

Abstract
Background
Anaplastic thyroid cancer (ATC) remains refractory to available surgical and medical interventions. Histone deacetylase (HDAC) inhibitors are an emerging targeted therapy with anti-proliferative activity in a variety of thyroid cancer cell lines. Thailandepsin A (TDP-A) is a novel class I HDAC inhibitor whose efficacy remains largely unknown in ATC. Therefore, we aimed to characterize the effect of TDP-A on ATC.
Methods
Human-derived ATC cells were treated with TDP-A. IC50 was determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) rapid colorimetric assay and cell proliferation was measured by viable cell count. Molecular mechanisms of cell growth inhibition were investigated by Western blot analysis of canonical apoptosis markers, intrinsic and extrinsic apoptosis regulators, and cell cycle regulatory proteins. Cell cycle staging was determined with propidium iodide flow cytometry.
Results
TDP-A dose- and time-dependently reduced cell proliferation. Increased cleavage of the apoptosis markers Caspase-9, Caspase-3, and poly ADP ribose polymerase (PARP) were observed with TDP-A treatment. Levels of the intrinsic apoptosis pathway proteins BAD, Bcl-XL, and BAX remained unchanged. Importantly, the extrinsic apoptosis activator cleaved Caspase-8 increased dose-dependently and the anti-apoptotic proteins Survivin and Bcl-2 decreased. Among the cell cycle regulatory proteins, levels of CDK inhibitors p21/WAF1 and p27/KIP increased. Flow cytometry showed that ATC cells were arrested in G2/M phase with diminished S phase following TDP-A treatment.
Conclusion
TDP-A induces a notable dose- and time-dependent anti-proliferative effect on ATC, which is mainly attributed to extrinsic apoptosis with concomitant cell cycle arrest. TDP-A therefore warrants further preclinical and clinical investigations.
Keywords: thyroid cancer, anaplastic, HDAC inhibitor, thailandepsin A, TDP-A, growth inhibition
Introduction
Anaplastic thyroid carcinoma (ATC) is one of the most aggressive human malignancies. ATC is an undifferentiated thyroid cancer that is responsible for over half of all thyroid cancer deaths despite accounting for <2% of thyroid cancer incidence [1,2]. ATC displays highly invasive behavior. Extrathyroidal extension and lymphatic metastasis afflicts 40% of ATC patients while the remaining 60% of patients have distant metastases [3]. The aggressive phenotype and poor prognosis associated with ATC form the basis for its automatic classification as TNM stage IV regardless of tumor burden [4].
Treatment options for ATC are mainly palliative due to the disease's aggression and resiliency. Gross resection is recommended in nearly all cases [5] and thyroidectomy can relieve airway compression, but curative resection is often impossible [6]. Lacking differentiated features [7], ATC is resistant to therapeutic radioactive iodine. Outcomes from radiotherapy and chemotherapy alone have shown limited curative potential [2]. Current recommendations support multimodal interventions that employ adjuvant and neoadjuvant chemoradiation in combination with surgery to improve control of locoregional and metastatic disease [5]. Despite these measures, ATC continues to carry a median survival of less than 6 months [8] and a one year survival rate of less than 20% [4], prompting investigation into novel targeted chemotherapies [9–11].
Histone deacetylases (HDACs) are key epigenetic controllers implicated in oncogenesis since epigenetic dysregulation can produce malignant transformation [12]. HDAC inhibitors target this dysregulated epigenetic control and have therefore garnered interest as antineoplastic agents. HDAC inhibitors are capable of inducing apoptosis, cell cycle arrest, and differentiation in transformed cancers including ATC [13–15].
Thailandepsin A (TDP-A) is a recently identified HDAC inhibitor with promising therapeutic potential. Discovered by genome mining of the Gram-negative bacterium Burkholderia thailandensis E264 [16], TDP-A is a despipeptide class I HDAC inhibitor that has shown marked anti-proliferative activity in a variety of cancer cell lines [16,17]. Importantly, the anti-proliferative activity occurs at low nanomolar concentrations [16,17]. Improved potency and targeted class I activity identify TDP-A as a promising novel chemotherapeutic agent for resistant and aggressive malignancies. However, little is known about the drug efficacy of TDP-A in ATC.
