–ligand complexes in which there is clear evidence for the presence of a fourth iron, the allyl group-containing ligands 3–59,12,20 and the enolate of 8(21) bind to (or very close to) the apical iron. This suggests that the fourth iron is stabilized by such ligand binding, and indeed with the complexes of 3 and the enolate of 8, the [Fe4S4] cluster is remarkably stable under aerobic conditions. However, the IspH products 1 and 2 crystallize as [Fe3S4] complexes, presumably due to weak product bonding (facilitating product release). Second, in previous work with Aquifex aeolicus IspH we found that 10 is a better inhibitor than are the remaining compounds investigated here.23 If 11–13 caused cluster degradation, they would presumably be very potent inhibitors, which they are not.23 We thus next consider the structures of 11–13 in more detail.
Figure 4.
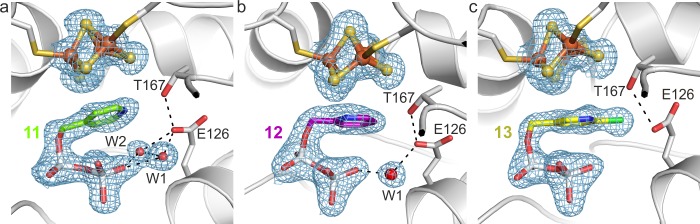
Crystal structures of IspH in complex with (a) (pyridin-4-yl)methyl diphosphate, 11; (b) (pyridin-2-yl)methyl diphosphate, 12; (c) (6-chloropyridin-3-yl) methyl diphosphate), 13. Orientation and representation are according to Figure 3.
Based on all of the X-ray structures determined here it seems unlikely that a para-N in a pyridine ring would bind to the apical Fe. The X-ray structure of IspH–11 indicates that the pyridine ring is positioned close to the [Fe3S4] cluster creating a space for two water molecules. Comparison between the structures of ligands 10 and 12 is intriguing since the nitrogen and the carbon atom that participate in the proposed η2 coordination in the IspH–10 structure are “switched” (Figure 5). The difference electron-density (FO – FC) map and metal site validation show, however, that the occupancy of the apical site is zero. Additionally, a B factor analysis suggests that the pyridine ring is in the same orientation as seen with 10 (both nitrogen atoms point in the same direction). The refinement with the aromatic ring flipped to the opposite site leads to significantly increased B factors, especially for the nitrogen atom. The preference of the ring for this orientation might be a result of the increased hydrophobicity of one part of the active site pocket with residues Val15, Val40, Ala73, and Val99 versus the more hydrophilic site with residues Glu126, Thr167, Thr168, Thr200, and Asn227. The 6-Cl analogue of 10, 13, would not be expected to have any significant σ-donor or H-bonding capacity due to the electron-withdrawing chlorine atom, expected to result in a decrease in pKa of ∼4.8 units for the conjugate acid, and as expected the fourth iron of the cluster is absent in the IspH–13 complex structure.
Figure 5.
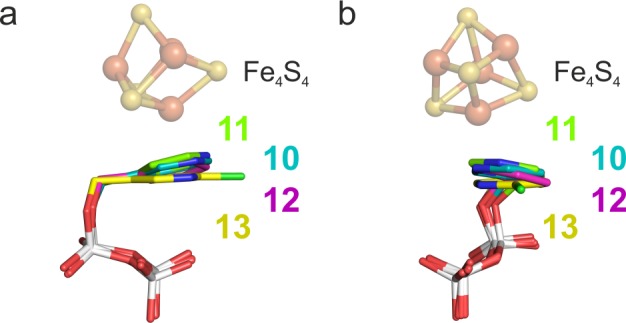
Structural superposition of IspH in complex with ligands 10–12. (a) Front view of the pyridine ring and the side chains of Thr167 as well as E126. (b) Side view of the pyridine rings.
