

Abstract
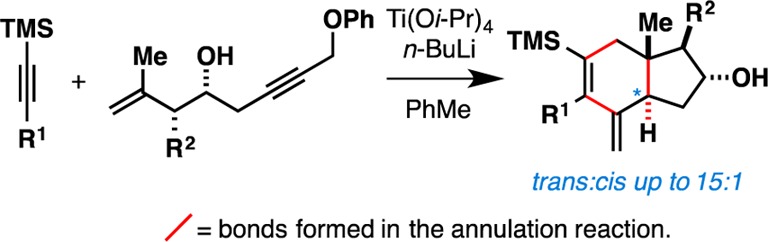
Angularly substituted trans-fused hydroindanes are now accessible by the direct and convergent union of trimethylsilyl (TMS)-alkynes with 4-hydroxy-1,6-enynes by a process that forges three C–C bonds, one C–H bond, and two new stereocenters. The annulation is proposed to proceed by initial formation of a Ti–alkyne complex (with a TMS-alkyne) followed by regioselective alkoxide-directed coupling with the enyne, stereoselective intramolecular cycloaddition, elimination of phenoxide, 1,3-metallotropic shift, and stereoselective protonation of the penultimate allylic organometallic intermediate. Several examples are given to demonstrate the compatibility of this reaction with substrates bearing aromatic and aliphatic substituents, and an empirical model is presented to accompany the stereochemical observations.
Functionalized carbocycles remain challenging targets in modern chemical synthesis despite decades of effort focused on their synthesis. In fact, campaigns to target such systems often abandon total synthesis campaigns altogether in favor of natural product derivatization (semisynthesis).1 Among the great variety of ubiquitous carbocyclic motifs in natural products and medicinal agents that continue to represent a significant challenge are angularly substituted trans-fused hydroindanes (bicyclo[4.3.0]nonanes; Figure 1). Synthetic strategies for such systems typically embrace key reactions such as cation–olefin cyclization,2 Robinson annulation,3 and cycloaddition,4 all of which suffer from substantial challenges associated with either the synthesis of the appropriately functionalized substrate or the requirement of numerous reactions after ring formation to establish the stereochemistry of ring fusion. Thus, a simple reaction process for the direct and convergent synthesis of highly substituted trans-fused hydroindanes would be a welcome alternative to these classic strategies. Here we describe the realization of such a method that proceeds from the intermolecular union of simple acyclic starting materials and delivers trans-fused hydroindanes bearing up to six substituents, including an angular methyl group.
Figure 1.
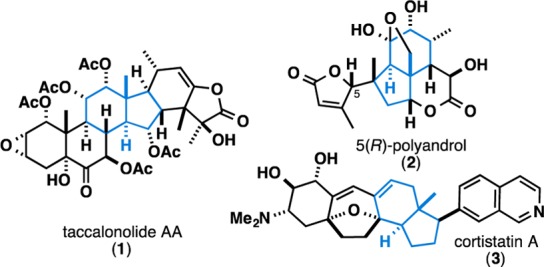
Representative examples of natural products that possess angularly substituted trans-fused hydroindanes.
We recently described a metallacycle-mediated coupling reaction between 4-hydroxy-1,6-enynes and internal alkynes that delivers dihydroindanes possessing two endocyclic tetrasubstituted alkenes.5 While this method proved useful for the stereoselective assembly of a variety of substituted carbocyclic structures where the stereochemistry of the angular alkyl group (C13, steroid numbering) was reliably set anti to the hydroxyl group at C16, the annulation reaction did not address the ring fusion stereochemistry (C14 remained sp2-hybridized). As illustrated in Figure 2, our efforts to address this different and more demanding stereochemical challenge (i.e., establishing the stereochemistry of C14 in concert with the ring-forming process) have resulted in the discovery of a metallacycle-mediated annulation reaction that delivers angularly substituted trans-fused hydroindanes 6 (ds up to 15:1).