Therefore, we investigated the effects of TDP-A on anaplastic thyroid cancer in vitro. In addition to establishing an IC50 at low nanomolar concentrations, we have shown that TDP-A has a dose- and time-dependent anti-proliferative effect on ATC that is associated with G2/M cell cycle arrest and increased extrinsic apoptosis.
Materials and Methods
Cell Culture and Reagents
The human-derived ATC cell line 8505C used in the current study was kindly provided by Dr. Daniel Ruan (Brigham and Women's Hospital, Boston, MA). It has been well documented as a unique ATC-derived cell line genotyped using a short tandem repeat profiling method [18]. Cells were maintained in RPMI 1640 medium (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St Louis, MO), 100 IU/ml penicillin, and 100μg/ml streptomycin (Invitrogen) and grown in a humidified environment of 5% CO2 at 37°C. TDP-A (chemical structure shown in Figure 1A) was discovered, patented, and provided by the Cheng group [19]. TDP-A was dissolved in dimethylsulfoxide (DMSO, Fischer Scientific, Pittsburg, PA) at a stock concentration of 10μM and stored at 4°C. Fresh dilutions were prepared in media prior to each experiment with the gross amount of DMSO standardized across treatments.
Figure 1. Effect of thailandepsin A (TDP-A) on ATC cell viability.
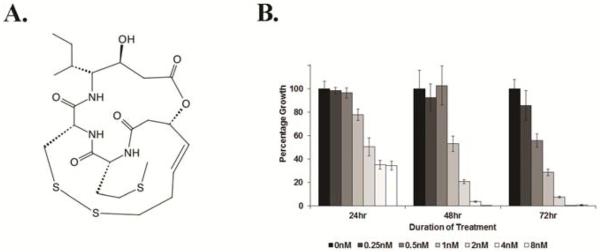
A), Chemical structure of TDP-A. B), TDP-A treatment resulted in dose- and time-dependent reduction of growth in ATC cells. 8505C cells were treated with TDP-A (0-8nM) for up to 72 hours and cell viability was measured by viable cell counts every 24 hours after treatment. Data are presented as mean percentage growth ± standard error, calculated by mean viable cell count normalized to DMSO treated cells (representative of 3 experiments).
Cellular Proliferation Assays
Drug sensitivity of ATC cells growing in a monolayer was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma Aldrich) rapid colorimetric assay as previously described [20]. Briefly, cells were plated in quadruplicate and incubated overnight under standard conditions to allow cell attachment. TDP-A (0–128nM) was added in serial dilutions in fresh medium and the plates were incubated for another 72 hours. Cells treated with vehicle control of DMSO only were regarded as negative controls with 0nM of TDP-A treatment. After the treatment medium was removed, serum-free medium containing 0.5mg/mL MTT was added to each well and incubated for 4 hours at 37°C. MTT formazan crystals were dissolved by adding DMSO in each well and the absorbance was measured at 540nm in a spectrophotometer (μQuant; Bio-Tek Instruments, Winooski, VT).
ATC cell proliferation was measured by viable cell count. An equal amount of 8505C cells were seeded in 10cm petri dishes and incubated overnight to allow cell attachment. Cells were then treated with TDP-A at various concentrations or DMSO vehicle control for a period of 24, 48, or 72 hours. On the day of measurement, cells were collected by trypsinization after washing with PBS. After centrifuging, the cell pellets were re-suspended in equal volumes of medium and stained with Trypan Blue (0.05% solution, Bio-Rad Laboratories, Hercules, CA). Trypan Blue dye exclusion was used to measure cell viability in duplicate per sample using a TC10 Automated Cell Counter (Bio-Rad Laboratories). Each experiment was repeated at least three times.