Quantum Chemical Calculations
In order to investigate the nature of the proposed iron–pyridine interaction in more detail, we carried out a series of quantum mechanical (QM) calculations. It should be noted that both the redox state of the cluster and the protonation state of the ligand are formally unknown, since oxidized clusters can be reduced by the X-ray beam, and protons are not detected in protein X-ray crystallography. Therefore, we investigated the four possible combinations of oxidized (Ox, S = 0) and reduced (Red, S = 1/2) iron–sulfur clusters with neutral pyridine (pyr) or pyridinium (pyrH+) ligands, including 180 degree pyridine ring-flipped isomers. We also studied four different structural models in order to see how the protein environment might affect bonding. In all cases the initial geometries were taken from the X-ray structures determined here, and we used the quantum chemical methods and program reported previously for other [Fe4S4] systems.23,30,31
At the so-called Small-Fopt level, the models consist of oxidized or reduced [Fe4S4(SCH3)4]n− (Small) clusters with the protein environment being reduced to the cysteine side chains truncated at the Cβ position. Both pyr and pyrH+ ligands were considered, in which all atoms were fully optimized (=F in Fopt) without any restraints. As shown in Table S3 in the Supporting Information, in these models, the final coordination modes in the optimized structures depend only on the protonation state of the pyridine and not on the redox state of the cluster. These results show that, in such unrestricted optimizations with no protein environment, pyridine prefers end-on coordination through the nitrogen’s lone pair, as expected. The pyridinium on the other hand can stably adopt the side-on coordination, because of the unavailability of the nitrogen’s lone pair due to protonation. Interestingly, when the three cysteine Cβ atoms and the two carbons neighboring the iron-bonded N and C atoms are fixed at the X-ray structure during geometry optimizations (called Small-Popt1, where P = Partial), the pyridine can now adopt side-on coordination, albeit only in the reduced state of the [Fe4S4] cluster (Table S3 in the Supporting Information). However, once the unique fourth iron is also fixed at the crystal structure position (Small-Popt2 models in Table S3 in the Supporting Information), even with the oxidized cluster, the side-on mode is possible for pyridine. The average energy penalties due to fixing iron between Small-Popt1 and Small-Popt2 models are ca. 2 kcal/mol. These results suggest that the protein environment may play a role in determining the coordination mode. To probe this possibility in a more direct way, we next investigated models containing Thr167, which may form a hydrogen bond to the nitrogen of the pyridine moiety in 10. To better mimic the protein environment effect32 we also extended the residue models up to Cα for Thr167 and the three coordinated cysteine residues, with only the Cα atoms being fixed at the X-ray structure positions. The pyridine was also modified to include the attached CH2OH group (mimicking the CH2OPP group), with only the oxygen fixed at the X-ray position. The results obtained using this structural model are summarized in Table 1 and are displayed in Figure 6.
Table 1. Geometric and Spectroscopic Properties of the Structural Models Shown in Figure 6.
| coord mode | RFeN/C (Å) | RFeC (Å) | AisoN (MHz) | NQCCN (MHz) | ||||
|---|---|---|---|---|---|---|---|---|
| expta | η2 | 2.3 | 2.4 | |||||
| exptb | reduced | 7.4 | 3.0 | |||||
| calc | 1 | oxidized | pyr | η1 | 2.1 | 3.0 | 3.0 | |
| 2 | oxidized | pyrH+ | η2 | 2.1 | 2.1 | 1.7 | ||
| 3 | reduced | pyr | η2 | 2.0 | 2.2 | 6.6 | 3.5 | |
| 4 | reduced | pyrH+ | η2 | 2.1 | 2.0 | 1.2 | 2.4 | |
| 5 | oxidized | pyr (flipped) | η1 | 2.3 | 2.8 | 5.0 | ||
| 6 | oxidized | pyrH+ (flipped) | η2 | 2.1 | 2.1 | 2.6 | ||
| 7 | reduced | pyr (flipped) | η2 | 2.2 | 2.2 | 0.2 | 4.5 | |
| 8 | reduced | pyrH+ (flipped) | η2 | 2.1 | 2.1 | 0.5 | 3.3 |
This work.
Reference (26).
Figure 6.