Figure 2.
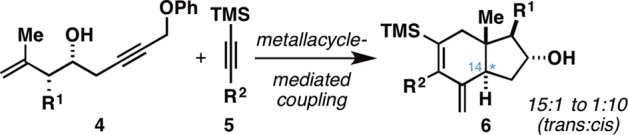
Trans-fused hydroindanes by stereoselective metallacycle-mediated cross-coupling.
Our investigations began with an exploration of the metallacycle-mediated union of enyne 7(5) with trimethylsilyl (TMS)-substituted phenylacetylene (8) (Figure 3). Treatment of 8 with ClTi(Oi-Pr)3 and an ethereal solution of c-C5H9MgCl (commercially available from Aldrich) in PhMe (−78 to −30 °C) was followed by addition of the lithium alkoxide of 7 as a solution in Et2O. After the mixture was slowly warmed from −78 °C to rt, this reaction was terminated by addition to a −78 °C solution containing a proton source. To our great surprise and delight, the nature of the alcohol used to quench the presumed organometallic intermediate had a profound effect on the stereoselectivity of the annulation. Quenching with t-BuOH led to a 3:2 mixture of annulation products (9a:9b) in 68% yield, while quenching with i-PrOH showed a significant preference for the formation of the cis-fused hydroindane 9b (ds = 10:1).7 Finally, quenching of the reaction with MeOH resulted in selective formation of the more strained trans-fused hydroindane product 9a (ds = 6:1).8 While the mechanistic underpinnings associated with this shift in stereoselection as a function of the alcohol employed to terminate the process remain poorly understood, the core components of the annulation mechanism are proposed as depicted in the bottom portion of Figure 3. Alkoxide-directed alkyne–alkyne coupling is reasoned to be an essential first step in the process,6 delivering the 2-silyl-substituted titanacyclopentadiene intermediate II. Subsequent ligand exchange to afford III and stereoselective intramolecular [4 + 2] cycloaddition is then proposed to deliver bridged bicyclic metallacyclopentene intermediate IV. Elimination of the distal phenyl ether then results in the formation of V. Isomerization of the resulting tertiary allylic metal species (to generate VI) is then followed by stereoselective protonation via VII or VIII, which generates the C14 stereocenter and establishes the nature of the ring fusion (trans or cis). It was recognized that the ability to selectively establish the stereochemistry of the ring fusion in concert with annulation represents a significant advance in annulation chemistry targeting this ubiquitous carbocyclic motif, and furthermore, the discovery of a path to selectively access the higher-energy trans-fused isomer was identified as a particularly compelling characteristic of this reaction process.8
Figure 3.

Stereoselective access to trans- and cis-fused angularly substituted hydroindanes by coupling of TMS-alkynes with 1,6-enynes.
Moving on from these initial observations, our first goal was to define a reaction procedure that could be run without careful control of the reaction temperature. While the procedure summarized in Figure 3 led to the discovery of a process for selective generation of trans-fused hydroindanes, the experimental details of the reaction were suboptimal. In short, the temperature profile required (−78 to −30 °C and prolonged stirring at −30 °C) resulted in a challenging experimental protocol that was difficult to reproduce. Other characteristics of this procedure that were unappealing included the use of a mixed PhMe/Et2O solvent system and commercial solutions of ClTi(Oi-Pr)3. With these concerns in mind, we developed an alternative procedure that employs Ti(Oi-Pr)4/n-BuLi to execute the metallacycle-mediated annulation reaction in PhMe.9 These modified reaction conditions proceed between −78 and 50 °C and avoid prolonged stirring at −30 °C.