Western Blot Analysis
Following 48-hour treatment with DMSO or TDP-A, total cellular proteins were isolated as previously described [21], and the protein concentration was quantified by BCA Protein Assay Kit (Thermo Scientific, Waltham, MA) following the manufacturer's instructions. Equal amounts of denatured protein from each sample were resolved by electrophoresis on 4%–15% Criterion TGX precast gels (Bio-Rad Laboratories) and then transferred to nitrocellulose membrane (Bio-Rad Laboratories). After blocking in 5% nonfat milk solution, protein-bound membranes were incubated with the appropriate primary antibodies (all from Cell Signaling Technology, Beverly, MA) overnight at 4°C. Cleaved poly ADP ribose polymerase (PARP), Caspase-3, Caspase-7, Caspase-8, Caspase-9, Bad, Bax, Bcl-2, Survivin, Cyclin D1, Cyclin B1, CDK2, p21/WAF1, acetylhistone H4 and β-actin primary antibodies were diluted 1:1000. Bcl-XL and p27/Kip1 primary antibodies were diluted 1:500. Cyclin A2 and CDK4 primary antibodies were diluted 1:2000. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primary antibody was diluted 1:10000. Membranes were washed the next day and incubated for one hour at room temperature with horseradish peroxidase-conjugated secondary antibodies (1:2000, Cell Signaling Technology). Immunoreactive protein bands were visualized by the detection systems of Immunstar (Bio-Rad Laboratories), SuperSignal West Pico, or SuperSignal West Femto (Pierce Biotechnology, Rockford, IL). Immunoblot analyses were repeated at least twice and the expression levels of β-actin or GAPDH were used as the loading control.
Detection of Cell Cycle by Flow Cytometry Analysis
ATC cells were collected after 48 hours of treatment with TDP-A at selective concentrations or DMSO. After overnight fixation with ice-cold absolute ethanol, cells were washed twice with PBS and incubated in staining solution containing 0.33mg/mL propidium iodide (PI; Sigma Aldrich) and 75μg/mL RNAse (Sigma Aldrich) at 4°C overnight. Samples were then filtered and analyzed by FACSCalibur (Becton Dickinson, San Jose, CA). The percentage of cells in the G0/G1 and G2/M phases was assessed by ModFit LT software (Verity Software House, Topsham, ME).
Statistical Analysis
Statistical Analyses were performed utilizing the Statistical Package for the Social Sciences (SPSS, version 17, IBM SPSS). Unless specifically noted, all data are presented as mean ± standard error. Two-tailed Student's t-test was used to determine statistical significance. A P value<0.05 was considered significant.
Results
TDP-A suppressed ATC cell proliferation in a dose- and time-dependent manner
The IC50 of TDP-A on 8505C cells was 4.45nM (95% CI 3.67 – 5.41), determined by an MTT assay with treatment drug concentrations ranging from 0 to 128nM. TDP-A treatment for up to 72 hours resulted in a dose- and time-dependent reduction in 8505C cell proliferation (Figure 1). Notably, cell growth was reduced by 22%, 47%, and 72% at 24, 48, and 72 hours, respectively, by 1nM TDP-A treatment compared with control. To validate TDP-A as an HDAC inhibitor, 8505C cells were treated with selective doses below IC50 and the levels of histone H4 were measured by Western blot. TDP-A treatment resulted in up-regulation of histone H4 which was acetylated at Lys8 (Figure S1), suggesting that TDP-A inhibited HDAC.
TDP-A caused G2/M cell cycle arrest
Having established a dose- and time-dependent effect of TDP-A on ATC cell viability and proliferation, cell cycle arrest was investigated as a potential mechanism of growth inhibition. Western blot analysis was performed for Cyclin D1, Cyclin A2, and Cyclin B1, cyclin-dependent kinases CDK2 and CDK4, and the CDK/cyclin inhibitors p21/WAF1 and p27/Kip1 following 48 hours of treatment with TDP-A. TDP-A induced a dose-dependent increase in p21/WAF1 and p27/Kip1 (Figure 2). Notably, CDK4, a target of p21/WAF1, decreased dose-dependently (Figure 2), while Cyclin D1 increased with TDP-A treatment. A decrease was observed in Cyclin A2 and Cyclin B1 at 4nM, but no change occurred in protein levels of CDK2.
Figure 2. Effect of TDP-A on cell cycle regulatory proteins.
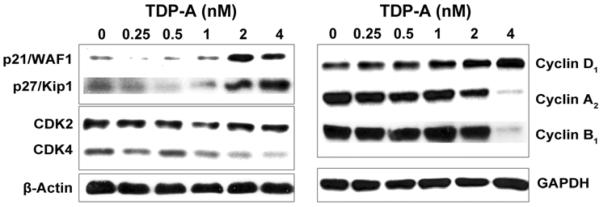
Detection of cell cycle regulatory proteins including p21/WAF1, p27/Kip1 and Cyclins by Western blot in 8505C cells treated with TDP-A or vehicle control (DMSO) for 48 hours. Concentrations of TDP-A are listed across the top. Cells treated with DMSO are defined as negative controls with 0nM of TDP-A treatment. Equal loading was confirmed with β-actin or GAPDH.