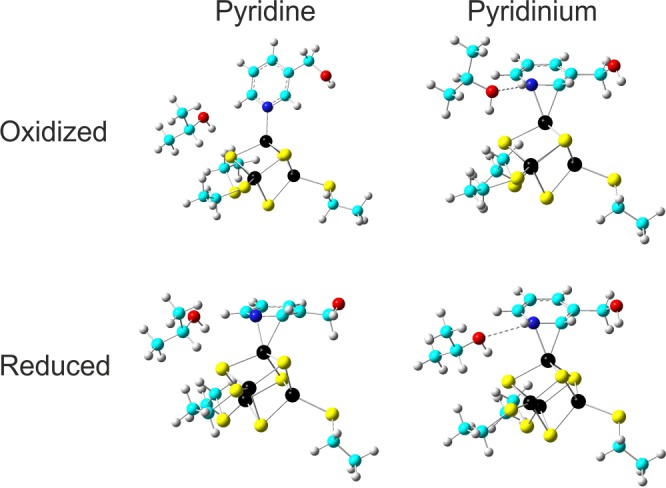
Optimized structures for oxidized and reduced models with pyridine or pyridinium ligands. Color scheme: C, cyan; Fe, black; S, yellow; O, red; H, gray.
For the oxidized cluster, only the pyridinium model is consistent with the X-ray structure. However, if the cluster is reduced (in the X-ray beam, as reported earlier for 3),9 then both the pyridine and the pyridinium structures have Fe–N and Fe–C distances close to the experimentally observed values. Based on a comparison with the previously reported experimental 14N isotropic hyperfine coupling (AisoN) and nuclear quadrupole coupling constant (NQCC = e2qQ/h) results for 10 bound to A. aeolicus IspH26, the pyridine model is clearly preferred over the pyridinium model. Both the predicted AisoN and NQCCN values are in excellent agreement with experiment, while the AisoN value for the pyridinium structure is poorly predicted (Table 1). Specifically, the error in AisoN is only ∼16% and that for the NQCC is ∼11%. Moreover, calculations based on the ring-flipped isomers (in which N is not bonded to the fourth Fe) result in very small 14N hyperfine couplings (∼0.2 MHz, to be compared with 6.6 MHz for the alternate conformer and 7.4 MHz from experiment). These results all suggest that 10 binds to the reduced cluster in a side-on pyridine conformation, as found with other pyridine complexes.27,28
Overview of Ligand Binding to IspH
The structures reported here are of general interest since they suggest another unusual type of metal–ligand bonding in IspH. In earlier work it was found that the substrate 3 binds to oxidized IspH via its 1-OH group, as illustrated in Scheme 4. The same type of η1 bonding is seen in the amino and thiol analogues of 3 bound to oxidized IspH, as well as in the enolate intermediate that forms from the acetylene 6 on hydration. Higher (η2) coordination numbers are now apparent in the pyridine complex 10, and it has also been proposed that η2 and η3 species14 are involved as reactive intermediates in IspH catalysis, based on the results of HYSCORE and electron–nuclear double resonance (ENDOR) spectroscopies and of DFT calculations.14,33 Plus, a putative ferraoxetane reactive intermediate (containing Fe–C and Fe–O bonds) in the IspG reaction15 (in which methyl-erythritol cyclo-diphosphate is converted to 3) has been proposed, again based on the results of HYSCORE and ENDOR spectroscopies and DFT calculations.34 These structures are in many cases of interest from a mechanism of action/inhibition perspective, and via protein engineering, they may lead to new chemistries.
Scheme 4. Overview of the Observed or Proposed Binding Modes of Various Ligands to the Unique Fourth Iron of the [Fe4S4] Cluster of IspH (or for 15, IspG).

The binding modes can be subdivided into (a) η1-, (b) η2-, and (c), η3-coordination.
Conclusions
We report the first crystal structures of E. coli IspH bound to pyridine ligands. Of the four structures obtained, only IspH bound to 10 contains a [Fe4S4] cluster (the others contain [Fe3S4] clusters) and 10 was previously reported to be the most potent inhibitor of A. aeolicus IspH.23 Two atoms in the ligand’s pyridine ring bind close (2.3–2.4 Å) to the fourth iron in the cluster, and the general pose is similar to that predicted earlier based on docking calculations (a 0.89 Å rmsd for all ligand non-hydrogen atoms). QM results enable very good predictions of previous 14N hyperfine coupling (AisoN = 7.4 MHz, experiment; 6.6 MHz, calculated) as well as the 14N quadrupole coupling constant results (NQCC = 3.0 MHz experiment; 3.5 MHz, calculated) from HYSCORE spectroscopy, and strongly suggest that the [Fe4S4] cluster is in a reduced state with the pyridine involved in side-on η2 coordination. Overall, the results contribute to the growing database of bio-organometallic complexes of IspH, of interest not only from a drug discovery perspective but potentially from a synthetic chemistry perspective as well.