As illustrated by eq 1 in Figure 4, initial experiments examined the reaction of TMS-phenylacetylene 8 with enyne 7. Ti-mediated coupling and quenching with MeOH delivered hydroindane products in 67% yield, favoring the formation of the trans-fused isomer 9 (ds = 10:1).10 As we have seen in related annulation reactions of 4-hydroxy-1,6-enynes,5 the control of stereochemistry at C13 is quite high (all of the hydroindane products had a C13 β-Me group; this stereocenter is predictably set anti to the C16 hydroxy group). Also related to previous observations, the nature of the protic quench did have an impact on the product ratio, although the effect here was not as dramatic as that seen in the annulation reactions promoted by the combination of ClTi(Oi-Pr)3 and c-C5H9MgCl (Figure 3). In short, quenching with i-PrOH delivered a 1:1 mixture of stereoisomers, while quenching with H2O restored the selectivity for the trans-fused isomer, albeit with a modest drop in stereoselection (to 5:1). In no case were we able to invert the selectivity in favor of the cis-fused isomer.
Figure 4.

Annulation for the convergent synthesis of trans-fused hydroindanes. In eqs 6 and 7, the ratio of products obtained after purification (dr) is given because the 1H NMR spectrum of the crude material could not be resolved to determine an accurate ds.
The annulation method is effective with substituted TMS-phenylacetylenes 10 and 12, delivering trans-fused isomers 11 and 13 with selectivities of 15:1 and 12:1, respectively (eqs 2 and 3). As illustrated in eq 4, C17-furyl substitution, a common structural feature of liminoids, can be addressed in a straightforward fashion. Here the coupling between alkyne 8 and enyne 14 selectively delivers the trans-fused isomer 15 (ds = 10:1; 64% combined yield).
Notably, aromatic substitution on the β-face of the hydroindane at C17 is not required in order to achieve stereoselection in this annulation reaction. As illustrated in eq 5, the union of TMS-alkyne 8 with C17-methyl-substituted enyne 16 delivers the hydroindane products in 66% yield with 11:1 selectivity in favor of trans-fused product 17. Notably, substrates devoid of C17 substitution can also be advanced to trans-fused hydroindanes with this method. As depicted in eq 6, the union of 8 with enyne 18 proceeds in 52% yield and delivers the trans-fused product 19 with 5:1 dr.
The TMS-alkyne coupling partner can contain substituents other than Ph and substituted Ph. As illustrated in eq 7, the reaction of the furyl-substituted alkyne 20 with enyne 7 delivers the trans-fused hydroindane product 21 with 4:1 dr. Also, as illustrated in eq 8, coupling of the isopropyl-substituted TMS-alkyne 22 with enyne 14 delivers the trans-fused hydroindane product 23 in 46% yield (ds = 11:1).
The stereoselectivity observed in this annulation reaction can be rationalized as depicted in Figure 5. The central issue is thought to be preferential positioning of the Ti species on one of the two faces of the cyclohexadiene, setting up the stereoselective syn SE′ addition of MeOH. Because of the rather rigid conformation of the fused bicyclic structure, the substituent at C9 (steroid numbering) occupies a position in space that is slightly above the plane of the hydroindane (positioned on the β-face), and bond rotation is sterically impeded by the presence of the proximal TMS substituent. Thus, we speculate that a “gearing effect” would result in preferred positioning of the Ti center (along with its sterically demanding alkoxide ligands–not shown) on the α-face, anti to the Ph group (as in structure A). Subsequent quenching with MeOH by initial coordination to Ti followed by syn SE′ addition would then deliver the trans-fused isomer as the major product.11
Figure 5.