To further determine the associated changes in cell cycle distribution, ATC cells were labeled with PI 48 hours after TDP-A treatment for flow cytometry analysis. TDPA treatment induced a dose-dependent decrease in S phase with a concomitant significant increase in G2/M phase (Figure 3). G1/G0 was unaffected despite these changes, suggesting an increase in the proportion of cells in G2/M at the expense of S phase.
Figure 3. Cell cycle arrest in ATC caused by TDP-A treatment.

TDP-A caused G2/M phase arrest in 8505C cells. 8505C cells were treated with selective concentrations of TDP-A for 48 hours, collected and stained with PI for flow cytometry analysis. Cells treated with DMSO are defined as negative controls with 0nM of TDP-A treatment. Data from three repeated experiments were summarized in bar graph as mean percentage of cell population in G0/G1, S, or G2/M phase± standard error. n=3. *p<0.05. ATC cells accumulated in G2/M phase with diminished S phase with TDP-A treatment.
TDP-A induced apoptosis
To investigate the effect of TDP-A on apoptosis, Western blot analysis was performed for apoptotic markers including cleaved Caspase-3, PARP, Caspase-9, and Caspase-7. Treatment with TDP-A resulted in cleavage of Caspase-9, Caspase-3, and PARP (Figure 4).
Figure 4. Apoptosis in ATC induced by TDP-A treatment.
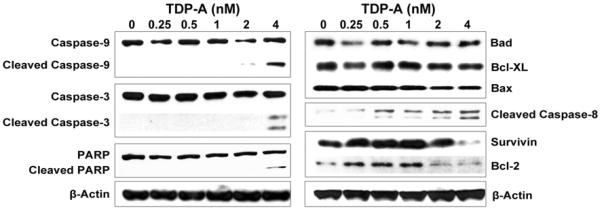
Detection of apoptotic markers including cleaved-PARP, cleaved Caspases, Bcl-2 and Survivin by Western blot in 8505C cells treated with TDP-A or vehicle control (DMSO) for 48 hours. Cells treated with DMSO are defined as negative controls with 0nM of TDP-A treatment. Concentrations of TDP-A are listed across the top. Equal loading was confirmed with β-actin.
Next, the intrinsic and extrinsic apoptosis pathways were both investigated. Intrinsic pathway proteins Bad, Bcl-XL, Bax and extrinsic pathway activator cleaved Caspase-8 were detected by Western blot. Protein levels of Bcl-2 and Survivin, anti-apoptotic proteins with distinct functions common to both intrinsic and extrinsic pathways, were also investigated. A dose-dependent reduction in protein levels of Survivin and Bcl-2 were observed with increasing concentrations of TDP-A treatment. No change was seen in upstream factors in the intrinsic apoptosis pathway (BAD, Bcl-XL, and BAX), but a dose-dependent increase in the 41/43kD remnant of cleaved Caspase-8, a major effector of the extrinsic pathway, was observed (Figure 4). No change was seen in total or cleaved Caspase-7 (data not shown).
Discussion
ATC is a deadly and aggressive cancer, and HDAC inhibitors represent a promising targeted therapy for ATC treatment. In the current study, we investigated a novel HDAC inhibitor with improved potency and class I HDAC selectivity. We found that the novel HDAC inhibitor TDP-A reduced ATC cell viability and cell growth in a dose- and time-dependent manner. Furthermore, we found that growth inhibition was mainly due to cell cycle arrest at low doses and apoptosis with higher concentrations.
Growth suppression was attained at concentrations of 1nM and higher; these concentrations showed significant changes in cell cycle distribution and levels of cell cycle regulators. Importantly, TDP-A increased levels of p21/WAF1 and p27/Kip1. These members of the Cip/Kip family can associate with all CDK/cyclin complexes to inhibit their activity at G1/S and G2/M checkpoints [22] and produce differential G1 and G2/M cell cycle arrest [23]. Accordingly, TDP-A increased p21/WAF1 and p27/Kip1, which correlated with an increase in the proportion of cells in the G2/M phase, suggestive of G2/M arrest. This cytostatic effect can be attributed to the observed decrease in Cyclin B1, which is necessary for successful M phase progression [24]. Cyclin A2 is responsible for S phase exit when conjugated to CDK2 [25] and TDP-A decreased Cyclin A2 without affecting CDK2. The proportion of S-phase cells decreased with TDP-A treatment, suggesting that CDK2 levels were sufficient to allow S phase exit despite reduced Cyclin A2.