Experimental Section
Protein purification
E. coli IspH containing a His6 tag (encoded in the pQE30 plasmid) was coexpressed with isc proteins (encoded in the pACYC184 plasmid) in XL-1 blue cells. Cells were grown at 30 °C for 20 h in Terrific Broth supplemented with ampicillin (180 mg/L), chloramphenicol (25 mg/L), l-cysteine (167 mg/L), and ammonium iron(III) citrate (32 mg/L). Cells were then harvested by centrifugation, washed with 0.9% aqueous sodium chloride, and stored at −80 °C. All further steps were performed under anaerobic conditions in a Coy Vinyl Anaerobic Chamber under an N2/H2 (95%/5%) atmosphere. The cell pellet was resuspended in five volumes of 100 mM Tris hydrochloride, pH 8, containing 500 mM sodium chloride, 20 mM imidazole, and 5 mM sodium dithionite. The mixture was passed through a French Press and was subsequently centrifuged. The resulting crude extract was loaded onto a Ni2+ Chelating Sepharose Fast Flow column (20 mL), which had been equilibrated in 100 mM Tris hydrochloride, pH 8, containing 500 mM NaCl and 20 mM imidazole. The protein was eluted with 100 mM Tris hydrochloride, pH 8, containing 500 mM NaCl and 150 mM imidazole. The solution was dialyzed against 50 mM Tris hydrochloride, pH 8.
Crystallization
E. coli IspH (18.3 mg/mL) was incubated with an aqueous solution of the ligand (final concentration 1 mM) for 20 min prior to setting up crystal trays. Brown crystals were obtained by using the sitting-drop vapor diffusion method with a 1:1 ratio of protein and reservoir solution at 20 °C. The precipitant consisted of 100 mM BisTris/HCl, pH 6.5, 200 mM ammonium sulfate, and 25% polyethylene glycol 3350. Crystals were soaked with cryoprotectant solution (50% aqueous polyethylene glycol 400) for 1 min, mounted on loops, and flash frozen in a stream of nitrogen gas at 100 K (Oxford Cryo Systems).
Data Collection and Structure Determination
Native data sets were collected using synchrotron radiation at the X06SA-beamline at the Swiss Light Source, Villigen, Switzerland. The coordinates of E. coli IspH bound to the substrate HMBPP (PDB ID 3KE8) were used for phasing by molecular replacement.16,35 Data sets were processed using the program package XDS,36 and anisotropy of diffraction was corrected using TLS37,38 refinement. Model building and refinement were performed with Coot39 and Refmac.38,40 The occupancy of the apical iron site was refined using phenix.refine41 and validated with CheckMyMetal web server.42 Electron density maps were calculated using FFT,38,43 Ramachandran plots were calculated with PROCHECK,38,44 and figures were prepared using PyMOL.45 For more details see Table S1 in the Supporting Information.
Accession Numbers
The atomic coordinates for IspH bound to the ligands 10, 11, 12, and 13 have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics at Rutgers University (IspH–10 PDB ID 4MUX, IspH–11 PDB ID 4MUY, IspH–12 PDB ID 4MV0, and IspH–13 PDB ID 4MV5).
Computational Details
To investigate the binding of 10 to the iron–sulfur cluster, we used density functional theory. All calculations were performed with the Gaussian 09 program46 using a Wachters’ basis for Fe,47 6-311G(d) for other heavy atoms, 6-31G(d) for hydrogens, with the BPW9148,49 functional, as reported previously.23,30,31,49 This approach23,50−56 has enabled accurate predictions of Mößbauer and EPR/nuclear magnetic resonance (NMR) hyperfine properties in a diverse range of iron-containing proteins and model systems. The predicted isotropic EPR hyperfine coupling and nuclear quadrupole coupling constant results (Table S3 in the Supporting Information) are from the directly calculated values from the quantum chemical calculations, which were scaled based on the regression lines between theory and experiment, as described in previous reports.26,31 The coordinates of all of the optimized structures described in the text are provided in Tables S4–S23 in the Supporting Information.