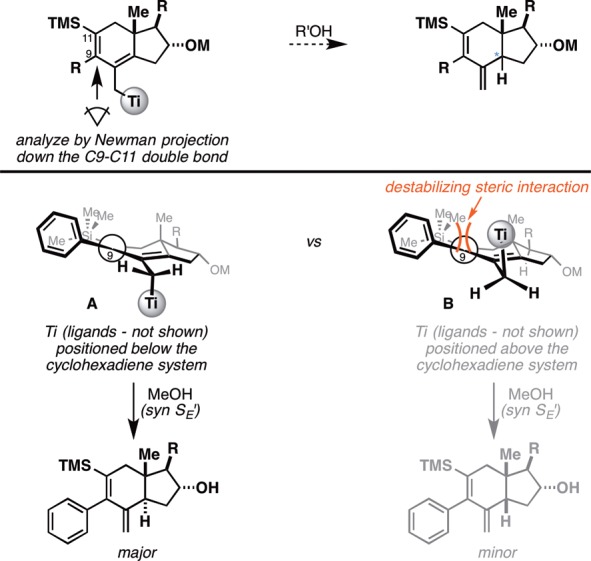
Empirical model for the annulation en route to trans-fused hydroindanes. Note: The Ph-substituted species was arbitrarily selected for discussion in the context of the empirical model presented above.
While the Ti-mediated annulation proved to be generally selective for the preparation of trans-fused hydroindanes when TMS-alkynes containing propargylic branching were employed, TMS-alkynes lacking this branching were observed to undergo coupling with a different sense of stereoselection. As illustrated in Figure 6, the use of benzyl-substituted TMS-acetylene (24) in coupling reactions with enynes 18 and 7 were moderately selective for the corresponding cis-fused products 25 and 26 (cis:trans = 3:1; eqs 9 and 10). As demonstrated in eq 11, C9 substitution of a steric nature more substantial than Me appears to be required for any stereoselectivity in the protic quench. Here the coupling of TMS-propyne (27) with enyne 14 delivered a 1:1 mixture of stereoisomeric hydroindanes (28).
Figure 6.
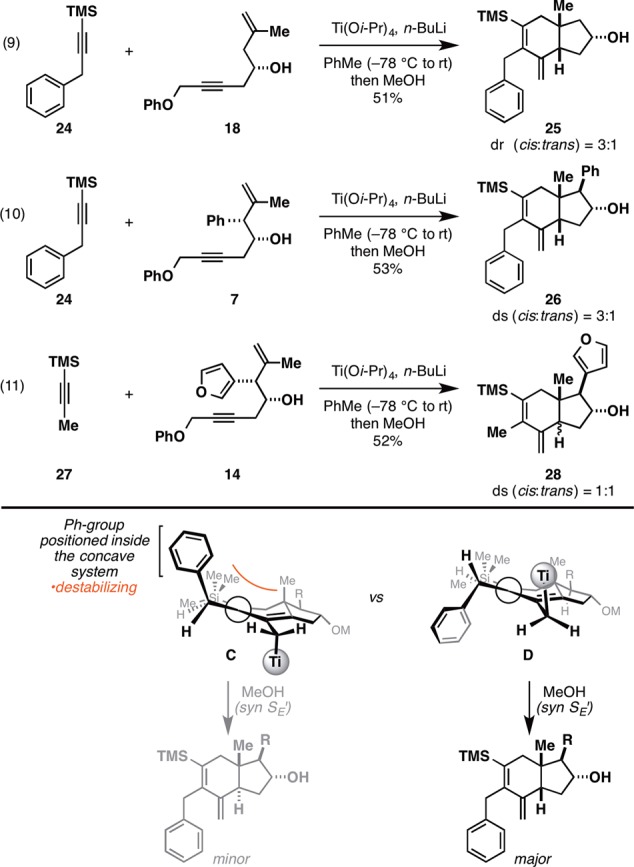
Metallacycle-mediated annulation reaction for the convergent synthesis of cis-fused hydroindanes.
An empirical model for the cis-selective annulation reaction is depicted in Figure 6 and is based on the preferential disposition of the Ph group on the α-face of the cyclohexadiene system (as in D). Subsequent “gearing” of the metal to avoid steric interactions with the Ph group is thought to result in positioning of the Ti center on the β-face, en route to the cis-fused hydroindane product by the syn SE′ reaction with MeOH.