Furthermore, TDP-A treatment resulted in a dose-dependent increase in p21/WAF1 and Cyclin D1 levels with a complementary decrease in CDK4. Overexpression of p21/WAF1 has been shown to increase Cyclin D levels and decrease associated CDK activity [23]. Cyclin A2 expression is a target of Cyclin D/CDK4 [24], so the decrease in Cyclin A2 levels may be due to a p21/WAF1-mediated decrease in Cyclin D/CDK4 activity.
TDP-A also induced apoptosis in ATC cells. Apoptosis is mediated via extrinsic or intrinsic pathways and executed by cysteine-aspartic proteases (Caspases). TDP-A treatment resulted in cleavage of the executioner Caspase-3, which is sufficient to induce apoptosis [26]. During apoptosis, Caspase-3 commonly cleaves PARP [27] and concomitant PARP cleavage was observed with Caspase-3 activation induced by TDP-A. Caspase-7 may also cleave PARP [27], but cleavage of Caspase-7 was apparently not induced by TDP-A. However, Caspase-7 is not essential to execute apoptosis [26].
HDAC inhibitors have also been shown to activate the intrinsic and extrinsic apoptotic pathways. In this study, TDP-A activated Caspase-9 cleavage. Caspase-9 is cleaved following intrinsic pathway mitochondrial outer membrane permeablization initiated by Bak and Bax [28]. Mechanisms of HDAC inhibitor-induced apoptosis involve up-regulation of Bax or Bak [29] [30] or reduction in the Bax inhibitor Bcl-XL [31]. However, TDP-A did not change the levels of Bcl-XL or Bax, or the related BH3-only inhibitor of Bcl-XL, Bad. Therefore, increased cleavage of Caspase-9 and Caspase-3 is unlikely to be mediated by a direct effect of TDP-A on Bax-mediated intrinsic apoptosis.
Importantly, TDP-A exposure induced cleavage of Caspase-8, the major effector protein of the extrinsic apoptosis pathway. Cleaved Caspase-8 can directly cleave Caspase-3 or activate the Bax and Bak mediated apoptosis pathway, via Bid [32], to produce Caspase-9 and Caspase-3 cleavage. Although the preferred downstream pathway of cleaved Caspase-8 in ATC is not known, the dose-dependent increase in Caspase-8 cleavage suggests that extrinsic apoptosis is activated during TDP-A induced cell death.
The pro-apoptotic activity of TDP-A is further potentiated by a reduction in the pro-survival proteins Bcl-2 and Survivin. Bcl-2 inhibits Bax/Bak-mediated apoptosis by sequestering pro-apoptotic BH3-only proteins [33] and Survivin inhibits Caspase-3 [34]. Decreasing levels of these inhibitors would increase Caspase-9 and Caspase-3 activity, increasing apoptosis and enhancing the pro-apoptotic effects of Caspase-8. Extrinsic apoptosis with concomitant Caspase-9 activation is thus both activated and potentiated following TDP-A exposure. This pattern of activation is most likely a major mechanism of TDP-A induced apoptosis in ATC.
The effect of TDP-A on apoptosis regulators is of considerable clinical importance. In addition to the potent in vitro efficacy of monoagent TDP-A demonstrated in the current study, TDP-A may be useful in combination with cytotoxic therapies. Previous studies have shown that HDAC inhibitors including valproic acid (VPA), suberoylanilide hydroxamic acid (SAHA), and m-carboxycinnamic acid bishydroxamide sensitize ATC cells to cytotoxic drugs such as doxorubicin [14,15], and this activity has been linked to activation of the extrinsic apoptosis pathway [14]. In this study we have shown that TDP-A activates Caspase-8, the main regulator of the extrinsic apoptosis pathway. Furthermore, Caspase-8 was cleaved at doses sufficient to reduce levels of Bcl-2 and Survivin. Extrinsic pathway activation and inhibitor suppression may render ATC cells more susceptible to additional activation of apoptosis pathways. Therefore, TDP-A may potentiate cytotoxic therapies such as doxorubicin or radiotherapy that promote apoptosis via extrinsic and canonical pathways.