Acknowledgments
This work was supported by the TUM Graduate School, the Hans-Fischer Gesellschaft, DFG Grant GR1861/5-1, the United States Public Health Service (NIH Grants GM085774 to Y.Z. and GM065307 to E.O.), and National Science Foundation Grant CHE-1300912 to Y.Z.. We would like to thank the staff of the X06SA-beamline at the Paul Scherer Institute (Swiss Light Source in Villigen, Switzerland) for support during data collection, as well as Professor Jared C. Lewis for helpful comments.
Supporting Information Available
Additional tables with data collection and refinement statistics; geometric and spectroscopic properties of various QM models; coordinates of all quantum chemically optimized models. Supplementary figures and references. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
‡ Department of Molecular Biosciences, Northwestern University, 2205 Tech Drive, Evanston, Illinois 60208, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ruzicka L. Cell. Mol. Life Sci. 1953, 9, 357. [Google Scholar]
- Bloch K. Steroids 1992, 57, 378. [DOI] [PubMed] [Google Scholar]
- Rohmer M.; Knani M.; Simonin P.; Sutter B.; Sahm H. Biochem. J. 1993, 295Pt 2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohdich F.; Zepeck F.; Adam P.; Hecht S.; Kaiser J.; Laupitz R.; Gräwert T.; Amslinger S.; Eisenreich W.; Bacher A.; Arigoni D. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam P.; Hecht S.; Eisenreich W.; Kaiser J.; Gräwert T.; Arigoni D.; Bacher A.; Rohdich F. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altincicek B.; Duin E. C.; Reichenberg A.; Hedderich R.; Kollas A. K.; Hintz M.; Wagner S.; Wiesner J.; Beck E.; Jomaa H. FEBS Lett. 2002, 532, 437. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Liu P. Angew. Chem., Int. Ed. 2008, 47, 9722. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Zhao Z. K.; Liu P. J. Am. Chem. Soc. 2008, 130, 2164. [DOI] [PubMed] [Google Scholar]
- Gräwert T.; Span I.; Eisenreich W.; Rohdich F.; Eppinger J.; Bacher A.; Groll M. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Wang K.; Liu Y. L.; No J. H.; Li J.; Nilges M. J.; Oldfield E. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Chang W. C.; Liu H. W.; Liu P. Org. Lett. 2011, 13, 5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Span I.; Gräwert T.; Bacher A.; Eisenreich W.; Groll M. J. Mol. Biol. 2012, 416, 1. [DOI] [PubMed] [Google Scholar]
- Citron C. A.; Brock N. L.; Rabe P.; Dickschat J. S. Angew. Chem., Int. Ed. 2012, 51, 4053. [DOI] [PubMed] [Google Scholar]
- Xu W.; Lees N. S.; Hall D.; Welideniya D.; Hoffman B. M.; Duin E. C. Biochemistry 2012, 51, 4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekittke I.; Wiesner J.; Rohrich R.; Demmer U.; Warkentin E.; Xu W.; Troschke K.; Hintz M.; No J. H.; Duin E. C.; Oldfield E.; Jomaa H.; Ermler U. J. Am. Chem. Soc. 2008, 130, 17206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräwert T.; Rohdich F.; Span I.; Bacher A.; Eisenreich W.; Eppinger J.; Groll M. Angew. Chem., Int. Ed. 2009, 48, 5756. [DOI] [PubMed] [Google Scholar]
- Kent T. A.; Dreyer J. L.; Kennedy M. C.; Huynh B. H.; Emptage M. H.; Beinert H.; Munck E. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer G.; Heinz D. W.; Jahn D.; Schubert W.-D. Curr. Opin. Chem. Biol. 2004, 8, 468. [DOI] [PubMed] [Google Scholar]
- Tamarit J.; Gerez C.; Meier C.; Mulliez E.; Trautwein A.; Fontecave M. J. Biol. Chem. 2000, 275, 15669. [DOI] [PubMed] [Google Scholar]