Finally, while propargylic branching in the TMS-alkyne has been shown to be an important structural component to enable trans selectivity in the hydroindane-forming process, we note that long-range steric effects can also be employed in place of propargylic branching to regain selectivity for the trans-fused product. For example, as illustrated in Figure 7, the functionalized benzyl-substituted TMS-alkyne 29 engages enyne 7 in a trans-selective annulation despite not having propargylic branching. We speculate that this is due to conformational biasing of the aryl substituent that results in preferential positioning on the β-face of the cyclohexadiene. To avoid further nonbonded steric interactions, the Ti center adopts a position on the α-face of the carbocycle (as depicted in E), setting up the syn SE′ reaction with MeOH en route to the trans-fused product 30.
Figure 7.
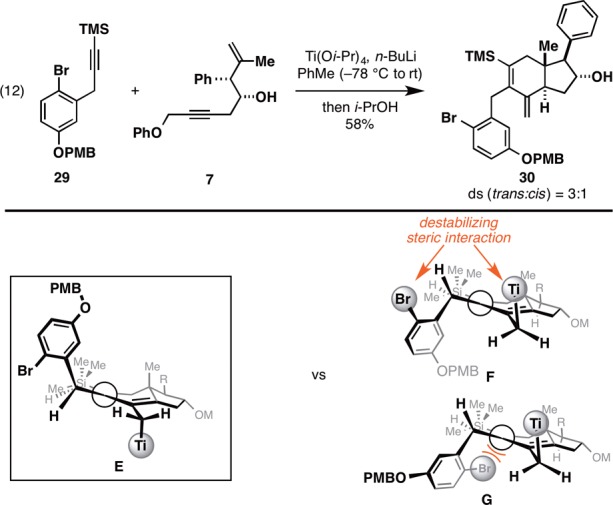
Long-range steric effects for selective access to trans-fused hydroindanes.
In conclusion, to the best of our knowledge, we have described the first convergent and stereoselective process for the direct synthesis of trans-fused hydroindanes from acyclic precursors. The reaction is thought to proceed by a complex metallacycle-mediated coupling reaction that features (1) initial alkoxide-directed coupling between a Ti–alkyne complex and a homopropargylic alkoxide, (2) stereoselective intramolecular [4 + 2] cycloaddition to generate a complex bridged tricyclic organometallic intermediate, (3) elimination of phenoxide, (4) metallatropic shift, and (5) stereoselective protonation. Our studies have demonstrated that densely functionalized hydroindanes with a variety of substituents at C17 and C9 (steroid numbering) can be easily accessed. These studies have revealed an important relationship between the structure of the TMS-alkyne employed and the ring fusion stereochemistry established in the product. Finally, an empirical model has been presented to support the stereochemical observations made. It is based on the position of the Ti center in the penultimate organometallic intermediate and protonation via a syn SE′ mechanism.
Acknowledgments
We thank the NIH NIGMS (GM080266) for financial support, the Uehara Memorial Foundation for a postdoctoral fellowship to H.M, and Prof. Peter Jacobi for helpful suggestions during preparation of the manuscript.