TDP-A has been shown in our study that it inhibits HDAC and up-regulates the expression of acetyl-histone H4. In addition to the effects on histone transcription factors, a lot of HDAC inhibitors are known that they can affect non-histone transcription factors including p53 and tubulin [35,36]. In thyroid cancers, HDAC inhibitors sodium butyrate and trichostatin A have been reported to inhibit the growth of anaplastic thyroid cancer cells due to the promotion of apoptosis and the induction of cell cycle arrest, both of which are independent of p53 status [37]. This is consistent with our observation in the current study as we found that p53 levels remained similar among the cells with and without treatment of TDP-A (data not shown). In one of our previous studies, we demonstrated that Notch1 mediated growth suppression of papillary and follicular thyroid cancer cells by HDAC inhibitors VPA and SAHA [38]. It is possible that TDP-A may also inhibit thyroid cancer growth via the regulation of Notch1 pathway.
In summary, we have demonstrated that TDP-A is effective against ATC at low nanomolar concentrations and reduces cell proliferation in a dose- and time-dependent manner. This activity is characterized by G2/M cell cycle arrest and activation of extrinsic apoptosis with concomitant Caspase-9 activation. Accordingly, TDP-A may be considered for further preclinical and clinical investigations on its effect against ATC as a monotherapy or its synergetic effects with other well-categorized cytotoxic chemo- and radiotherapies for ATC patients.
Supplementary Material
Figure S1. Effect of TDP-A on cell histone acetylation status. Detection of upregulation of histone H4 acetylated at Lys8 by Western blot in 8505C cells treated with various doses of TDP-A for 48 hours. Concentrations of TDP-A are listed across the top. Cells treated with DMSO are defined as negative controls with 0nM of TDP-A treatment. Equal loading was confirmed with β-actin.
Ackowledgements
The authors would like to thank Jon Blake Matsumura and Harpreet Gill for their assistance during data collection, and Maria Georgen for her technical assistance.
Financial Support: American Cancer Society Research Scholar Grant (H. Chen); American Cancer Society MEN2 Thyroid Cancer Professorship (H. Chen); R01 CA121115 (H. Chen); R01 CA152212 (Y-Q. Cheng); and NIH T35 DK062709-08 Surgery Summer Research Experience for Medical Students (E. Weinlander)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement: No competing financial interests exist.
Contribution of Each Author • Conception and design: Yu XM and Chen H
• Development of methodology: Weinlander E, Yu XM, Cheng YQ and Wang C
• Acquisition of data: Weinlander E, Somnay Y, Harrison AH
• Analysis and interpretation of data: Weinlander E, Somnay Y, Harrison AH
• Writing, review, and/or revision of the manuscript: Weinlander E, Harrison AH, Jaskula-Sztul R and Yu XM
• Administrative, technical, or material support: Cheng YQ, Wang C and Chen H
• Study supervision: Yu XM and Chen H
REFERENCES
- 1.Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Database Report on 53,856 Cases of Thyroid Carcinoma Treated in the U.S., 1985–1995. Cancer. 1998;83:2638–48. doi: 10.1002/(sici)1097-0142(19981215)83:12<2638::aid-cncr31>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 2.Are C, Shaha AR. Anaplastic Thyroid Carcinoma: Biology, Pathogenesis, Prognostic Factors, and Treatment Approaches. Annals of Surgical Oncology. 2006;13:453–64. doi: 10.1245/ASO.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Tward J, Shrieve DC, Hitchcock YJ. Surgery and Radiotherapy Improves Survival in Patients With Anaplastic Thyroid Carcinoma. Analysis of the Surveillance, Epidemiology, and End Results 1983–2002. American Journal of Clinical Oncology. 2008;31:460–4. doi: 10.1097/COC.0b013e31816a61f3. [DOI] [PubMed] [Google Scholar]
- 4.O'Neill JP, Shaha AR. Oral Oncology. 2013. Anaplastic Thyroid Cancer. [DOI] [PubMed] [Google Scholar]
- 5.Smallridge RC, Ain KB, Asa SL, et al. American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid. 2012;22 doi: 10.1089/thy.2012.0302. [DOI] [PubMed] [Google Scholar]