- Span I.; Wang K.; Wang W.; Jauch J.; Eisenreich W.; Bacher A.; Oldfield E.; Groll M. Angew. Chem., Int. Ed. 2013, 52, 2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Span I.; Wang K.; Wang W.; Zhang Y.; Bacher A.; Eisenreich W.; Li K.; Schulz C.; Oldfield E.; Groll M. Nat. Commun. 2012, 3, 1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield E. Acc. Chem. Res. 2010, 43, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.; Wang W.; No J. H.; Zhang Y.; Oldfield E. J. Am. Chem. Soc. 2010, 132, 6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janthawornpong K.; Krasutsky S.; Chaignon P.; Rohmer M.; Poulter C. D.; Seemann M. J. Am. Chem. Soc. 2013, 135, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens-Botzong A.; Janthawornpong K.; Wolny J. A.; Tambou E. N.; Rohmer M.; Krasutsky S.; Poulter C. D.; Schünemann V.; Seemann M. Angew. Chem., Int. Ed. 2011, 50, 11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Li J.; Wang K.; Smirnova T. I.; Oldfield E. J. Am. Chem. Soc. 2011, 133, 6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert K. J.; Neithamer D. R.; Zonnevylle M. C.; LaPointe R. E.; Schaller C. P.; Wolczanski P. T. Inorg. Chem. 1991, 30, 2494. [Google Scholar]
- Neithamer D. R.; Parkanyi L.; Mitchell J. F.; Wolczanski P. T. J. Am. Chem. Soc. 1988, 110, 4421. [Google Scholar]
- Wiedemann S. H.; Lewis J. C.; Ellman J. A.; Bergman R. G. J. Am. Chem. Soc. 2006, 128, 2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. L.; Guerra F.; Wang K.; Wang W.; Li J.; Huang C.; Zhu W.; Houlihan K.; Li Z.; Zhang Y.; Nair S. K.; Oldfield E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Wang K.; Smirnova T. I.; Khade R. L.; Zhang Y.; Oldfield E. Angew. Chem., Int. Ed. 2013, 52, 6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Ling Y.; Zhang Y. J. Am. Chem. Soc. 2011, 133, 13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Wang K.; Span I.; Jauch J.; Bacher A.; Groll M.; Oldfield E. J. Am. Chem. Soc. 2012, 134, 11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Li J.; Wang K.; Huang C.; Zhang Y.; Oldfield E. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein F. C.; Koetzle T. F.; Williams G. J.; Meyer E. F. Jr.; Brice M. D.; Rodgers J. R.; Kennard O.; Shimanouchi T.; Tasumi M. Eur. J. Biochem. 1977, 80, 319. [DOI] [PubMed] [Google Scholar]
- Kabsch W. J. Appl. Crystallogr. 1993, 26, 795. [Google Scholar]
- Winn M. D.; Isupov M. N.; Murshudov G. N. Acta Crystallogr. D 2001, 57, 122. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Acta Crystallogr. D 2011, 67, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Acta Crystallogr. D 2004, 60, 2126. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Acta Crystallogr. D 1997, 53, 240. [DOI] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriatry N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Acta Crystallogr. D 2012, 68, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.; Chordia M. D.; Cooper D. R.; Chruszcz M.; Müller P.; Sheldrick G. M.; Minor W. Nat. Protoc. 2014, 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read R. J.; Schierbeek A. J. J. Appl. Crystallogr. 1988, 21, 490. [Google Scholar]
- Laskowski R. A.; Rullmannn J. A.; MacArthur M. W.; Kaptein R.; Thornton J. M. J. Biomol. NMR 1996, 8, 477. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.3; Schrödinger, LLC. [Google Scholar]
- Frisch M. J.et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, 2010. [Google Scholar]
- http://www.emsl.pnl.gov/forms/basisform.html.
- Becke A. D. Phys. Rev. A 1988, 38, 3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Wang Y. Phys. Rev. B 1996, 54, 16533. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Oldfield E. J. Am. Chem. Soc. 2004, 126, 4470. [DOI] [PubMed] [Google Scholar]
- Mao J.; Zhang Y.; Oldfield E. J. Am. Chem. Soc. 2002, 124, 13911. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Mao J.; Godbout N.; Oldfield E. J. Am. Chem. Soc. 2002, 124, 13921. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Mao J.; Oldfield E. J. Am. Chem. Soc. 2002, 124, 7829. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Gossman W.; Oldfield E. J. Am. Chem. Soc. 2003, 125, 16387. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Oldfield E. J. Phys. Chem. A 2003, 107, 4147. [Google Scholar]
- Zhang Y.; Oldfield E. J. Phys. Chem. B 2003, 107, 7180. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.