Supporting Information Available
Procedures and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For notable recent examples, see:; a Shenvi R. A.; Guerrero C. A.; Shi J.; Li C.-C.; Baran P. S. J. Am. Chem. Soc. 2008, 130, 7241. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Renata H.; Zhou Q.; Baran P. S. Science 2013, 339, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Snider B. B.; Kirk T. C. J. Am. Chem. Soc. 1983, 105, 2364. [Google Scholar]; b Johnson W. S.; Elliot J. D.; Hanson G. J. J. Am. Chem. Soc. 1984, 106, 1138. [Google Scholar]; c Hatakeyama S.; Numata H.; Osanai K.; Takano S. J. Chem. Soc., Chem. Commun. 1989, 1893. [Google Scholar]; d Browder C. C.; West F. G. Synlett 1999, 1363. [Google Scholar]
- a Hajos Z. G.; Parrish D. R. J. Org. Chem. 1974, 39, 1612. [Google Scholar]; b Stork G.; Shiner C. S.; Winkler J. D. J. Am. Chem. Soc. 1982, 104, 310. [Google Scholar]; c Stork G.; Winkler J. D.; Shiner C. S. J. Am. Chem. Soc. 1982, 104, 3767. [Google Scholar]; d Yamamoto K.; Iijima M.; Ogimura Y.; Tsuji J. Tetrahedron Lett. 1984, 25, 2813. [Google Scholar]; e Kotoku N.; Sumii Y.; Hayashi T.; Kobayashi M. Tetrahedron Lett. 2008, 49, 7078. [Google Scholar]
- a Parker K. A.; Iqbal T. J. Org. Chem. 1982, 47, 337. [Google Scholar]; b Wilson S. R.; Jacob L. J. Org. Chem. 1992, 57, 4380. [Google Scholar]; c Wender P. A.; Smith T. E. Tetrahedron 1998, 54, 1255. [Google Scholar]; d Taber D. F.; Song Y. J. Org. Chem. 1996, 61, 7508. [DOI] [PubMed] [Google Scholar]; e Kim W. H.; Lee J. H.; Aussedat B.; Danishefsky S. J. Tetrahedron 2010, 66, 6391. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Kim W. H.; Lee J. H.; Danishefsky S. J. J. Am. Chem. Soc. 2009, 131, 12576. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an intramolecular double Michael type of reaction for trans-hydroindanes, see:; g Ihara M.; Suzuki S.; Taniguchi N.; Fukumoto K.; Kabuto C. J. Chem. Soc., Chem. Commun. 1991, 1168. [Google Scholar]; For Pd-catalyzed reduction of allylic formats, see:; h Mandai T.; Matsumoto T.; Kawada M.; Tsuji J. J. Org. Chem. 1992, 57, 1326. [Google Scholar]
- Greszler S. N.; Reichard H. A.; Micalizio G. C. J. Am. Chem. Soc. 2012, 134, 2766. [DOI] [PMC free article] [PubMed] [Google Scholar]; The enyne coupling partners used are easily prepared in a stepwise fashion from epoxy alcohol derivatives or epichlorohydrin [see the Supporting Information (SI) for details].
- Ryan J.; Micalizio G. C. J. Am. Chem. Soc. 2006, 128, 2764. [DOI] [PubMed] [Google Scholar]
- In our initial study aimed at exploring the mechanism of the Ti-mediated annulation reaction en route to hydroindanes possessing two endocyclic alkenes,5 an annulation related to that in Figure 3 was presented in support of a [4 + 2] mechanism for the annulation (rather than alkene insertion into a metallacyclopentadiene). In that work, quenching with sat. aq. NH4Cl resulted in cis- and trans-fused hydroindanes without stereoselection.
- AM1 calculations (Spartan 2008) predict that the cis-fused isomer 9b is 3.5 kcal/mol lower in energy than the trans-fused isomer 9a.
- a Obora Y.; Moriya H.; Tokunaga M.; Tsuji Y. Chem. Commun. 2003, 2820. [DOI] [PubMed] [Google Scholar]; b Rassadin V. A.; Six Y. Tetrahedron 2014, 70, 787. [Google Scholar]; For the use of n-BuLi en route to Ti–imine complexes, see:; c Tarselli M. A.; Micalizio G. C. Org. Lett. 2009, 11, 4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In addition to the trans- and cis-fused isomers (formed in 64% yield), a small amount of an endocyclic tetrasubstituted alkene annulation product was formed (18%). Thus, the combined yield of all annulation products was 82%. In all of the reported examples, a small amount of this “endo” isomer was also produced; the yields given are only for the combination of the trans- and cis- hydroindanes. See the SI for details.
- This empirical model is proposed only to help understand/predict the selectivity for annulation reactions. We do not intend to imply that stereoselective quenching of this intermediate proceeds in a non-Curtin–Hammett manner.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.