- 6.Pinchot SN, Sippel RS, Chen H. Multi-targeted approach in the treatment of thyroid cancer. Therapeutics and Clinical Risk Management. 2008;4:935–47. doi: 10.2147/tcrm.s3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spitzweg C, Harrington KJ, A PL, Vile RG, C MJ. The Sodium Iodide Symporter and Its Potential Role in Cancer Therapy. The Journal of Clinical Endocrinology & Metabolism. 2001;86:3327–35. doi: 10.1210/jcem.86.7.7641. [DOI] [PubMed] [Google Scholar]
- 8.Smallridge RC, Copland JA. Anaplastic Thyroid Carcinoma: Pathogenesis and Emerging Therapies. Clinical Oncology. 2010;22:486–97. doi: 10.1016/j.clon.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deshpande HA, Roman S, Sosa JA. New targeted therapies and other advances in the management of anaplastic thyroid cancer. Current Opinion in Oncology. 2013;25:44–9. doi: 10.1097/CCO.0b013e32835a448c. [DOI] [PubMed] [Google Scholar]
- 10.Yu XM, Phan T, Patel PN, Jaskula-Sztul R, Chen H. Chrysin activates Notch1 signaling and suppresses tumor growth of anaplastic thyroid carcinoma in vitro and in vivo. Cancer. 2013;119:774–81. doi: 10.1002/cncr.27742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu XM, Jaskula-Sztul R, Ahmed K, Harrison AD, Kunnimalaiyaan M, Chen H. Resveratrol induces differentiation markers expression in anaplastic thyroid carcinoma via activation of Notch1 signaling and suppresses cell growth. Mol Cancer Ther. 2013;12:1276–87. doi: 10.1158/1535-7163.MCT-12-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones PA, Baylin SB. The Epigenomics of Cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marks PA, Richon VM, Rifkind RA. Histone Deacetylase Inhibitors: Inducers of Differentiation or Apoptosis of Transformed Cells. Journal of the National Cancer Institute. 2000;92:1210–6. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 14.Mitsiades CS, Poulaki V, McMullan C, et al. Novel Histone Deacetylase Inhibitors in theTreatment of Thyroid Cancer. Clinical Cancer Research. 2005;11 doi: 10.1158/1078-0432.CCR-03-0776. [DOI] [PubMed] [Google Scholar]
- 15.Catalano MG, Fortunati N, Pugliese M, et al. Valproic acid, a histone deacetylase inhibitor, enhances sensitivity to doxorubicin in anaplastic thyroid cancer cells. Journal of Endocrinology. 2006;191:465–72. doi: 10.1677/joe.1.06970. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Henkes LM, Doughty LB, et al. Thailandepsins: Bacterial Products with Potent Histone Deacetylase Inhibitory Activities and Broad-Spectrum Antiproliferative Activities. Journal of Natural Products. 2011;74:2031–8. doi: 10.1021/np200324x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson AJ, Cheng Y-Q, Khabele D. Thailandepsins are new small molecule class I HDAC inhibitors with potent cytotoxic activity in ovarian cancer cells: a preclinical study of epigenetic ovarian cancer therapy. Journal of Ovarian Research. 2012;5 doi: 10.1186/1757-2215-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schweppe RE, Klopper JP, Korch C, et al. Deoxyribonucleic Acid Profiling Analysis of 40 Human Thyroid Cancer Cell Lines Reveals Cross-Contamination Resulting in Cell Line Redundancy and Misidentification. The Journal of Clinical Endocrinology & Metabolism. 2008;93:4331–41. doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C, Henkes LM, Doughty LB, et al. Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities. J Nat Prod. 2011;74:2031–8. doi: 10.1021/np200324x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunnimalaiyann M, Ndiaye M, Chen H. Apoptosis-mediated medullary thyroid cancer growth suppression by the PI3K inhibitor LY294002. Surgery. 2006;140:1009–15. doi: 10.1016/j.surg.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 21.Somnay Y, Simon K, Harrison AD, Kunnimalaiyaan S, Chen H, Kunnimalaiyann M. Neuroendocrine Phenotype Alteration and Growth Suppression Through Apoptosis by MK-2206, an Allosteriv Inhibitor of AKT, in Carcinoid Cell Lines In Vitro. Anticancer Drugs. 2013;24:66–72. doi: 10.1097/CAD.0b013e3283584f75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitrea DM, Yoon M-K, Kriwacki RW. Disorder-function Relationships for the Cell Cycle Regulatory Proteins p21 and p27. The Journal of Biological Chemistry. 2012;393:259–74. doi: 10.1515/hsz-2011-0254. [DOI] [PubMed] [Google Scholar]
- 23.Niculescu AB, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21Cip1/Waf1 at Both the G1/S and the G2/M Cell Cycle Transitions: pRb Is a Critical Determinant in Blocking DNA Replication and in Preventing Endoreduplication. Molecular and Cellular Biology. 1998:629–43. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Proliferation. 2003;36:131–49. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is Required at Two Points in the Human Cell Cycle. The EMBO Journal. 1992;11:961–71. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slee E, Adrain C, Martin SJ. Executioner Caspase-3, -6, and -7 Perform Distinct, Non-redundant Roles during the Demoltion Phase of Apoptosis. The Journal of Biological Chemistry. 2001;276:7320–6. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z-B, Liu Y-Q, Cui Y-F. Pathways to caspase activation. Cell Biology International. 2005;29:489–96. doi: 10.1016/j.cellbi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Wei MC, Zong W-X, Cheng EH-YA, et al. Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang XD, Gillespie SK, Borrow JM, Hersey P. The Histone Deacetylase Inhibitor Suberic Bishydroxamate: a Potential Sensitizer of Melanoma to TNF-related Apoptosis-inducing Ligand (TRAIL) Induced Apoptosis. Biochemical Pharmacology. 2003;66:1537–45. doi: 10.1016/s0006-2952(03)00509-4. [DOI] [PubMed] [Google Scholar]
- 30.Waby JS, Chirakkal H, Yu C, et al. Sp1 acetylation is associated with loss of DNA binding at promoters associated with cell cycle arrest and cell death in a colon cell line. Molecular Cancer. 2010;9 doi: 10.1186/1476-4598-9-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neuzil J, Swettenham E, Gellert N. Sensitization of mesothelioma to TRAIL apoptosis by inhibition of histone deacetylase: role of Bcl-xL down-regulation. Biochemical and Biophysical Research Communications. 2004;314:186–91. doi: 10.1016/j.bbrc.2003.12.074. [DOI] [PubMed] [Google Scholar]
- 32.Cory S, Huang DCS, Adams JM. The Bcl-2 Family: Roles in Cell Survival and Oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 33.Cheng EH-YA, Wei MC, Weiler S, et al. BCL-2, BCL-XL Sequester BH3 Domain-Only Molecules Preventing BAX- and BAK-Mediated Mitochondrial Apoptosis. Molecular Cell. 2001;8:705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 34.Shin S, Sung B-J, Cho Y-S, et al. An Anti-apoptotic Protein Human Survivin Is a Direct Inhibitor of Caspase-3 and -7. Biochemistry. 2001;40:1117–23. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- 35.Sonnemann J, Marx C, Becker S, et al. p53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. Br J Cancer. 2014;110:656–67. doi: 10.1038/bjc.2013.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao B, Li J, Zhu J, et al. The antiparasitic clioquinol induces apoptosis in leukemia and myeloma cells by inhibiting histone deacetylase activity. J Biol Chem. 2013;288:34181–9. doi: 10.1074/jbc.M113.472563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenberg VL, Williams JM, Cogswell JP, Mendenhall M, Zimmer SG. Histone deacetylase inhibitors promote apoptosis and differential cell cycle arrest in anaplastic thyroid cancer cells. Thyroid. 2001;11:315–25. doi: 10.1089/10507250152039046. [DOI] [PubMed] [Google Scholar]
- 38.Xiao X, Ning L, Chen H. Notch1 mediates growth suppression of papillary and follicular thyroid cancer cells by histone deacetylase inhibitors. Mol Cancer Ther. 2009;8:350–6. doi: 10.1158/1535-7163.MCT-08-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of TDP-A on cell histone acetylation status. Detection of upregulation of histone H4 acetylated at Lys8 by Western blot in 8505C cells treated with various doses of TDP-A for 48 hours. Concentrations of TDP-A are listed across the top. Cells treated with DMSO are defined as negative controls with 0nM of TDP-A treatment. Equal loading was confirmed with β-actin.